Introduction
Hemoglobinopathies, including sickle cell anemia and
β-thalassemia, are highly prevalent monogenic gene disorders in
Saudi Arabia (1–10). β-thalassemia is caused by point
sequence variations or large sequence deletions, that are heritable
and either prevent the synthesis of the β-globin chain completely
(β0 variants) or alters the function of β-globin chain
(β+ variants) (1–10).
The phenotype of β-thalassemia in the Saudi population is highly
varied, ranging from asymptomatic to severe transfusion-dependent
anemia (1,4,9,11).
Furthermore, it has been previously demonstrated that the α-globin
gene deletions, gene conversion [hemoglobin-α 12 (HBA12)] and point
mutations, are also highly prevalent in the Saudi population,
particularly in the densely populated regions of Qatif and Al-Ahssa
in the Eastern Province (13.41% of sickle cell disease carriers;
5.9% β-thalassemia carriers) of Saudi Arabia (1–6,9,10,12,13).
Previous studies have investigated the prevalence of the α-globin
gene deletion in Arab populations, however, very little has been
determined with regards to the prevalence of the α-globin gene
deletion in Saudi patients with thalassemia (14–20).
It has previously been revealed that the coinheritance of
α-thalassemia in patients with heterozygous β-thalassemia results
in an increase of the mean corpuscular volume (MCV) and mean
corpuscular hemoglobin (MCH) blood concentration, however, carriers
of β-thalassemia mutations have an increased blood concentration of
hemoglobin A2 (HbA2) (21). However, not all carriers of
β-thalassemia mutations exhibit this phenotype, and some may in
fact exhibit normal HbA2 blood concentrations (22), which has previously been attributed
to a number of factors, including α-globin gene deletions (23). A pre-marriage screening for
β-thalassemia mutations in these regions depends on the
determination of HbA2 blood concentrations (24,25).
The most common α globin gene deletion is the 3.7 kb rightward
deletion (−α3.7), which is caused by the breakage of DNA
molecules in the α globin genes (HBA2 and HBA1) region and
rejoining of the broken ends by leaving α globin genes region with
single functional gene. The present study aimed to determine the
effect of α-globin deletion on fetal hemoglobin (HbF) and
HbA2 blood concentrations in Saudi populations.
Materials and methods
Patient enrollment
A total of 503 Saudi individuals (Age 12.98±11.08;
80 female and 86 male patients with transfusion-dependent
β-thalassemia, and 133 female and 204 male healthy patients)
attending major hospitals in the Eastern Province of Saudi Arabia
were included in the present study. The study was performed over a
5-year period between February 2012 and February 2017. The study
was approved by the University of Dammam Institutional Review Board
and Committee for Biological and Medical Ethics (CBME2012032;
IRB-2013-08-030; Dammam, Saudi Arabia).
Determination of hematological
parameters
Following receipt of informed consent from all
participants, blood samples (5 ml) were collected in EDTA-coated
vacutainers. VARIANT™ II Hemoglobin Testing System
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and Coulter Micro
Diff II (Beckman Coulter, Inc., Brea, CA, USA) were used in order
to measure all the hematological parameters.
DNA extraction and polymerase chain
reaction (PCR)
DNA was extracted (QIAamp DNA blood mini kit; Qiagen
GmbH, Hilden, Germany) from the blood samples, and the
−α3.7, −α4.2, −−FIL,
−−SEA, −−MED and −−(20.5) gene
deletions were identified using multiplex α-globin deletion PCR as
described previously (26,27). Samples positive for the
−α3.7 deletion were subjected to amplification of the
region around the deletion using primers according to methods
previously described (28).
Forward and reverse primers were used separately for PCR using the
BigDye Terminator Cycle Sequencing Kit (Thermo Fisher Scientific,
Inc., Waltham, MA, USA), and then purified and electrophoresed
using the Series Genetic Analyzer 3500 (Thermo Fisher Scientific,
Inc.). From the total number of samples, PCR analysis revealed that
5% of the samples were positive for the −α3.7 deletion,
and this was confirmed by Sanger sequencing at the Department of
Genetic Research, Institute for Research and Medical Consultation,
Imam Abdulrahman Bin Faisal University (Dammam, Saudi Arabia).
