Introduction
Hereditary spherocytosis (HS) and uridine
diphosphate glucuronosyltransferase 1A1 (UGT1A1) deficiency are
common inherited conditions characterized by unconjugated
hyperbilirubinemia. HS is an inherited hemolytic anemia caused by
constitutional erythrocyte membrane defects, and characterized
clinically by anemia with variable severity, jaundice and
splenomegaly (1). Diagnosis is
usually established based on the presence of spherocytes, increased
erythrocyte osmotic fragility and the absence of any other cause of
the hemolytic anemia (1). UGT1A1
is responsible for bilirubin glucuronidation, and reduced activity
of UGT1A1 is associated with an increased level of unconjugated
bilirubin (2). UGT1A1 deficiency
is a common hereditary condition of bilirubin metabolism in the
general population, particularly in Asian communities (2,3).
As the prevalence of HS is 1 in 2,000 (4), and that of UGT1A1 caused by G211A
mutation deficiency is much higher at 13–23% in Asian newborns
(2,3), diagnoses of their coexistence should
not be rare; however, the number of documented cases is low
(5,6). This underdiagnosis may be attributed
to one condition masking another (7), or to a lack of conclusive evidence
for their coexistence. In the present case report, atypical HS
combined with UGT1A1 partial deficiency was diagnosed by pathogenic
gene analysis. A novel c.G828T (Y276X) mutation in spectrin β
(SPTB), which is causative for HS, along with compound
heterozygous mutations in UGT1A1, T3279G and G211A, which
are pathogenic for UGT1A1 partial deficiency, were identified. In
addition, the individual pathogenesis of hyperbilirubinemia was
investigated for this patient.
Materials and methods
Patient
The present study was approved by the Ethics
Committee of the Second Xiangya Hospital of Central South
University (Changsha, China), and written informed consent was
obtained from the patient and the patient's mother. On July 12,
2016, a 20-year-old female with congenital jaundice and anemia for
~20 years was re-hospitalized to confirm the patient's condition.
The proband was diagnosed with neonatal jaundice several days
following birth. In 2003, when the patient was 7 years old,
splenomegaly, anemia and jaundice were detected. Between 2004 and
2005, hyperplastic anemia (hemoglobin, 96 g/l; reticulocytes,
0.05), splenomegaly, elevated serum bilirubin [total bilirubin
(TBIL), 36.9 µmol/l; direct bilirubin, 11.3 µmol/l] and detectable
spherocytes in blood smear were identified, and HS was suspected.
However, the erythrocyte osmotic fragility test, which is a crucial
test for HS diagnosis, was repeatedly normal. Furthermore, the
elevated serum bilirubin concentration decreased to normal level
following phenobarbitone treatment, which suggested UGT1A1
deficiency, which would not contribute to hyperplastic anemia and
splenomegaly. Subsequently, the patient has been interviewed
regularly. Upon infection or overexertion, the patient's serum
bilirubin increased significantly, mainly by unconjugated
bilirubin. Notably, bilirubin levels gradually increased over time,
whereas the anemia was mild. In 2013, when the patient was 17 years
old, the patient's serum TBIL rose to >80 µmol/l and hemoglobin
was >100 g/l. There was no history of alcoholism, hepatitis,
drug ingestion or drug abuse. The proband's father had a history of
cholelithiasis, and succumbed to cirrhosis in his twenties. The
patient's father's brother also died early, and presented with
jaundice at his death.
Clinical analysis
The proband received a detailed clinical evaluation
at the Second Xiangya Hospital of Central South University.
Extensive laboratory testing was performed, including complete
blood count, liver functional tests, lactate dehydrogenase, vitamin
B12, folate, iron studies, rheumatoid factor and anti-nuclear
antibodies testing, erythrocyte osmotic fragility test, hemoglobin
electrophoresis, Ham test, glucose-6-phasphate dehydrogenase
activity, Coombs' direct and indirect tests, flow cytometric test
using eosin-5′-maleimide (EMA) labeling of red blood cells
(1,8) as well as abdominal computed
tomography (CT) scans. Additionally, bone marrow and blood smears
of the patients were stained by Wright-Giemsa staining for 30 min
at room temperature. Then the smears were observed using a light
microscope, erythrocyte morphology was evaluated, and the
proportion of spherocytes was counted among 500 erythrocytes.
