Introduction
Pancreatic cancer (PC), which arises when pancreatic
cells proliferate abnormally, is characterized by yellow skin,
weight loss, back or abdominal pain, dark urine, light-colored
stools, and appetite loss (1). The
early symptoms of PC are not obvious, thus newly diagnosed PC cases
have usually reached an advanced stage (2). PC is primarily induced by tobacco
smoking, diabetes, obesity and genetic conditions (3,4).
Globally, PC is the seventh leading cause of cancer-associated
mortality, resulting in 330,000 fatalities in 2012 (5). The prognosis of PC is usually very
poor, with 5% of people surviving for five years and 25% surviving
just one year after diagnosis (5,6).
Pancreatic adenocarcinoma is the most common type of PC and
accounts for 85% of all PC cases (3). Almost all cases of pancreatic
adenocarcinoma originate from the ducts of the pancreas, and are
termed pancreatic ductal adenocarcinoma (PDAC) (7). Therefore, revealing the underlying
mechanisms of PDAC is significant for developing novel
treatments.
Yamazaki et al (8) demonstrated that SMAD family member 3
contributes to the malignant potential of PDAC by inducing
epithelial-mesenchymal transition (EMT) in tumor cells and thus
presents as a promising biomarker of poor prognosis (8). Masugi et al (9) identified that integrin β4 functions
in regulating EMT and cancer invasion, and its overexpression has
clinicopathological and prognostic significance in PDAC (9). Sex determining region Y-box 9
(SOX9) and phosphorylated-v-Akt murine thymoma viral
oncogene homolog 1 (p-AKT) are reported to be associated
with proliferation and distant metastasis, indicating that
SOX9 and p-AKT may potentially predict the prognosis
of PDAC (10). The downregulation
of anterior gradient 2 is induced by EMT and serves as a novel
prognostic marker in patients with PDAC (11). In PDAC patients who undergo
pancreaticoduodenectomy, high expression levels of transforming
growth factor β-1 may inhibit the poorer survival that is
associated with high proliferation (12). However, the genes involved in the
prognosis of PDAC have not been fully reported.
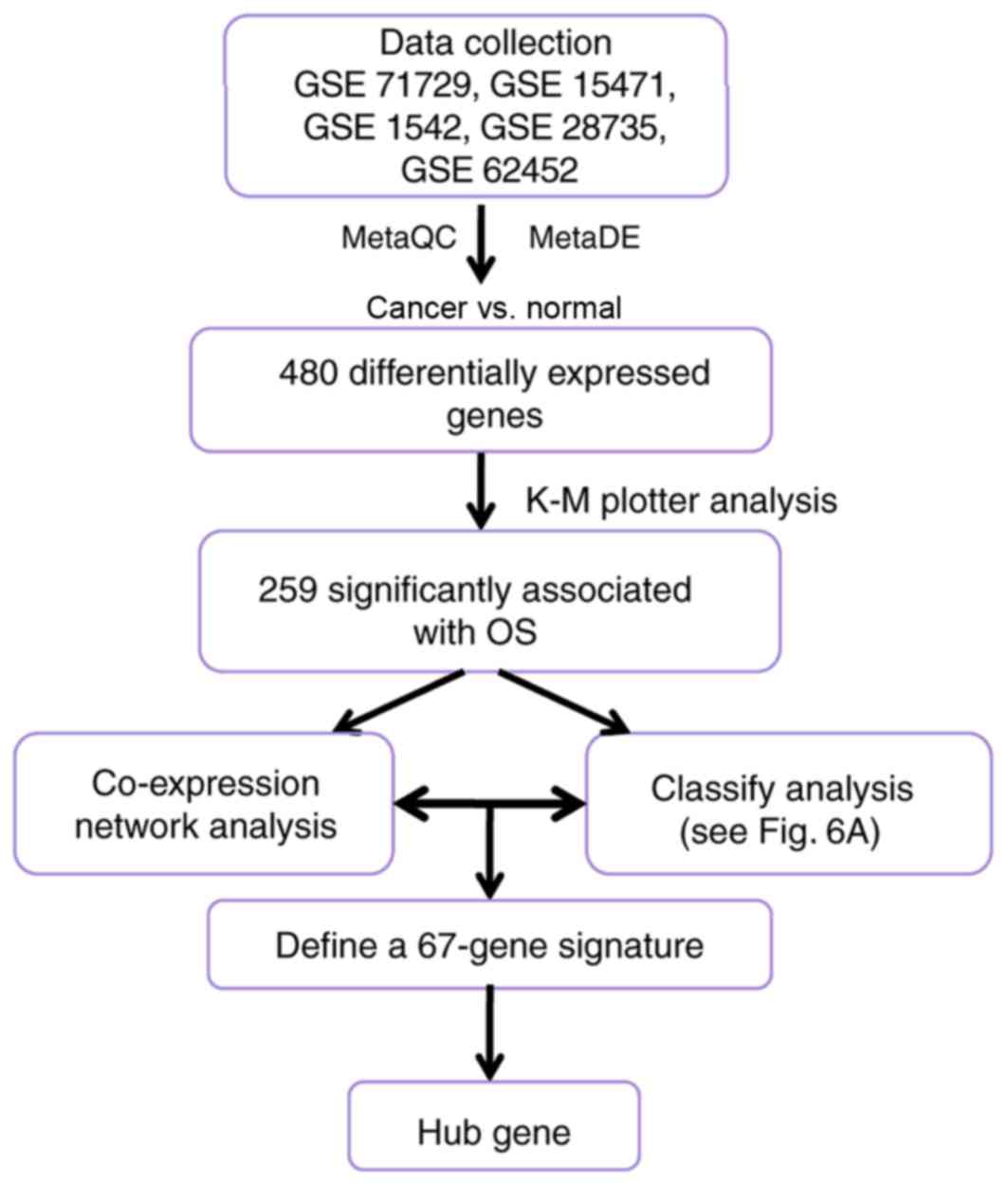
In the current study, public databases were searched
for microarray datasets associated with PDAC in homo sapiens.
Subsequently, comprehensive bioinformatics analyses [including
identification of differentially expressed genes (DEGs) and
prognosis-associated genes, co-expression network and module
analyses, construction and validation of prognostic prediction
system, and enrichment analysis] were performed successively to
investigate the key prognosis-associated genes in PDAC. The current
study may contribute to developing targeted therapeutic strategies
for improving the prognosis of PDAC.
Data and methods
Expression profile data
Using ‘Pancreatic ductal adenocarcinoma’ and ‘homo
sapiens’ as keywords, the gene expression profiles in the Gene
Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) database were
searched. The datasets that met the following criteria were
included: i) The dataset was associated with PDAC; ii) the dataset
included PDAC tissue samples and normal control samples. Finally, a
total of five datasets were included in the current study,
including GSE71729 (based on the GPL20769 platform, including 145
PDAC samples and 46 normal samples), GSE15471 (based on the GPL570
platform, including 39 PDAC samples and 39 normal samples), GSE1542
(based on the GPL96 platform, including 24 PDAC samples and 25
normal samples), GSE28735 (based on the GPL9644 platform, including
45 PDAC samples and 45 normal samples) and GSE62452 (based on the
GPL9644 platform, including 69 PDAC samples and 69 normal
samples).