Hemoglobin subunit α1 (HBA1) and hemoglobin subunit α2 (HBA2) genes
were also sequenced as previously described (10). Electropherograms were analyzed
using DNA sequencing analysis software v5.3 (Applied Biosystems;
Thermo Fisher Scientific, Inc.). A multiple alignment program
(MAFFT, v.7; https://mafft.cbrc.jp/alignment/server/) was used for
HBA1, HBA2 and 3.7 fusion gene sequence alignment. Patients were
classed into four groups [αα/αα, −α3.7/αα,
−α3.7/−α3.7 and -−/−α3.7 where
(−−, indicates the SEA, MED or FIL deletion)] depending on the
presence of their −α3.7 deletion genotypes (Fig. 1).
Statistical analysis
Statistical analyses between two groups were
performed using the Student's t test (SPSS statistical package
version 19; IBM, Corp, Armonk, NY, SA). The data are presented as
the mean ± standard deviation. Analysis of variance (ANOVA)
combined with post hoc Tukey test, Bonferroni and Holm multiple
comparison tests; were performed in order to demonstrate
statistically significant differences between multiple groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
The results of the present study demonstrated that
the −α3.7 gene deletion is the most prevalent (43.5%)
among the population of the Eastern province of Saudi Arabia. The
second most prevalent gene deletion was the HBA2:
c.95+2_95+6het_delTGAGG (α2HphI) deletion,
with a prevalence score of 24.3%. The −α3.7 gene
deletion is characterized by the deletion of 3,804 base pairs
(Fig. 2). Further α-globin gene
mutations revealed to be present in the tested population were:
α1–4.2 (1.78%),
α1polyA-1α2 1.78% and double gene deletions
were at a prevalence rate of 1.39% for
−α2(20.5) and <1% for −-FIL and
−-MED. The prevalence of the recently identified α12
(HBA12) allele was demonstrated to be 3.78% in the investigated
population. A number of genotypes, namely
−3.7α2/α1α2,
−3.7α2/α1α12,
−3.7α2/−3.7α2,