Genetic analysis
A total of 5 ml peripheral vein blood was drawn from
the proband and the proband's mother. The genomic DNA was extracted
using QIAamp DNA Blood Mini kit (cat. no. 51104; Qiagen GmbH,
Hilden, Germany) according to the manufacturer's protocol,
quantified using an Eppendorf BioPhotometer® D30
(Eppendorf, Hamburg, Germany) and stored at −20°C until use. Then
the genomic DNA library was constructed according to the Agilent's
protocol for Preparing Samples for Sequencing Genomic DNA. Using
the SureSelectXT Reagent kits (cat. no. G9611B; Agilent
Technologies, Inc., Santa Clara, CA, USA), 3 µg high-quality gDNA
was sheared by the Bioruptor® Plus sonication device
(Diagenode S.A., Seraing, Belgium) with 175 peak incident power,
end-repaired at 20°C (30 min), adenylated at 37°C (30 min) and
ligated with an adaptor at 20°C (15 min) using a next-generation
high throughout sequencer (HiSeq 2000 v3; Illumina, Inc., San
Diego, CA, USA). The DNA fragments were purified at each step with
AMPure XP beads DNA Purification kit (cat. no. A63880; Beckman
Coulter, Inc., Brea, CA, USA) according to the manufacturer's
protocol.
The 5 known genes associated with inherited
spherocytosis include ankyrin 1, SPTB, spectrin α
erythrocytic 11, solute carrier family 4 member 1 and erythrocyte
membrane protein band 4.2 gene. To amplify and capture the 5 known
genes a costumed Agilent Comparative Genomic Hybridization Array
for the exons and adjacent intron regions (50 bp) of the 5 genes
and SureSelect Focused Exome kit (cat. no. 5190-7788, Agilent
Technologies, Inc.) were used according to the manufacturer's
protocol. The adaptor-ligated library was amplified in 25 µl
reaction using 5X Herculase II Reaction Buffer (5 µl), SureSelect
Primer and SureSelect ILM Indexing Pre-Capture PCR Reverse Primer
(0.625 µl each), 100 mM dNTP mix (0.25 µl), Herculase II Fusion DNA
Polymerase (0.5 µl) and adaptor-ligated DNA (18 µl). The thermal
cycler conditions were as follows: Initial denaturation at 98°C for
2 min, 4–6 cycles of denaturation at 98°C (30 sec), annealing at
65°C (30 sec) and extension at 72°C (1 min), and final extension at
72°C (10 min).
To capture the target DNA fragments, 500 ng total
library, which was concentrated by a vacuum concentrator
(Concentrator Plus; Eppendorf), was hybridized with
streptavidin-coated beads, at 65°C for 24 h. Then the captured gene
fragments were sequenced by the next-generation high throughput
sequencer. The results demonstrated that 88.61% of target bases
were covered to a total depth of >20X with high quality (Q20)
reads. Reads were aligned to the reference sequence University of
California Santa Cruz, human genome assembly 19 (UCSC.hg19;
genome.ucsc.edu/). Variants, including
single-nucleotide polymorphisms (SNPs) and indels, were identified
and called with the VCFtools program of the SAMTools software,
version 0.1.16 (samtools.sourceforge.net/). Amino acid substitutions
that affected protein function were annotated with polymorphism
phenotyping v2 (PolyPhen-2; genetics.bwh.harvard.edu/pph2) and MutationTaster2
(mutationtaster.org). Pathogenicity of
the variants was interpreted according to the American College of
Medical Genetics (ACMG) guidelines (9).
The detected mutation in SPTB was confirmed
by direct sequencing of the polymerase chain reaction (PCR)
products amplified from the genomic DNA samples of the patient and
her mother, which were extracted from their blood as mentioned
above. Genome DNA samples were amplified in 25 µl reactions using
2X Power Taq PCR MasterMix (12.5 µl; BioTeke Corporation, Beijing,
China), nuclease-free water (11 µl), 10 pmol/µl forward and reverse
primers (0.5 µl each), and 100 ng/µl template (0.5 µl). The
thermocycling conditions were as follows: Initial denaturation at
95°C for 3 min, 30 cycles of denaturation at 95°C (30 sec),
annealing at 57°C (30 sec) and extension at 72°C (1 min), and final
extension at 72°C (7 min). Primers for PCR were designed using
primer3 software (primer3.ut.ee): SPTB forward,
5′-TGCTCTGTTGGTTGTCACTTG-3′ and reverse,
5′-AGCCATCAATGTTGCCAAGG-3′. PCR products were examined on 1%
agarose gels containing 0.5 µg/ml ethidium bromide and were
subsequently sequenced on an ABI 3730 DNA Analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Sequence comparisons and analysis were performed using
Phred-Phrap-Consed version 12.0 software (phrap.org/phredphrapconsed.html). Sequencing results
were compared with the reference sequence for SPTB (GenBank
accession no. NM_001024858) in the UCSC database to confirm
potential mutation. Then the detected mutation in SPTB
(G828T) was absent in the 1,000 Genomes Project database and Exome
Aggregation (10).