Data preprocessing
Using the Affy package (http://www.bioconductor.org/packages/release/bioc/html/affy.html)
(13) in R language, background
correction and normalization of raw data of GSE15471 and GSE1542
were conducted. For the datasets of GSE71729, GSE28735 and
GSE62452, probes were corresponded to gene symbols based upon
platform annotation information. After the unloaded probes were
removed, the average value of the probes mapped to the same gene
was obtained as the initial gene expression value. Finally, the
data were normalized by the linear models for microarray data using
the limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html)
(14) in R language.
Meta-analysis to screen characteristic
factors
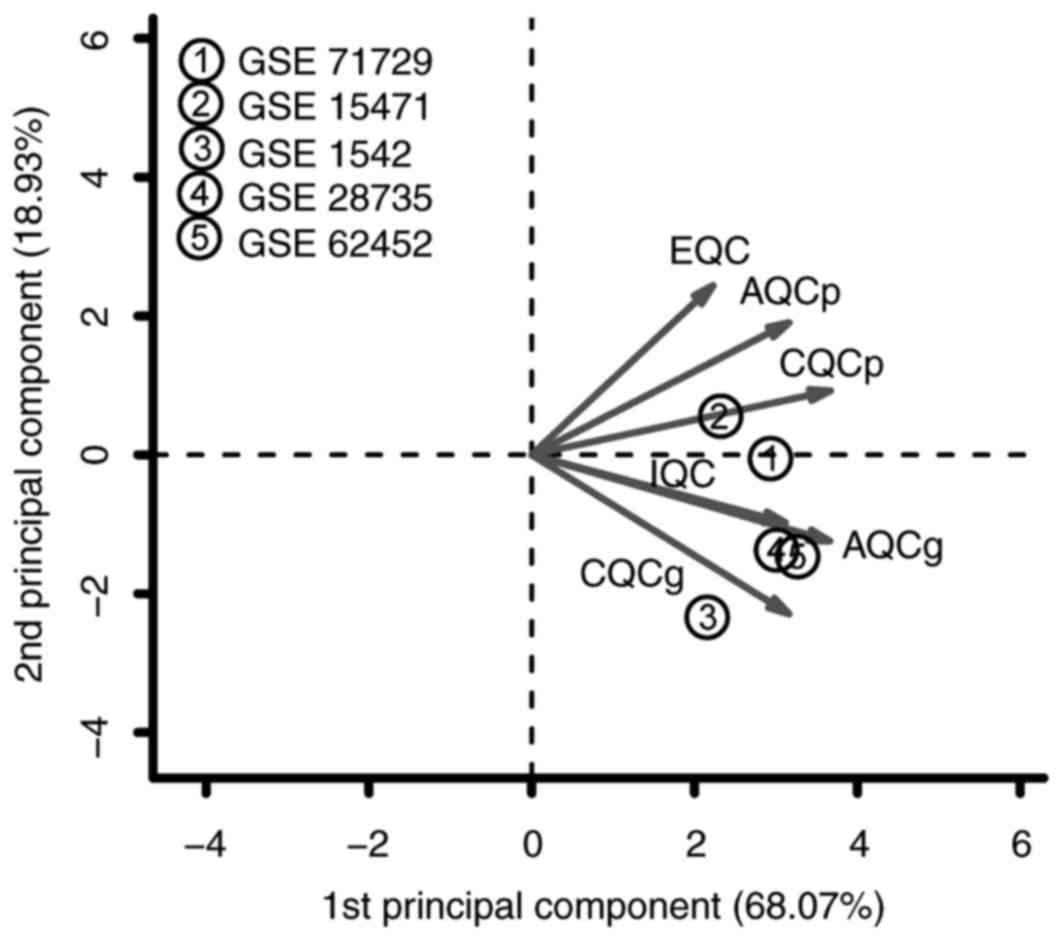
Using the quality control standards [including
external quality control, internal quality control, consistency
quality control (CQCg and CQCp) and accuracy quality control (AQCg
and AQCp)] in the MetaQC package (15), quality control was performed for
the datasets. To identify reliable datasets, the datasets were
further assessed and screened using the two-dimensional diagram of
principal component analysis (PCA) and standardized mean rank
scores. Based on the MetaDE.ES method in the MetaDE package
(15), homogeneous unbiased genes
were identified using a heterogeneity test [thresholds:
Tau2=0 and Qpval (statistical parameter representing
heterogeneity of the dataset) >0.05] and the DEGs were screened
[threshold: False discovery rate (FDR) <0.05].
Identification of prognosis-associated
genes
Among the included datasets, GSE28735 and GSE62452
were based upon the GPL9644 platform and contained sample survival
information. Thus, the data of the two datasets were merged and
prognosis-associated genes were identified using the Cox regression
analysis in survival package, with P<0.05 set as the significant
threshold (https://github.com/therneau/survival). After
significant P-values were obtained using the log-rank test,
Kaplan-Meier (KM) survival analysis was performed on the top six
genes with higher-logRank (P-values) (16).
Co-expression network and module
analyses
The expression values of the prognosis-associated
genes were extracted from the datasets and the COR function
(17) in R language was used to
calculate their correlation coefficients. The co-expression pairs
with correlation coefficient |r|≥0.6 and P<0.05 were selected
for constructing a co-expression network using Cytoscape software
(version 3.5.0; http://www.cytoscape.org/) (18). Furthermore, module analysis was
performed for the co-expression network using the GraphWeb tool
(http://biit.cs.ut.ee/graphweb/)
(19).
Construction of a prediction and
discrimination system for prognosis
The PDAC samples in GSE28735 and GSE62452 had
survival information, and thus were taken as the training dataset
for the prediction and discrimination system of prognosis. Firstly,
the samples were divided into alive and deceased groups according
to their survival states. Secondly, the samples were further
divided into groups with bad prognosis (deceased and alive survival
time <15 months) and good prognosis (alive and alive survival
time ≥15 months). After the genes in the co-expression network were
sorted in descending order according to their-logRank (P-values),
Baye's discriminant analysis was performed using the
discriminant.bayes function in the e1071 package (20). Genes were added one by one, and the
genes affecting discrimination accuracy were removed until the
highest discrimination accuracy was obtained. Under the highest
discrimination accuracy, the discrimination coefficient of each
sample, gene combination, and discrimination system were defined as
the prognostic score, signature gene and prognostic prediction
system, respectively.
Validation of the prognostic
prediction system
To detect the effect of the prognostic prediction
system, KM survival analysis (16)
was performed for GSE28735 and GSE62452 to verify the correlation
between the sample types recognized by the prognostic prediction
system, and the actual survival time and states. In addition, the
microarray data under E-MEXP-2780 (downloaded in January 5, 2017;
including 30 PDAC samples containing survival information) were
downloaded from the European Bioinformatics Institute (http://www.ebi.ac.uk/) database and used as an
independent validation dataset for the prognostic prediction
system. In addition, the PDAC dataset (downloaded in January 5,
2017; based on Illumina HiSeq 2000 RNA Sequencing platform;
including 183 PDAC samples, among which 163 samples had survival
information) in The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) database was also
downloaded. Subsequently, the expression values of the signature
genes were extracted from E-MEXP-2780 and the PDAC dataset was
downloaded from the TCGA database. After prognostic scores were
obtained based on the prognostic prediction system, the samples in
E-MEXP-2780 and the PDAC dataset downloaded from the TCGA database
were divided into groups with bad prognosis and good prognosis.