−3.7α2HphI/α1α2HphI,
−3.7α2/ α1- 4.2,
−3.7α2/α1polyA-1α2,
−3.7α12/α1α12,
−−FIL/−3.7α2 and
−3.7α2/−3.7α2Hb
Villiers le Bel were observed in the population. In addition,
>10% of the total population carried other types of α-gene
deletion, namely
−α2(20.5)/α1α2,
−−MED/α1α2HphI,
α1α2init/α1polyA-1α2,
α1–4.2/α1α1,
−−MED/α1α2,
−−FIL/α1α2,
−−FIL/α1polyA-1α2,
α1α2/α1α2HphI,
α1α2HphI/α1α2HphI,
α1α2/α1α2Hb
Handsworth and
α1polyA-1α2/α1α2.
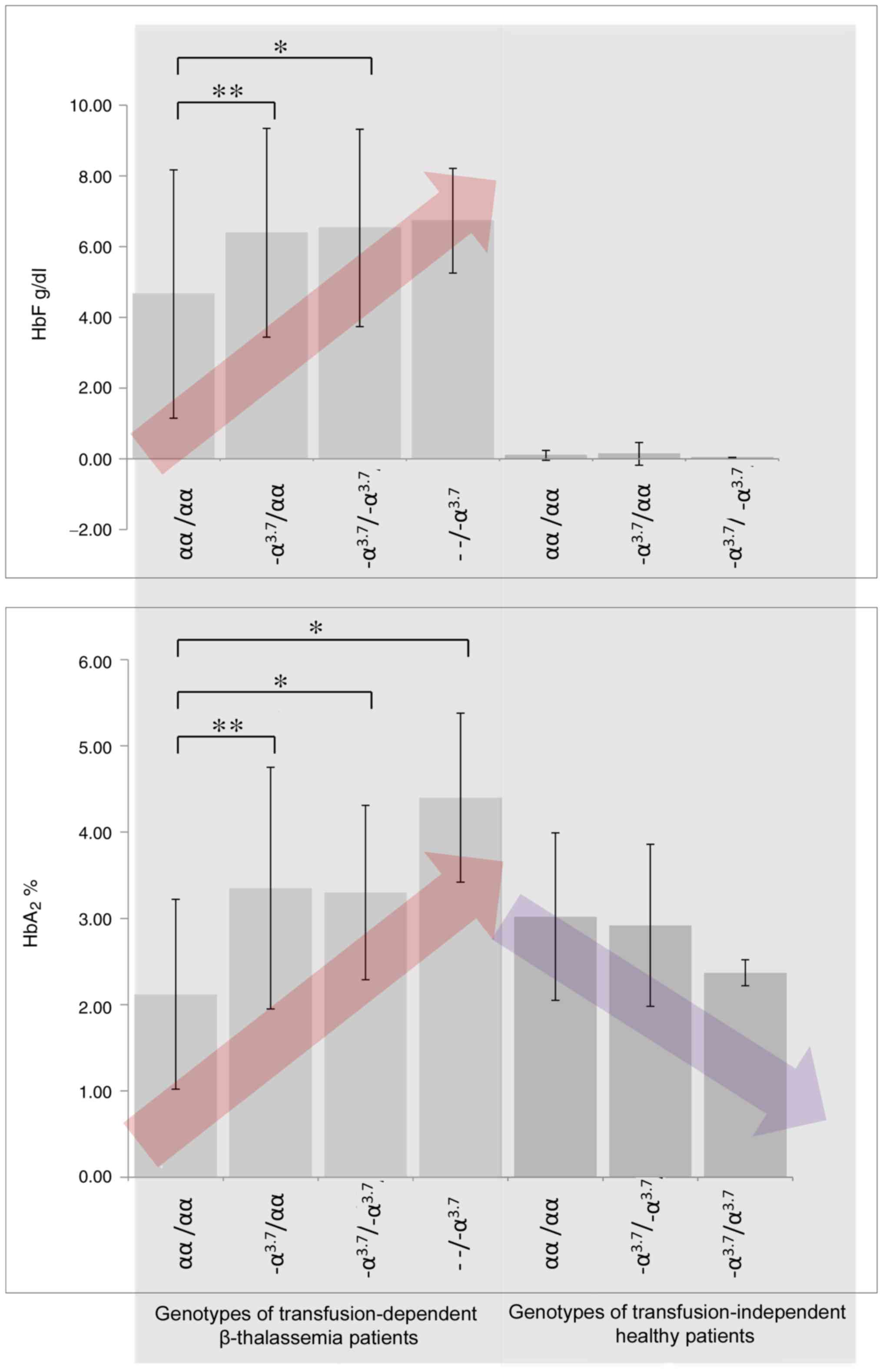
The concentrations of HbF and HbA2 in the
blood are presented in Table I and
Fig. 1. A gradual increase in the
levels of both HbF and HbA2 in the β thalassemia patient
groups was demonstrated as the number of α-gene deletions increased
(Fig. 1 and Table II; P<0.05). However, in healthy
patients, the concentration of HbA2 was revealed to
decrease as the number of gene deletions increased. The post-hoc
Tukey, Bonferroni and Holm multiple comparison tests revealed
significant differences with regards to the blood concentration of
HbF (f-ratio=3.42806; P=0.020334 for ANOVA of all data groups) and
HbA2 (f-ratio=9.72308; P=0.000012 for ANOVA of all data
groups; Table III), in different
patient groups.
 | Table I.Genotypes of −α3.7 deletion and blood
concentration of HbF and HbA2. |
Table I.
Genotypes of −α3.7 deletion and blood
concentration of HbF and HbA2.
| Genotype | Number of
patients | HbF (g/dl) | HbA2 (%) |
|---|
| Patients with
transfusion-dependent β-thalassemia |
|
|
|
|
αα/αα | 63 | 4.66±3.51 | 2.12±1.1 |
|
−α3.7/αα | 55 | 6.39±2.95 | 3.35±1.4 |
|
−α3.7/−α3.7 | 11 | 6.53±2.79 | 3.3±1.01 |
|
−−/−α3.7 | 3 | 6.73±1.48 | 4.4±0.98 |
| Healthy
patients |
|
|
|
|
αα/αα | 162 | 0.1±0.14 | 3.02±0.97 |
|
−α3.7/αα | 142 | 0.14±0.32 | 2.92±0.94 |
|
−α3.7/−α3.7 | 4 | 0.04±0.006 | 2.37±0.15 |
|
−−/−α3.7 | 0 | − | − |
| Table II.Significance of −α3.7
deletion on blood concentrations of HbF and HbA2. |
Table II.