Amplification of the exons, the promoter region and
the enhancer region [that is, the phenobarbital responsive enhancer
module (PBREM)] of UGT1A1 was performed by PCR. Genome DNA
samples were amplified in reactions as mentioned above.
Thermocycling conditions were as follows: Initial denaturation at
95°C for 3 min, 30 cycles of denaturation at 95°C (30 sec),
annealing temperatures as shown in Table I (30 sec), extension at 72°C (1
min), and final extension at 72°C (7 min). Primer sequences are
listed in Table I. Amplified DNA
fragments were sequenced as aforementioned. Mutations in
UGT1A1 were identified by comparing the sequencing results
with the reported reference sequence (GenBank accession no.
NM_000463).
 | Table I.Primers sequences of uridine
diphosphate glucuronosyltransferase 1A1 gene. |
Table I.
Primers sequences of uridine
diphosphate glucuronosyltransferase 1A1 gene.
| UGT1A1
exon | Primer sequence
(5→3′) | Amplicon size
(bp) | Annealing temperature
(°C) |
|---|
| Exon 1–1 | F:
TATAAGTAGGAGAGGGCGAACC | 588 | 57 |
|
| R:
TCAAATTCCAGGCTGCATG |
|
|
| Exon 1–2 | F:
GGCCTCCCTGGCAGAAAG | 617 | 60 |
|
| R:
ATGCCAAAGACAGACTCAAACC |
|
|
| Exon 2 | F:
AGGAACCCTTCCTCCTTTAGA | 402 | 59 |
|
| R:
GAAGCTGGAAGTCTGGGATTAG |
|
|
| Exon 3 | F:
CCTCAGAAGCCTTCACAGTTAC | 255 | 59 |
|
| R:
ATCCAATCCGCCCAACATAC |
|
|
| Exon 4 | F:
GTGTCCAGCTGTGAAACTCA | 323 | 55 |
|
| R:
TGAATGCCATGACCAAAGTATTC |
|
|
| Exon 5 | F:
CAACAGGGCAAGACTCTGTATC | 489 | 60 |
|
| R:
CCTTATTTCCCACCCACTTCTC |
|
|
| Promoter | F:
ACAGGTTTCCATGGCGAAAG | 782 | 56 |
|
| R:
TGTTTTGATCACACGCTGCA |
|
|
| PBREM | F:
GGTCACTCAATTCCAAGGGG | 598 | 61 |
|
| R:
GCATCCAAGCCAGCAAGTAA |
|
|
Results
Clinical diagnosis
Physical examination revealed cutaneous and icteric
sclera; spleen was palpable 4 cm below costal margin, and was firm
and non-tender. TBIL was 106.2 µmol/l and direct bilirubin was 7.5
µmol/l. The complete blood count revealed hemoglobin 103 g/l,
reticulocytes 0.358×1012/l, mean corpuscular volume 84.3
fl, mean corpuscular hemoglobin 30.6 pg and mean corpuscular
hemoglobin concentration 363 g/l, and white blood cell and platelet
counts were normal.
Extensive laboratory evaluation revealed normal
levels of alanine aminotransferase, aspartate transaminase, lactate
dehydrogenase, vitamin B12, folate, iron studies, rheumatoid factor
and anti-nuclear antibodies. A series of diagnostic tests for
hemolytic anemia, including erythrocyte osmotic fragility test,
hemoglobin electrophoresis, Ham's test, glucose-6-phosphate