Finally, KM survival analysis (16) was performed to analyze the
correlation between the sample types recognized by the prognostic
prediction system, and the actual survival time and states.
Construction of the co-expression
network for the signature genes and identification of the key
genes
The co-expression pairs of the signature genes were
identified from the above co-expression network, and the
co-expression networks for the signature genes were visualized.
Gene Ontology (GO) (21)
functional analysis and Kyoto Encyclopedia of Genes and Genomes
(KEGG) pathway (22) enrichment
analysis were performed on genes in the co-expression network using
the clusterProfiler package (http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
(23) in R. To further identify
the key genes associated with PDAC, the enriched terms were
integrated into the co-expression network and the genes involved in
multiple terms were selected.
Results
Identification of characteristic
factors and prognosis-associated genes
After the raw data of the five datasets were
normalized, quality control was further performed for the datasets
(Fig. 1; Table I). The two-dimensional diagram of
PCA for the five datasets is presented in Fig. 2. As a result, all five datasets
were in accordance with the quality control standards and were
included for the subsequent analysis. Based on the MetaDE.ES
method, a total of 480 DEGs were identified from the five
datasets.
 | Table I.Results of quality control measures
and SMRs. |
Table I.
Results of quality control measures
and SMRs.
| Dataset | IQC | EQC | CQCg | CQCp | AQCg | AQCp | SMR |
|---|
| GSE71729 | 3.85 | 4.83 | 264.65 | 133.86 | 32.71 | 82.01 | 2.42 |
| GSE15471 | 4.96 | 3.09 | 275.15 | 70.62 | 39.49 | 39.38 | 3.18 |
| GSE1542 | 4.09 | 4.34 | 242.36 | 103.51 | 101.54 | 58.61 | 3.92 |
| GSE28735 | 5.11 | 3.21 | 310.21 | 90.31 | 18.24 | 29.18 | 3.17 |
| GSE62452 | 5.42 | 3.23 | 307.54 | 75.54 | 19.03 | 29.94 | 3.08 |
GSE28735 and GSE62452 were merged and a total of 259
prognosis-associated genes were screened. Subsequently, KM survival
analysis was performed with the top six prognosis-associated genes,
including transketolase (TKT), basic transcription factor 3,
(BTF3), myomesin 1 (MYOM1), neurexophilin 4 (NXPH4);
serine/threonine kinase 24 (STK24), transducin β-like 3 (TBL3) with
the highest-logRank (P-values). The KM survival curves are
presented in Fig. 3.