Significance of −α3.7
deletion on blood concentrations of HbF and HbA2.
| Comparison between
associated groups |
|
|
|
|---|
|
|
|
|
|---|
| Group 1 | Group 2 | Number of
patients | HbF, P-value | HbA2,
P-value |
|---|
| −α3.7/αα
transfused | αα/αα
transfused | 55 vs. 63 |
0.007609a |
<0.00001b |
|
−α3.7/−α3.7 or
−α3.7/−α4.2 transfused | αα/αα
transfused | 11 vs. 63 |
0.048726a |
0.001384a |
| −−/−α3.7
transfused | αα/αα
transfused | 3 vs. 63 | 0.207171 |
0.001937a |
| −α3.7/αα
healthy patients | αα/αα healthy
patients | 144 vs. 162 | 0.110116 | 0.211731 |
|
−α3.7/−α3.7 or
−α3.7/−α4.2 healthy patients | αα/αα healthy
patients | 4 vs. 162 | 0.226603 | 0.071323 |
| Table III.Statistical analyses using multiple
comparison tests. |
Table III.
Statistical analyses using multiple
comparison tests.
| Treatment pair | Bonferroni and Holm
TT-statistic | Bonferroni
P-value | Bonferroni
inference | Holm P-value | Holm inference |
|---|
| HbF |
|
|
|
|
|
| A vs.
B | 2.5587 | 0.0364182 | Not
significant | 0.0364182 | P<0.05 |
| A vs.
C | 2.4523 | 0.0482490 | Not
significant | 0.0321660 | P<0.05 |
| A vs.
D | 0.9074 | 1.0996299 | Not
significant | 0.3665433 | Not
significant |
|
HbA2 |
|
|
|
|
|
| A vs.
B | 4.8279 | 0.0000156 | P<0.01 | 0.0000156 | P<0.01 |
| A vs.
C | 2.7093 | 0.0239577 | P<0.05 | 0.0159718 | P<0.05 |
| A vs.
D | 2.6306 | 0.0297893 | Not
significant | 0.0099298 | P<0.01 |
Discussion
The high prevalence of these disorders has
previously been attributed to the high endemicity of malaria in
affected areas (29).
β-thalassemia disorders represent a group of heterogeneous
hemoglobin disorders, characterized by either the absence or
reduced synthesis of the β-globin chain. Such disorders can be
classified into three groups according to the severity of their
associated clinical representation: β-thalassemia carrier (low
severity), thalassemia intermedia (moderate severity) and
thalassemia major (high severity). An excess of α-globin chain
production, which aggregates in red blood cell precursors forming
inclusion bodies, characterizes thalassemia major. This results in
the destruction of red blood cells in the bone marrow and
ineffective erythropoiesis, which results in the development of
anemia as well as intense proliferation and expansion of the bone
marrow (30).
The phenotypic variation exhibited by patients with
β-thalassemia is due to the heterogeneity in genetic mutations
associated with the disease. However, the phenotype can also be
modified by other genetic factors. In addition to the influence of
HbF concentration on the β-thalassemia phenotype, deletions of
α-globin genes have also been demonstrated as having a significant
effect on patient phenotype (20,21).
Furthermore, it has been revealed that the
coinheritance of α-thalassemia in individuals heterozygous for
β-thalassemia results in elevated levels of MCV and MCH (31). This most frequently occurs when two
α-globin genes are silenced due to gene deletion or another type of
mutation (32). These individuals
also exhibit an increased concentration of HbA2: A
characteristic that is exploited for the identification of
β-thalassemia carrier state.
Previously, we have demonstrated that the α-globin
gene deletion is highly prevalent in the populations of the
Al-Qatif and Al-Ahssa regions (4,9,10).
Furthermore, it has previously been demonstrated that the
association of the α-gene deletion with β-thalassemia and sickle
cell anemia leads to amelioration of disease severity (33).
The results of the present study suggest that the
frequency of α-gene deletions is increased in both normal and
β-thalassemia populations in the Eastern province compared with
other provinces, which is in agreement with the results of previous
studies, including reports on African populations (34). Furthermore, previous studies have
demonstrated that the prevalence of the α-gene deletion in Arab
populations is varied, ranging from 28% in Kuwait and the United
Arab Emirates to as high as 75% in Lebanon (14–20,35).