dehydrogenase activity, Coombs' direct and indirect tests were
normal. Bone marrow smear indicated hypercellular marrow with
myeloid-to-erythroid precursor ratio of 0.4:1. Blood smear results
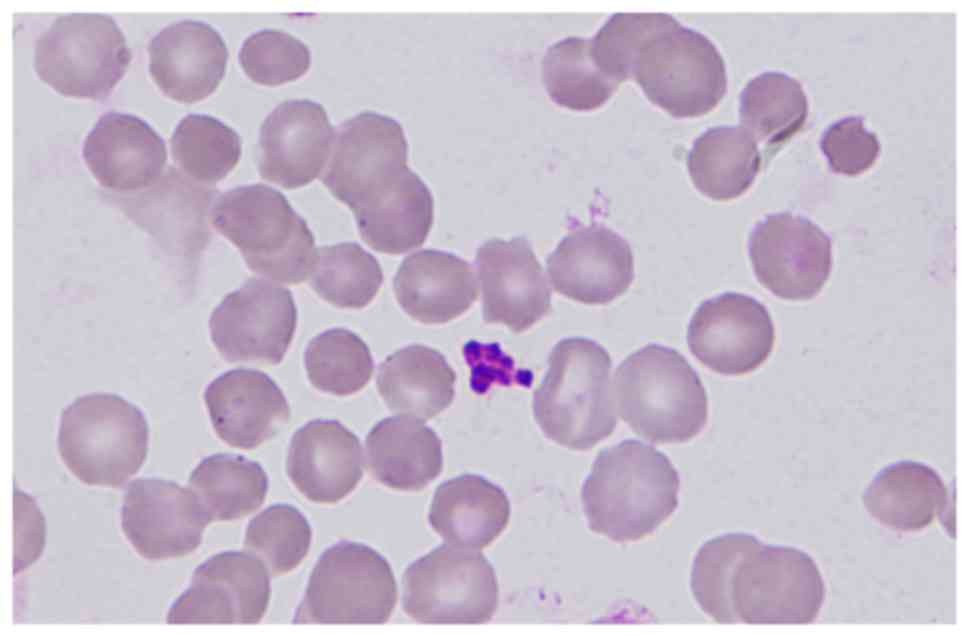
demonstrated that spherocytes accounted for 62 out of 500 red blood
cells (Fig. 1). EMA test of red
blood for the diagnosis of HS was positive. An abdominal CT scan
revealed splenomegaly without the combination of gallstone or
cholangiectasis.
HS was diagnosed clinically based on the presence of
hyperplastic anemia, splenomegaly and spherocytes, and
particularly, the positive EMA test. Although the erythrocyte
osmotic fragility test was repeatedly normal, the positive EMA test
supported the diagnosis of HS.
Pathogenic mutation analysis of
SPTB
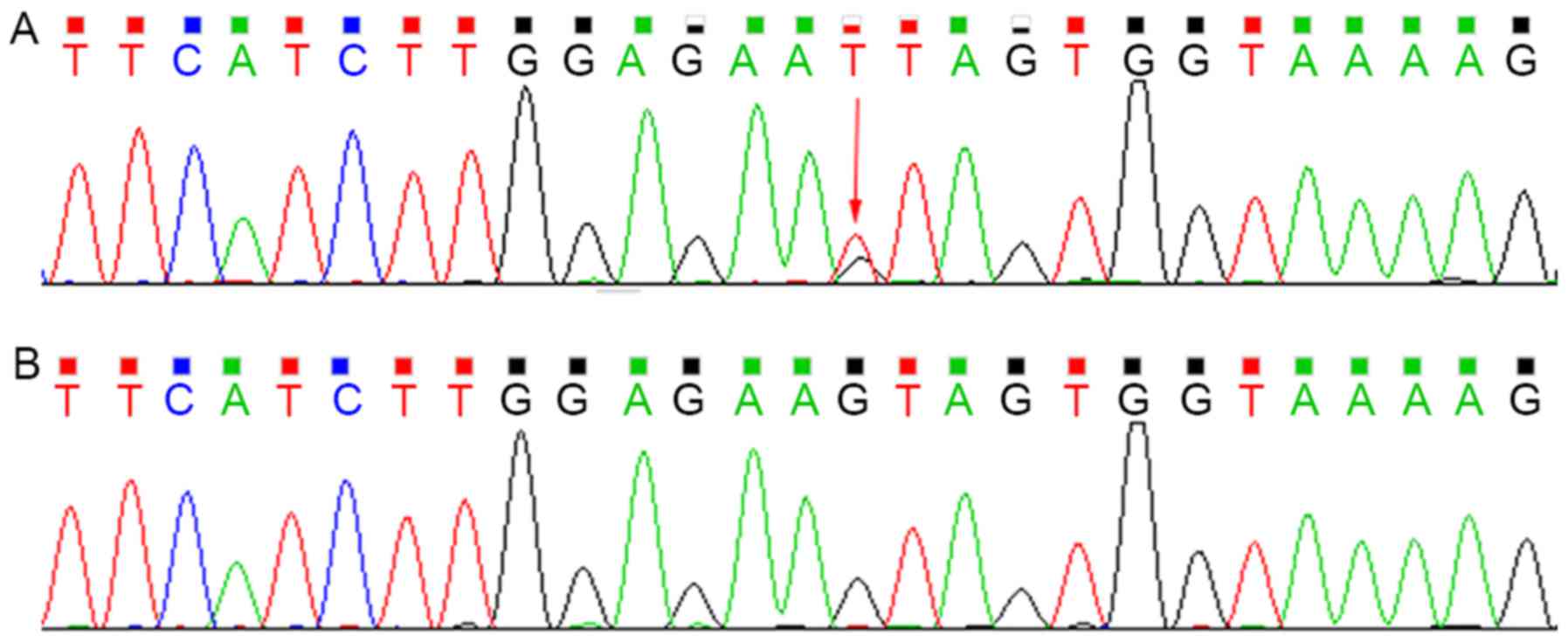
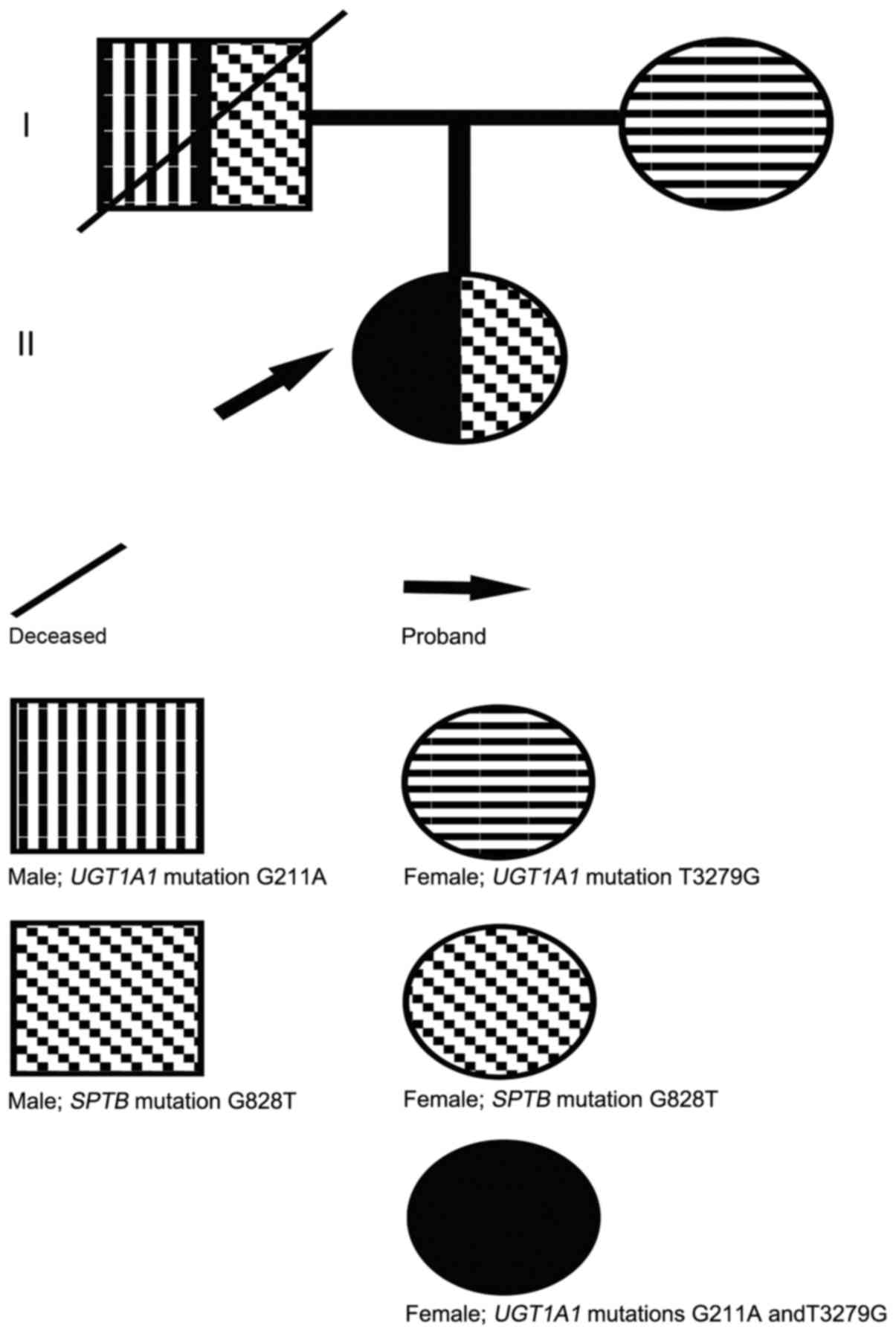
Sequencing analysis revealed that the proband was
heterozygous for the c.G828T (p.Y276X) mutation within exon 7 of
SPTB, which was not present in the mother (Fig. 2). This mutation introduced a
premature stop codon at amino acid residue 276, which created a
truncated protein and was predicted to be disease-causing by the
analytical software used. In addition, it had not been reported
previously and is a novel mutation.