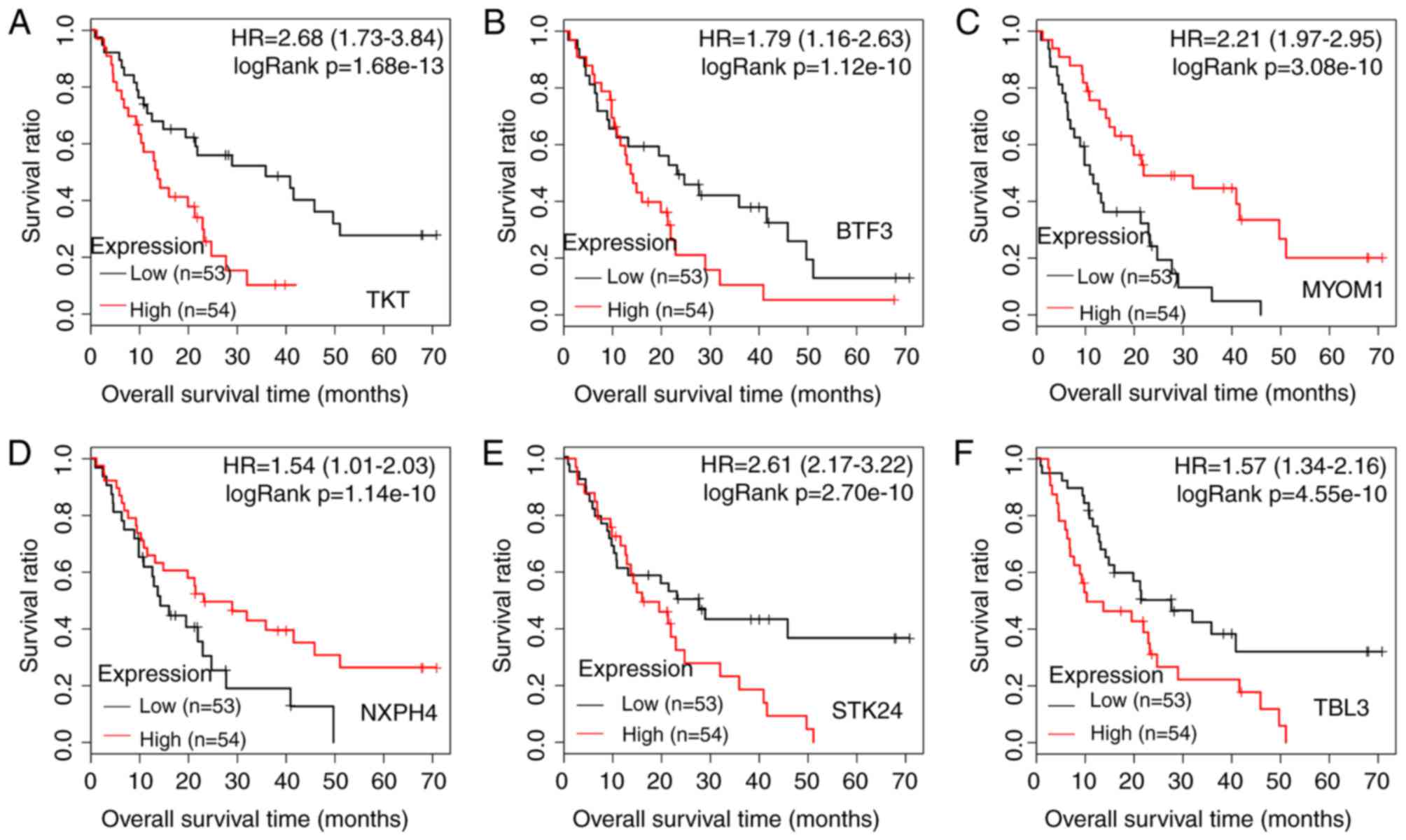 | Figure 3.Kaplan-Meier survival curves for the
top six prognosis-associated genes with higher -logRank (P-values):
(A) TKT, (B) BTF3, (C) MYOM1, (D)
NXPH4, (E) STK24 and (F) TBL3. The red and
black lines represent samples with high and low expression levels,
respectively. HR, hazard ratio; TKT, transketolase; BTF3, basic
transcription factor 3; MYOM1, myomesin 1; NXPH4, neurexophilin 4;
STK24, Serine/threonine-protein kinase 24; TBL3, transducin β-like
protein 3. |
Co-expression network and module
analyses
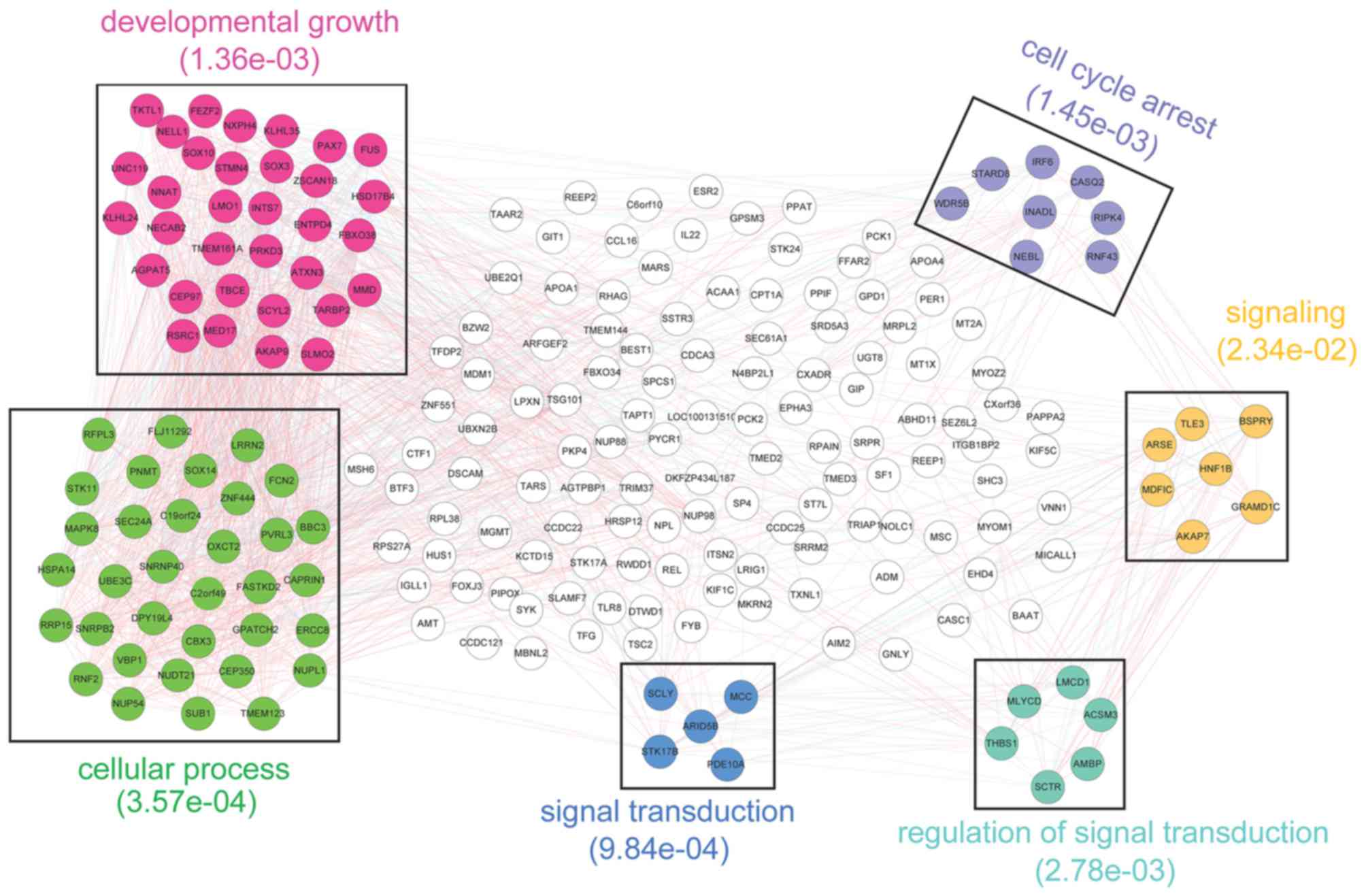
Under |r|≥0.6 and P<0.05, the co-expression pairs
among the prognosis-associated genes were identified and the
co-expression network (which had 213 nodes and 1,984 edges) was
constructed (Fig. 4).
Subsequently, a total of six modules were identified from the
co-expression network. Furthermore, GO functional enrichment
analysis was performed for the genes involved in each of the
modules. Additionally, the most significant GO terms enriched for
purple, yellow, cyan, blue, green, and red modules separately were
cell cycle arrest (P=1.45e-03), signaling (P=2.34e-02), regulation
of signal transduction (P=2.78e-03), signal transduction
(P=9.84e-04), cellular process (P=3.57e-04), and developmental
growth (P=1.36e-03).
Construction and validation of a
prognostic prediction system
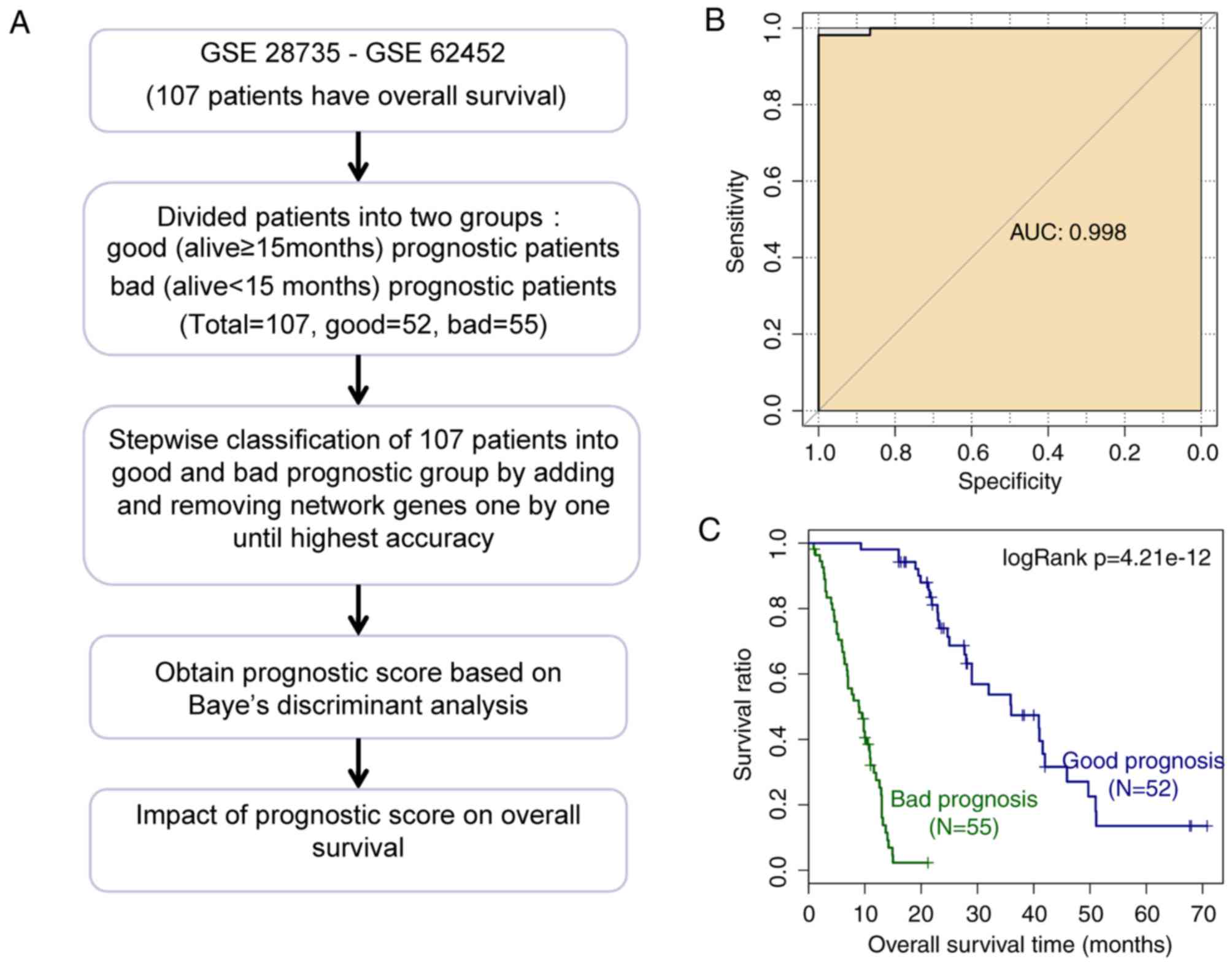
A total of 107 PDAC samples in GSE28735 and GSE62452
had survival information, and were divided into groups with good
(52 samples) and bad (55 samples) prognoses. According to the
process in Fig. 5A, the prognostic
prediction system composed of 67 signature genes [including
BTF3, serine/threonine kinase 11 (STK11),
thrombospondin 1 (THBS1), ribosomal protein L38
(RPL38) and secretin receptor, (SCTR)] was finally
constructed. The area under the receiver operating characteristic
(ROC) curve (AUC) demonstrating the discriminant accuracy of the
prognostic prediction system is presented in Fig. 5B. In addition, a discriminant
scoring system was constructed for the prognostic prediction
system, which was as follows:
prognostic
score=ai=167(Bayes'discriminant
analysis)=([3,0]~bad[–3,0]~good)
KM survival analysis was performed for GSE28735 and
GSE62452 to detect the effect of the prognostic prediction system,
identifying that the group with good prognosis had a significantly
higher survival rate when compared with the group with bad
prognosis (P=4.21e-12; Fig. 5C).