This difference in the reported prevalence frequencies of α-globin
gene deletions may be attributed to variation in the sample size as
well as the inclusion criteria of different studies. Genome-wide
association studies have revealed that the α-globin gene deletions
have a significant effect on the blood concentrations of HbA and
HbA2, whereas it has no significant effect on the
concentration of HbF (36). The
results of the present study suggest that there is an association
between the −α3.7 deletion and elevated concentrations
of HbA2, and is therefore in agreement with the
aforementioned study. In addition, the β-thalassemia mutations were
demonstrated as being associated with an increased concentration of
HbF (36–38). Therefore, it can be concluded that
the elevated HbF concentration in the present study is
predominantly associated with β-thalassemia mutations as opposed to
α-gene deletions. Furthermore, the results of the present study
also revealed new α-gene deletion genotypes prevalent in the
studied populations, namely:
α1α2/α1α2HphI,
α1α2HphI/α1α2HphI,
α1α2/α1α2Hb
Handsworth,
−3.7α2HphI/α1α2HphI,
−3.7α2/−3.7α2Hb
Villiers le Bel and
−−MED/α1α2HphI, which,
to the best of our knowledge, have not previously been
reported.
Acknowledgements
The present study was supported by The Deanship of
Scientific Research, University of Dammam (grant nos. 2012186,
2014024 and 2014051), the King Abdulaziz City for Science and
Technology (grant nos. LGP-35-204, LGP-32-3 and LGP-36-132) and the
National Science Technology and Innovation Plan (grant no.
12-MED-2798-46). The authors would like to extend their
appreciation to Mr. Tumbaga, Mr. Pacifico, Ms. Aquino, Mr.
Evangelista, Ms. Charmine and Mr. Al-Shamlan (Institute for
Research and Medical Consultation, Imam Abdulrahman Bin Faisal
University, Dammam, Saudi Arabia) for their technical support.
References
|
1
|
el-Hazmi MA: Alpha-thalassemia in Saudi
Arabia: Deletion pattern. Hum Genet. 76:196–198. 1987. View Article : Google Scholar
|
|
2
|
Nasserullah Z, Al Jame A, Srair Abu H, Al
Qatari G, Al Naim S, Al Aqib A and Mokhtar M: Neonatal screening
for sickle cell disease, glucose-6-phosphate dehydrogenase
deficiency and a-thalassemia in Qatif and Al Hasa. Ann Saudi Med.
18:289–292. 1998. View Article : Google Scholar
|
|
3
|
Al-Jaouni SK: Prevalence of thalassemia
disorders and hemoglobinopathies in Jeddah, Western Saudi Arabia. J
Appl Hematol. 1:43–46. 2010.
|
|
4
|
Akhtar MS, Qaw F, Borgio JF, Albuali W,
Suliman A, Nasserullah Z, Al-Jarrash S and Al-Ali A: Spectrum of
α-thalassemia mutations in transfusion-dependent β-thalassemia
patients from the Eastern Province of Saudi Arabia. Hemoglobin.
37:65–73. 2013. View Article : Google Scholar
|
|
5
|
Hamamy HA and Al-Allawi NA:
Epidemiological profile of common haemoglobinopathies in Arab
countries. J Community Genet. 4:147–167. 2013. View Article : Google Scholar
|
|
6
|
Alsultan A, Alabdulaali MK, Griffin PJ,
Alsuliman AM, Ghabbour HA, Sebastiani P, Albuali WH, Al-Ali AK,
Chui DH and Steinberg MH: Sickle cell disease in Saudi Arabia: The
phenotype in adults with the Arab-Indian haplotype is not benign.
Br J Haematol. 164:597–604. 2014. View Article : Google Scholar
|
|
7
|
Borgio JF, AbdulAzeez S, Naserullah ZA,
Al- S, Al-Ali RA, Al-Madan MS, Al-Muhanna F, Al-Suliman AM,
Al-Nafie A, Steinberg MH and Al-Ali AK: Mutations in the β-globin
gene from a Saudi population: An update. Int J Lab Hematol.
38:e38–e40. 2016. View Article : Google Scholar
|
|
8
|
Al-Nafie AN, Borgio JF, AbdulAzeez S,
Al-Suliman AM, Qaw FS, Naserullah ZA, Al-Jarrash S, Al-Madan MS,
Al-Ali RA, AlKhalifah MA, et al: Co-inheritance of novel ATRX gene
mutation and globin (α & β) gene mutations in transfusion
dependent beta-thalassemia patients. Blood Cells Mol Dis. 55:27–29.