Although SPTB c.G828T may be a novel
mutation, several nonsense mutations located downstream of it have
been previously identified as pathogenic mutations (11,12).
Therefore, combined with the patient's clinical diagnosis of HS,
the c.G828T SNP was identified as a pathogenic mutation, and was
probably causative for this HS development in this patient,
according to the ACMG guidelines. Notably, although previously
reported cases of HS that had nonsense mutations in SPTB
displayed a conspicuous spherocytosis with frequently encountered
dense spiculated red blood cells (11), the spherocytes of the present case
had not exhibited prominent surface projections (Fig. 1).
Pathogenic mutation analysis of
UGT1A1
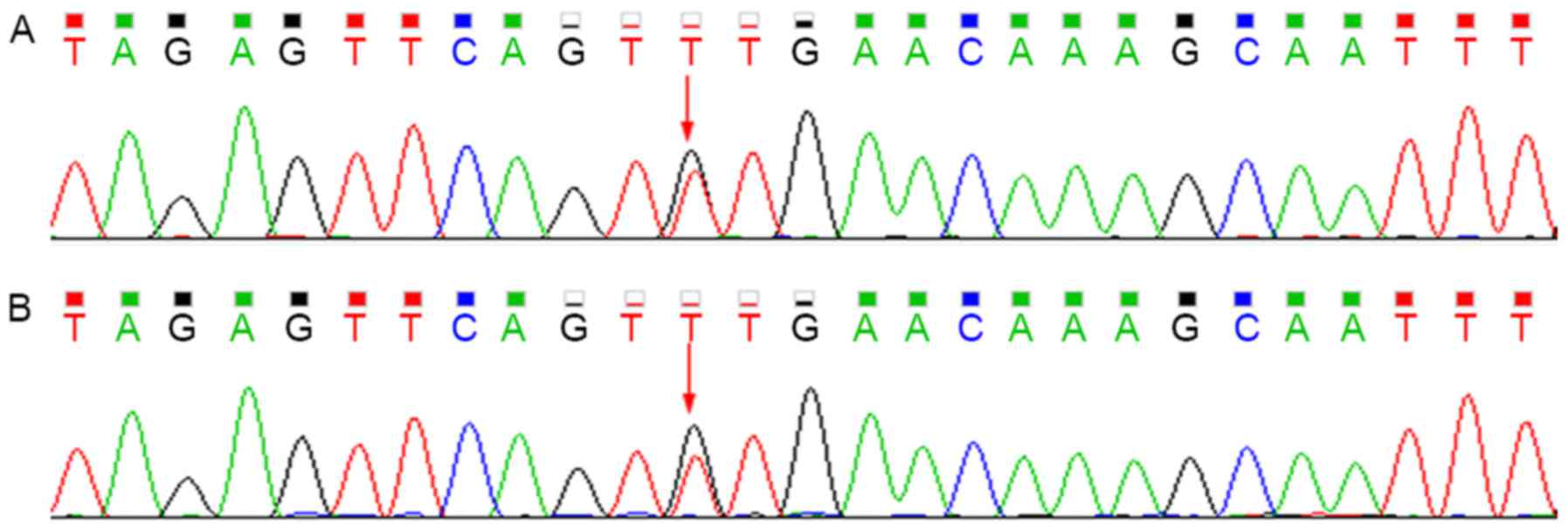
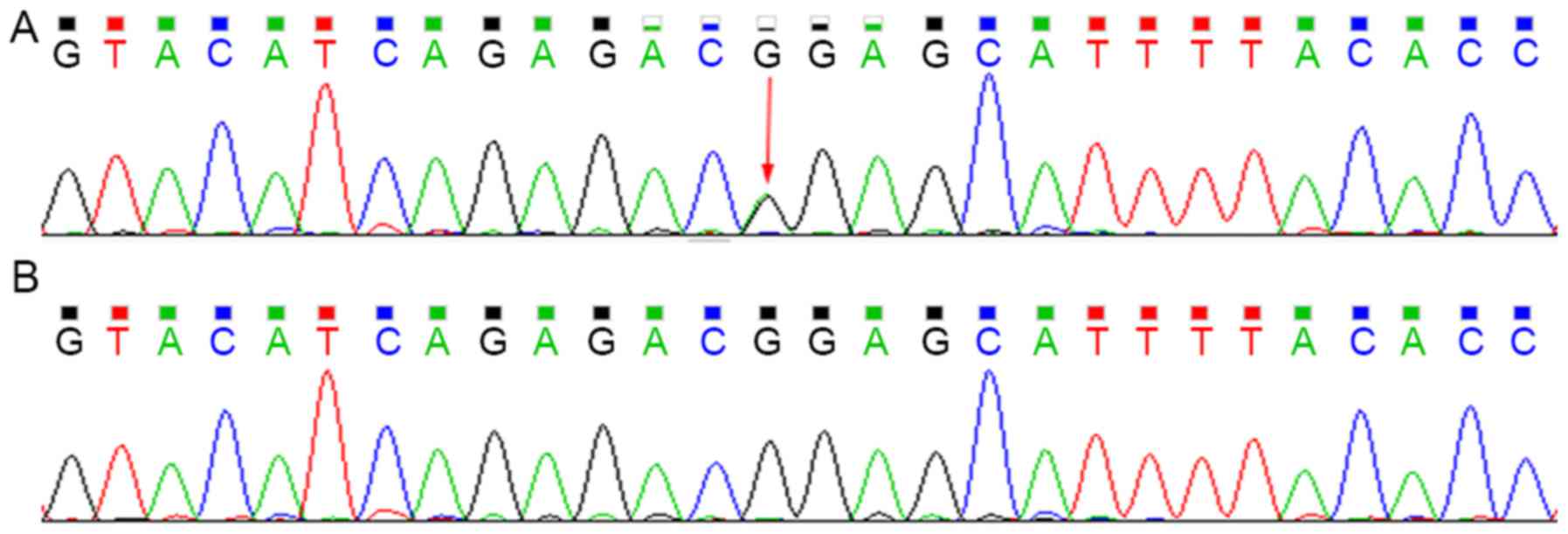
Sequencing analysis revealed that the patient
harbored the c.G211A (p.G71R) heterozygous mutation within exon 1
of UGT1A1 and the T3279G heterozygous mutation within the
PBREM of UGT1A1. The heterozygous T3279G mutation was also
detected in the proband's mother, whereas the G211A mutation was
not (Figs. 3 and 4).
In a previous study, the heterozygous G211A mutation
was reported to reduce UGT1A1 transferase activity to 60% of the
normal level, and the homozygous G211A mutation reduced the
activity to 30%, whereas the homozygous T3279G mutation reduced the
activity to approximately 60% (13). Therefore, the compound heterozygous
mutations may result in 30–60% activity of the normal UGT1A1 level.
Based on the present results, UGT1A1 partial deficiency was
diagnosed.
Discussion
HS and UGT1A1 deficiency are relatively common
causes of unconjugated hyperbilirubinemia, and their coexistence
may interfere with the proper diagnosis (2–4). The
present study described a female Chinese patient presenting with
congenital jaundice and anemia that was eventually diagnosed as
having HS combined with UGT1A1 deficiency following follow-up of