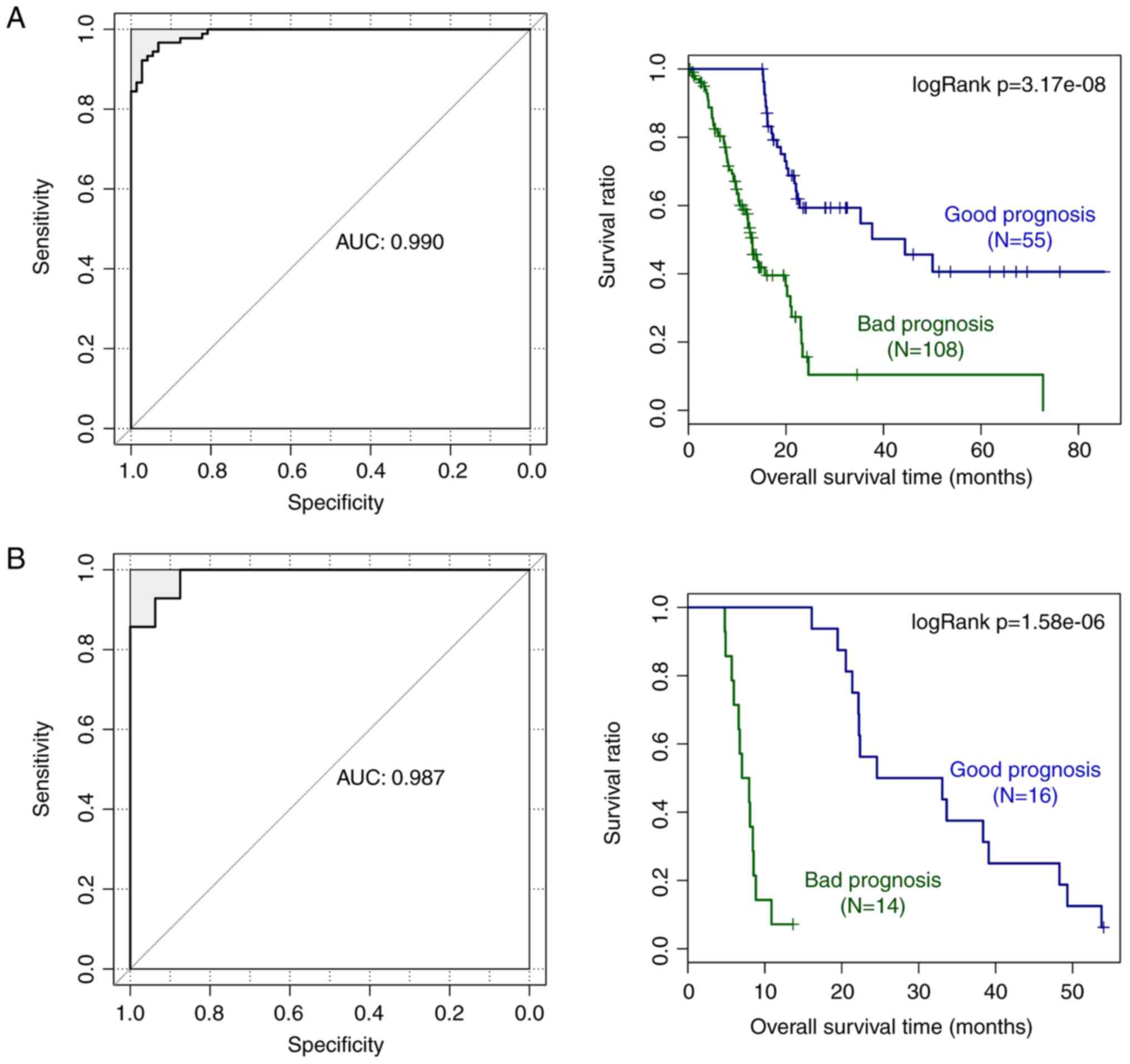
In addition, the PDAC dataset downloaded from the TCGA database
served as an independent validation dataset for the prognostic
prediction system. The AUC and KM survival curve in Fig. 6A demonstrates that the survival
rate of the good prognosis group was significantly higher than that
of the bad prognosis group (P=3.17e-08). Furthermore, the
microarray data of E-MEXP-2780 was used to validate the prognostic
prediction system, identifying that the group with good prognosis
exhibited a significantly higher survival rate when compared with
the bad prognosis group (P=1.58e-06; Fig. 6B). Therefore, the prognostic
prediction system could accurately classify the samples from
prognostic level.