2015. View Article : Google Scholar
|
|
9
|
Borgio JF: Molecular nature of
alpha-globin genes in the Saudi population. Saudi Med J.
36:1271–1276. 2015. View Article : Google Scholar :
|
|
10
|
Borgio JF, AbdulAzeez S, Al-Nafie AN,
Naserullah ZA, Al-Jarrash S, Al-Madan MS, Al-Muhanna F, Steinberg
MH and Al-Ali AK: A novel HBA2 gene conversion in cis or trans:
‘α12 allele’ in a Saudi population. Blood Cells Mol Dis.
53:199–203. 2014. View Article : Google Scholar
|
|
11
|
Harteveld CL and Higgs DR:
Alpha-thalassaemia. Orphanet J Rare Dis. 5:132010. View Article : Google Scholar :
|
|
12
|
Nasserullah Z, Alshammari A, Abbas MA,
Abu-Khamseen Y, Qadri M, Jafer SA and Wabel MA: Regional experience
with newborn screening for sickle cell disease, other
hemoglobinopathies and G6PD deficiency. Ann Saudi Med. 23:354–357.
2003. View Article : Google Scholar
|
|
13
|
AbdulAzeez S and Borgio JF: In-silico
computing of the most deleterious nsSNPs in HBA1 gene. PLoS One.
11:e01477022016. View Article : Google Scholar :
|
|
14
|
Adekile A and Haider M: Morbidity, beta S
haplotype and alpha-globin gene patterns among sickle cell anemia
patients in Kuwait. Acta Haematol. 96:150–154. 1996. View Article : Google Scholar
|
|
15
|
Neyshabouri M, Abbasi-Moheb L, Kahrizi K,
Keyhany E, Najmabad H, Elah Pourfath AA, Krugluger W and Oberkanins
CH: Alpha-thalassemia:deletion analysis in Iran. Arch Iranian Med.
4:160–164. 2001.
|
|
16
|
Baysal E: α-Thalassemia syndromes in the
United Arab Emirates. Hemoglobin. 35:574–580. 2011. View Article : Google Scholar
|
|
17
|
Gilad O, Dgany O, Noy-Lotan S, Krasnov T,
Elitzur S, Pissard S, Kventsel I, Yacobovich J and Tamary H:
Characterization of two unique α-globin gene cluster deletions
causing α-thalassemia in Israeli Arabs. Hemoglobin. 38:319–324.
2014. View Article : Google Scholar
|
|
18
|
Farra C, Badra R, Fares F, Muwakkit S,
Dbaibo G, Dabbous I, Ashkar H, Mounsef C and Abboud MR: Alpha
thalassemia allelic frequency in Lebanon. Pediatr Blood Cancer.
62:120–122. 2015. View Article : Google Scholar
|
|
19
|
Farra C, Daher R, Badra R, el Rafei R,
Bejjany R, Charafeddine L and Yunis K: Incidence of alpha-globin
gene defect in the lebanese population: A pilot study. Biomed Res
Int. 2015:5176792015. View Article : Google Scholar :
|
|
20
|
Miri-Moghaddam E, Bahrami S, Naderi M,
Bazi A and Karimipoor M: Molecular characterization of
β-thalassemia intermedia in southeast Iran. Hemoglobin. 40:173–178.
2016. View Article : Google Scholar
|
|
21
|
Cao A and Galanello R: Beta-thalassemia.
Genet Med. 12:61–76. 2010. View Article : Google Scholar
|
|
22
|
Giambona A, Passarello C, Vinciguerra M,
Li Muli R, Teresi P, Anzà M, Ruggeri G, Renda D and Maggio A:
Significance of borderline hemoglobin A2 values in an Italian
population with a high prevalence of beta-thalassemia.
Haematologica. 93:1380–1384. 2008. View Article : Google Scholar
|
|
23
|
Perseu L, Satta S, Moi P, Demartis FR,
Manunza L, Sollaino MC, Barella S, Cao A and Galanello R: KLF1 gene
mutations cause borderline HbA(2). Blood. 118:4454–4458. 2011.