>10 years.
In the present case, anemia, splenomegaly,
reticulocytosis, spherocytes, hypercellular bone marrow with
remarkable erythroid hyperplasia and positive family history had
led to the presumptive diagnosis of HS. However, the erythrocyte
osmotic fragility test was repeatedly normal, and there was no
cut-off value of spherocytes for the diagnosis of HS. Therefore,
confirmation of the diagnosis relied on the positive EMA test and
genetic analysis.
As the patient's clinical findings were not typical
for both HS and UGT1A1 deficiency, the diagnoses had been pending
for >10 years. Notably, the serum bilirubin levels increased
over time; while the patient's anemia became milder. Then, up to
puberty, the discrepancy in the severity of hyperbilirubinemia and
anemia became significant, which may be related to the increased
hemoglobin turnover around puberty (5). This discrepancy suggests that the
patient's HS coexists with other conditions associated with
unconjugated hyperbilirubinemia, especially UGT1A1 partial
deficiency. Therefore, for patients with similar discrepancies
(inappropriately high serum bilirubin level compared with the
degree of hemolysis), the possibility of the coexistence of UGT1A1
partial deficiency and HS should be considered.
HS is normally inherited in an autosomal dominant
manner, which may be induced by pathogenic mutations in ankyrin 1
on chromosome 8p11, SPTB on chromosome 14q23, spectrin
α erythrocytic 1 on chromosome 1q21, solute carrier family 4
member 1 on chromosome 17q21, or erythrocyte membrane protein band
4.2 on chromosome 15q15 (14). As
HS may develop from multiple pathogenic genes, genetic diagnosis
was not applicable prior to the clinical application of NGS. In
previously reported cases of HS combined with UGT1A1 deficiency,
the diagnosis of HS was based on typical clinical findings, such as
spherocytes with increased erythrocyte osmotic fragility (5,6).