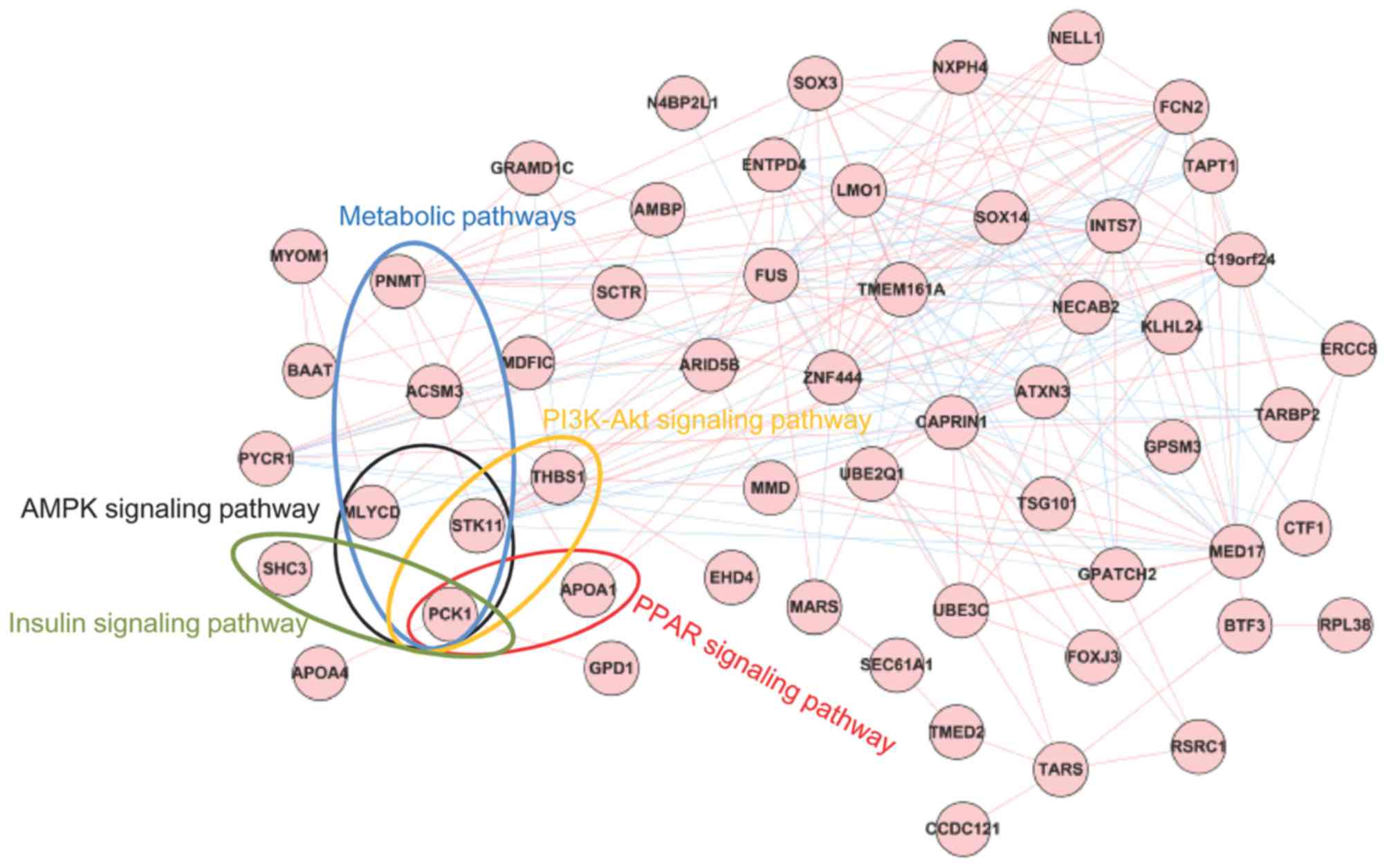
Construction of co-expression network
for the signature genes and identification of the key genes
The co-expression network constructed for the
signature genes included 56 signature genes and 237 edges (Fig. 7). A total of 14 GO_biological
process (BP) terms and five KEGG signaling pathways were enriched
for the genes involved in the co-expression network (Table II). Meanwhile, the five signaling
pathways were merged into the co-expression network for the
signature genes. As presented in Fig.
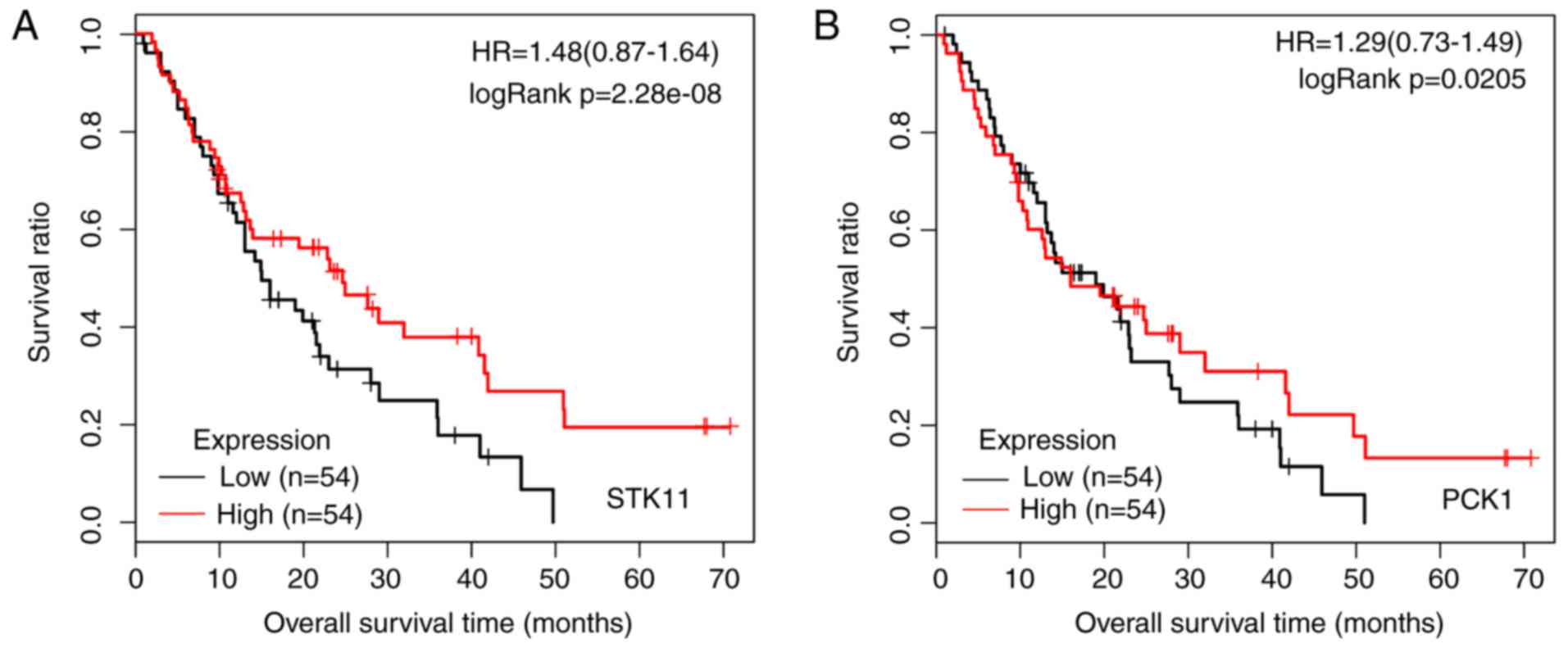
7, phosphoenolpyruvate carboxykinase 1 (PCK1) was
enriched in all five signaling pathways, and STK11 was
involved in three signaling pathways. In addition, THBS1 was
enriched in the phosphoinositide 3-kinase (PI3K)-Akt signaling
pathway (P=0.017456). In addition, KM survival analysis was
performed for STK11 (Fig.
8A) and PCK1 (Fig. 8B)
based on the GSE28735 and GSE62452 datasets.
| Table II.GO_ BP terms and signaling pathways
enriched for the signature genes involved in the co-expression
network. |
Table II.