View Article : Google Scholar
|
|
24
|
AlHamdan NA, AlMazrou YY, AlSwaidi FM and
Choudhry AJ: Premarital screening for thalassemia and sickle cell
disease in Saudi Arabia. Genet Med. 9:372–377. 2007. View Article : Google Scholar
|
|
25
|
AL-Shahrani M: Steps toward the prevention
of hemoglobinopathies in the kingdom of Saudi Arabia. Hemoglobin.
33 Suppl 1:S21–S24. 2009. View Article : Google Scholar
|
|
26
|
Liu YT, Old JM, Miles K, Fisher CA,
Weatherall DJ and Clegg JB: Rapid detection of alpha-thalassaemia
deletions and alpha-globin gene triplication by multiplex
polymerase chain reactions. Br J Haematol. 108:295–299. 2000.
View Article : Google Scholar
|
|
27
|
Bragós IM, Noguera NI, Raviola MP and
Milani AC: Triplication (/alphaalphaalpha anti3.7) or deletion
(-alpha3.7/) association in Argentinian beta-thalassemic carriers.
Ann Hematol. 82:696–698. 2003. View Article : Google Scholar
|
|
28
|
Chow A, Ghassemifar R and Finlayson J:
Alpha thalassaemia due to non-deletional mutations on the-3.7 alpha
globin fusion gene: Laboratory diagnosis and clinical importance.
Pathology. 45:591–594. 2013. View Article : Google Scholar
|
|
29
|
Malaria Genomic Epidemiology Network;
Malaria Genomic Epidemiology Network: Reappraisal of known malaria
resistance loci in a large multicenter study. Nat Genet.
46:1197–1204. 2014. View Article : Google Scholar :
|
|
30
|
Chao YH, Peng CT, Harn HJ, Chan CK and Wu
KH: Poor potential of proliferation and differentiation in bone
marrow mesenchymal stem cells derived from children with severe
aplastic anemia. Ann Hematol. 89:715–723. 2010. View Article : Google Scholar
|
|
31
|
Sin S, Ghosh A, Tang LC and Chan V: Ten
years' experience of antenatal mean corpuscular volume screening
and prenatal diagnosis for thalassaemias in Hong Kong. J Obstet
Gynaecol Res. 26:203–208. 2000. View Article : Google Scholar
|
|
32
|
Al-Awamy BH: Thalassemia syndromes in
Saudi Arabia. Meta-analysis of local studies. Saudi Med J. 21:8–17.
2000.
|
|
33
|
Hassan SM, Al Muslahi M, Al Riyami M,
Bakker E, Harteveld CL and Giordano PC: Sickle cell anemia and
α-thalassemia: A modulating factor in homozygous HbS/S patients in
Oman. Eur J Med Genet. 57:603–606. 2014. View Article : Google Scholar
|
|
34
|
Mouélé R, Pambou O, Feingold J and
Galactéros F: Alpha-thalassemia in Bantu population from
Congo-Brazzaville: Its interaction with sickle cell anemia. Hum
Hered. 50:118–125. 2000. View Article : Google Scholar
|
|
35
|
Al-Allawi NA, Jalal SD, Rasheed NS, Bayat
N, Imanian H, Najmabadi H and Faraj A: The spectrum of
α-thalassemia mutations in the Kurdish population of Northeastern
Iraq. Hemoglobin. 37:56–64. 2013. View Article : Google Scholar
|
|
36
|
Danjou F, Zoledziewska M, Sidore C, Steri
M, Busonero F, Maschio A, Mulas A, Perseu L, Barella S, Porcu E, et
al: Genome-wide association analyses based on whole-genome
sequencing in Sardinia provide insights into regulation of
hemoglobin levels. Nat Genet. 47:1264–1271. 2015. View Article : Google Scholar :
|
|
37
|
Lim WF, Muniandi L, George E, Sathar J,
Teh LK and Lai MI: HbF in HbE/β-thalassemia: A clinical and
laboratory correlation. Hematology. 20:349–353. 2015. View Article : Google Scholar
|
|
38
|
Sripichai O and Fucharoen S: Fetal
hemoglobin regulation in β-thalassemia: Heterogeneity, modifiers
and therapeutic approaches. Expert Rev Hematol. 9:1129–1137. 2016.
View Article : Google Scholar
|