GO_ BP terms and signaling pathways
enriched for the signature genes involved in the co-expression
network.
| Category | Description | Gene no. | P-value | Gene symbol |
|---|
| GO_BP |
GO:0010565~regulation of cellular ketone
metabolic process | 3 | 0.02095 | APOA4, MLYCD,
STK11 |
|
|
GO:0030300~regulation of intestinal
cholesterol absorption | 2 | 0.023282 | APOA4,
APOA1 |
|
|
GO:0046486~glycerolipid metabolic
process | 4 | 0.02552 | APOA4, GPD1,
APOA1, PCK1 |
|
| GO:0010873~positive
regulation of cholesterol esterification | 2 | 0.02711 | APOA4,
APOA1 |
|
|
GO:0046782~regulation of viral
transcription | 2 | 0.02711 | TARBP2,
MDFIC |
|
|
GO:0010872~regulation of cholesterol
esterification | 2 | 0.030924 | APOA4,
APOA1 |
|
| GO:0048524~positive
regulation of viral reproduction | 2 | 0.038507 | TARBP2,
MDFIC |
|
|
GO:0033700~phospholipid efflux | 2 | 0.038507 | APOA4,
APOA1 |
|
| GO:0051186~cofactor
metabolic process | 4 | 0.040832 | AMBP, GPD1,
BAAT, MLYCD |
|
| GO:0002683~negative
regulation of immune system process | 3 | 0.04189 | AMBP, TARBP2,
THBS1 |
|
|
GO:0044058~regulation of digestive system
process | 2 | 0.042277 | APOA4,
APOA1 |
|
| GO:0034377~plasma
lipoprotein particle assembly | 2 | 0.046032 | APOA4,
APOA1 |
|
|
GO:0065005~protein-lipid complex
assembly | 2 | 0.046032 | APOA4,
APOA1 |
|
|
GO:0007586~digestion | 3 | 0.049426 | APOA4, BAAT,
SCTR |
| Pathway | AMP-activated
protein kinase signaling pathway | 3 | 0.001119 | STK11, MLYCD,
PCK1 |
|
| Peroxisome
proliferator-activated receptor signaling pathway | 2 | 0.006183 | APOA1,
PCK1 |
|
| Metabolic
pathways | 5 | 0.013991 | ACSM3, PNMT,
MLYCD, PCK1, STK11 |
|
| Phosphoinositide
3-kinase-Akt signaling pathway | 3 | 0.017456 | STK11, THBS1,
PCK1 |
|
| Insulin signaling
pathway | 2 | 0.021115 | SHC3,
PCK1 |
Discussion
In the current study, a total of 480 DEGs were
identified from five datasets. In addition, 259
prognosis-associated genes were screened from GSE28735 and
GSE62452, and BTF3 was among the top six
prognosis-associated genes. Furthermore, a total of six modules
were identified from the co-expression network for the
prognosis-associated genes. A prognostic prediction system composed
of 67 signature genes, which included BTF3, STK11,
THBS1, RPL38 and SCTR, was finally constructed
and validated. Finally, the co-expression network for the signature
genes was constructed and the signature genes involved in the
co-expression network were enriched in five signaling pathways. In
particular, STK11 was involved in three of the signaling
pathways.
STK11/LKB1 causes somatic mutations in
intraductal papillary mucinous neoplasms (IPMNs), sporadic
pancreatic adenocarcinomas and biliary adenocarcinomas, and its
expression is abrogated in pancreatic and biliary neoplasms
(24,25). Sato et al (26) identified that the STK11/LKB1
gene is associated with the development and progression of certain
IPMNs (26). The LKB1-p21
axis cooperates with the Kirsten rat sarcoma viral oncogene homolog
(Kras) mutation to inhibit PDAC in vivo, and
downregulated LKB1 may function in promoting PC by replacing
the p53 mutation (27,28).
As a Peutz-Jeghers syndrome gene, LKB1 induces apoptosis of
PC cells in a p73-dependent manner (29). These studies indicate that
STK11 is implicated in the prognosis of PDAC.
Stromal expression levels of THBS1 are a
prognostic marker and invasive indicator in IPMN (30). By upregulating THBS1 and
caveolin-1 and downregulating cyclin D1, metronomic C2 and AL6
analogs perform antiangiogenic and antitumor roles in PC (31). THBS1 is implicated in cell
growth and metastasis of PC cells, and stromal THBS1
immunoreactivity may be used for predicting the prognosis of PC
patients (32). By promoting the
expression of matrix metalloproteinase-9, THBS1 is important
in mediating matrix remodeling in the invasion of pancreatic
adenocarcinoma (33,34). The signature gene, THBS1 was
enriched in the PI3K-Akt signaling pathway, indicating that
THBS1 may be involved in the prognosis of PDAC via the
PI3K-Akt signaling pathway.
Overexpressed BTF3 functions as a
transcriptional regulator by mediating the transcription of
tumor-associated genes in PDAC (35). RPL38, FOS-like antigen-1 and
uridine phosphorylase are highly expressed in PC cell lines;
therefore, they have the potential to serve as tumor markers or in
tumor targeting (36). SCTR
are key in regulating healthy pancreatic ductal epithelial cells,
and its silence may contribute to tumor growth and progression of
PC (37). SCTR is
overexpressed in non-neoplastic pancreas ducts and its isoforms may
be correlated with decreased secretin binding in pancreatic ductal
tumors, indicating that SCTRs may represent promising clinical
targets in pancreatic tumors (38). Therefore, BTF3,
RPL38, and SCTR may be important in the prognosis of
PDAC.
There were certain limitations of the present study.
The findings obtained from bioinformatics analysis require further
validation via experimental studies. However, the validation
experiment could not be performed in the current study due to being
limited by experimental conditions and sample sources. The present
results may provide valuable data for future investigations.
In conclusion, a total of 480 DEGs were identified
from five datasets, and 259 prognosis-associated genes were
screened from GSE28735 and GSE62452. Furthermore, the prognostic
prediction system composed of 67 signature genes was constructed
and validated. Notably, signature genes, including BTF3,
STK11, THBS1, RPL38 and SCTR may
function in the prognosis of PDAC.
References
|
1
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yadav D and Lowenfels AB: The epidemiology
of pancreatitis and pancreatic cancer. Gastroenterology.
144:1252–1261. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ryan DP, Hong TS and Bardeesy N:
Pancreatic adenocarcinoma. N Engl J Med. 371:1039–1049. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wolfgang CL, Herman JM, Laheru DA, Klein
AP, Erdek MA, Fishman EK and Hruban RH: Recent progress in
pancreatic cancer. CA Cancer J Clin. 63:318–348. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peto R, Lopez AD, Pan H and Thun MJ: World
Cancer Report. 2014.
|
|
6
|
Mohammed S, Van Buren G 2nd and Fisher WE:
Pancreatic cancer: advances in treatment. World J Gastroenterol.
20:9354–9360. 2014.PubMed/NCBI
|
|
7
|
Dunne RF and Hezel AF: Genetics and
Biology of Pancreatic Ductal Adenocarcinoma. Hematol Oncol Clin
North Am. 29:595–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamazaki K, Masugi Y, Effendi K, Tsujikawa
H, Hiraoka N, Kitago M, Shinoda M, Itano O, Tanabe M, Kitagawa Y
and Sakamoto M: Upregulated SMAD3 promotes epithelial-mesenchymal
transition and predicts poor prognosis in pancreatic ductal
adenocarcinoma. Lab Invest. 94:683–691. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Masugi Y, Yamazaki K, Emoto K, Effendi K,
Tsujikawa H, Kitago M, Itano O, Kitagawa Y and Sakamoto M:
Upregulation of integrin β4 promotes epithelial-mesenchymal
transition and is a novel prognostic marker in pancreatic ductal
adenocarcinoma. Lab Invest. 95:308–319. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xia S, Feng Z, Qi X, Yin Y, Jin J, Wu Y,
Wu H, Feng Y and Tao M: Clinical implication of Sox9 and activated
Akt expression in pancreatic ductal adenocarcinoma. Med Oncol.
32:3582015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizuuchi Y, Aishima S, Ohuchida K, Shindo
K, Fujino M, Hattori M, Miyazaki T, Mizumoto K, Tanaka M and Oda Y:
Anterior gradient 2 downregulation in a subset of pancreatic ductal
adenocarcinoma is a prognostic factor indicative of
epithelial-mesenchymal transition. Lab Invest. 95:193–206. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Glazer ES, Welsh E, Pimiento JM, Teer JK
and Malafa MP: TGFβ1 overexpression is associated with improved
survival and low tumor cell proliferation in patients with
early-stage pancreatic ductal adenocarcinoma. Oncotarget.
8:999–1006. 2017.PubMed/NCBI
|
|
13
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Smyth GK: Limma: Linear Models for
Microarray Data. Springer; New York: 2005
|
|
15
|
Wang X, Kang DD, Shen K, Song C, Lu S,
Chang LC, Liao SG, Huo Z, Tang S, Ding Y, et al: An R package suite
for microarray meta-analysis in quality control, differentially
expressed gene analysis and pathway enrichment detection.
Bioinformatics. 28:2534–2536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Porcher R: CORR insights (®):
Kaplan-meier survival analysis overestimates the risk of revision
arthroplasty: A meta-analysis. Clin Orthop Relat Res.
473:3443–3445. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nowicka-Zagrajek J and Weron R: COR:
MATLAB function to compute the correlation coefficients. Hsc
Software. 2008.
|
|
18
|
Kohl M, Wiese S and Warscheid B:
Cytoscape: Software for visualization and analysis of biological
networks. Methods Mol Biol. 696:291–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reimand J, Tooming L, Peterson H, Adler P
and Vilo J: GraphWeb: Mining heterogeneous biological networks for
gene modules with functional significance. Nucleic Acids Res.
36:W452–W459. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dimitriadou E, Hornik K, Leisch F and
Meyer D: The e1071 package. Ethnos J Anthropol. 23:55–56. 2006.
|
|
21
|
Smith B, Williams J and Schulze-Kremer S:
The ontology of the gene ontology. AMIA Annu Symp Proc. 609–613.
2003.PubMed/NCBI
|
|
22
|
Aoki KF and Kanehisa M: Using the KEGG
database resource. Curr Protoc Bioinformatics. Chapter.
1:Unit1.122012.
|
|
23
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sahin F, Maitra A, Argani P, Sato N,
Maehara N, Montgomery E, Goggins M, Hruban RH and Su GH: Loss of
Stk11/Lkb1 expression in pancreatic and biliary neoplasms. Mod
Pathol. 16:686–691. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hezel AF and Bardeesy N: LKB1; linking
cell structure and tumor suppression. Oncogene. 27:6908–6919. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sato N, Rosty C, Jansen M, Fukushima N,
Ueki T, Yeo CJ, Cameron JL, Iacobuzio-Donahue CA, Hruban RH and
Goggins M: STK11/LKB1 Peutz-Jeghers gene inactivation in
intraductal papillary-mucinous neoplasms of the pancreas. Am J
Pathol. 159:2017–2022. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Morton JP, Jamieson NB, Karim SA, Athineos
D, Ridgway RA, Nixon C, McKay CJ, Carter R, Brunton VG, Frame MC,
et al: LKB1 haploinsufficiency cooperates with kras to promote
pancreatic cancer through suppression of p21-dependent growth
arrest. Gastroenterology. 139(586–597): e1–6. 2010.
|
|
28
|
Wei C, Amos CI, Stephens LC, Campos I,
Deng JM, Behringer RR, Rashid A and Frazier ML: Mutation of Lkb1
and p53 genes exert a cooperative effect on tumorigenesis. Cancer
Res. 65:11297–11303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qanungo S, Haldar S and Basu A:
Restoration of silenced peutz-jeghers syndrome gene, LKB1, induces
apoptosis in pancreatic carcinoma cells. Neoplasia. 5:367–374.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Okada K, Hirabayashi K, Imaizumi T,
Matsuyama M, Yazawa N, Dowaki S, Tobita K, Ohtani Y, Tanaka M,
Inokuchi S and Makuuchi H: Stromal thrombospondin-1 expression is a
prognostic indicator and a new marker of invasiveness in
intraductal papillary-mucinous neoplasm of the pancreas. Biomed
Res. 31:13–19. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bocci G, Fioravanti A, Orlandi P, Di
Desidero T, Natale G, Fanelli G, Viacava P, Naccarato AG, Francia G
and Danesi R: Metronomic ceramide analogs inhibit angiogenesis in
pancreatic cancer through up-regulation of caveolin-1 and
thrombospondin-1 and down-regulation of cyclin D1. Neoplasia.
14:833–845. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tobita K, Kijima H, Dowaki S, Oida Y,
Kashiwagi H, Ishii M, Sugio Y, Sekka T, Ohtani Y, Tanaka M, et al:
Thrombospondin-1 expression as a prognostic predictor of pancreatic
ductal carcinoma. Int J Oncol. 21:1189–1195. 2002.PubMed/NCBI
|
|
33
|
Qian X, Rothman VL, Nicosia RF and
Tuszynski GP: Expression of thrombospondin-1 in human pancreatic
adenocarcinomas: Role in matrix metalloproteinase-9 production.
Pathol Oncol Res. 7:251–259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McElroy MK, Kaushal S, Tran Cao HS, Moossa
AR, Talamini MA, Hoffman RM and Bouvet M: Upregulation of
thrombospondin-1 and angiogenesis in an aggressive human pancreatic
cancer cell line selected for high metastasis. Mol Cancer Ther.
8:1779–1786. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kusumawidjaja G, Kayed H, Giese N, Bauer
A, Erkan M, Giese T, Hoheise JD, Friess H and Kleeff J: Basic
transcription factor 3 (BTF3) regulates transcription of
tumor-associated genes in pancreatic cancer cells. Cancer Biol
Ther. 6:367–676. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sahin F, Qiu W, Wilentz RE,
Iacobuziodonahue CA, Grosmark A and Su GH: RPL38, FOSL1 and UPP1
are predominantly expressed in the pancreatic ductal epithelium.
Pancreas. 30:158–167. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ding WQ, Cheng ZJ, McElhiney J, Kuntz SM
and Miller LJ: Silencing of secretin receptor function by
dimerization with a misspliced variant secretin receptor in ductal
pancreatic adenocarcinoma. Cancer Res. 62:5223–5229.
2002.PubMed/NCBI
|
|
38
|
Körner M, Hayes GM, Rehmann R, Zimmermann
A, Friess H, Miller LJ and Reubi JC: Secretin receptors in normal
and diseased human pancreas: Marked reduction of receptor binding
in ductal neoplasia. Am J Pathol. 167:959–968. 2005. View Article : Google Scholar : PubMed/NCBI
|