Thus, atypical cases with normal erythrocyte osmotic fragility may
have been underdiagnosed. The present study used NGS combined with
direct sequencing to identify a novel pathogenic mutation,
SPTB G828T, and the diagnosis of HS was confirmed in this
case study. The SPTB G828T mutation was not present in the
proband's mother, and was probably inherited from the deceased
father (Fig. 5).
UGT1A1 deficiency results from the causative
mutations in UGT1A1 gene, which is located on chromosome
2q37 (15). A number mutations or
SNPs of UGT1A1 that influence bilirubin glucuronidation have
been identified, which may cause Crigler-Najjar syndrome, Gilbert's
syndrome, neonatal hyperbilirubinemia or non-symptom based on the
severity of the decrease in enzymatic activity (16,17).
Therefore, genetic analysis was crucial to clarify the diagnosis
and severity of UGT1A1 deficiency. In the present case report,
compound heterozygous mutations (T3279G and G211A) probably led to
the reduced UGT1A1 activity, to 30–60% of normal activity; patients
with 30–60% UGT1A1 activity are typically asymptomatic (13), other than experiencing jaundice if
not in combination with other conditions such as HS. The T3279G
mutation of UGT1A1 in the proband was also detected in the
mother, but the G211A mutation was not, which suggested that the
patient inherited the T3279G mutation from the patient's mother and
the G211A mutation from the patient's deceased father (Fig. 5). Additionally, although the
proband did not develop gallstones, which is common in patients
with both HS and UGT1A1 deficiency (18), the patient was considered to be
at-risk for cholelithiasis and was advised to accept periodical
abdominal ultrasonic examination.
In conclusion, both HS and UGT1A1 deficiency are
associated with hyperbilirubinemia, and their coexistence is often
underdiagnosed. The discrepancy between the level of elevated serum
bilirubin and the degree of hemolysis suggested the possibility of
the coexistence. However, when the clinical or laboratory findings
were inconclusive, the diagnosis was questioned; thus, genetic
analysis was crucial to confirm the diagnosis and to avoid
underdiagnosis. Early diagnosis saves time-consuming clinical
reasoning and observation. Comprehensive NGS, as a new diagnostic
tool, may not only identify HS specifically and efficiently, but
also may contribute to expanding the mutation spectrum of
associated genes. In addition, genetic analysis combined with
evaluation of UGT1A1 activity may clarify the pathogenesis of the
associated hyperbilirubinemia and may explain the clinical
variability, which may aid patients in making informed medical and
personal decisions.
Acknowledgements
The present study was supported in part by the
National Natural Sciences Foundation of China (grant no.
81100360).
References
|
1
|
Bolton-Maggs PH, Langer JC, Iolascon A,
Tittensor P and King MJ: General Haematology Task Force of the
British Committee for Standards in Haematology: Guidelines for the
diagnosis and management of hereditary spherocytosis-2011 update.
Br J Haematol. 156:37–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Udomuksorn W, Elliot DJ, Lewis BC,
Machenzie PI, Yoovathaworn K and Miners JO: Influence of mutations
associated with Gilbert and Crigler-Najjar type II syndromes on the
glucuronidation kinetics of bilirubin and other
UDP-glucuronosyltransferase 1A substrates. Pharmacogenet Genomics.
17:1017–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akaba K, Kimura T, Sasaki A, Tanabe S,
Ikegami T, Hashimoto M, Umeda H, Yoshida H, Umetsu K, Chiba H, et
al: Neonatal hyperbilirubinemia and mutation of the bilirubin
uridine diphosphate-glucuronosyltransferase gene: A common missense
mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int.
46:21–26. 1998.PubMed/NCBI
|
|
4
|
Tse WT and Lux SE: Red blood cell membrane
disorders. Br J Haematol. 104:2–13. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garg PK, Kumar A, Teckchandani N and Hadke
NS: Hereditary spherocytosis coexisting with Gilbert's syndrome: A
diagnostic dilemma. Singapore Med J. 49:e308–e309. 2008.PubMed/NCBI
|
|
6
|
Iijima S, Ohzeki T and Maruo Y: Hereditary
spherocytosis coexisting with UDP-glucuronosyltransferase
deficiency highly suggestive of Crigler-Najjar syndrome type II.
Yonsei Med J. 52:369–372. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sharma S, Vukelja SJ and Kadakia S:
Gilbert's syndrome co-existing with and masking hereditary
spherocytosis. Ann Hematol. 74:287–289. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Girodon F, Garçon L, Bergoin E, Largier M,
Delaunay J, Fénéant-Thibault M, Maynadié M, Couillaud G, Moreira S
and Cynober T: Usefulness of the eosin-5′-maleimide cytometric
method as a first-line screening test for the diagnosis of
hereditary spherocytosis: Comparison with ektacytometry and protein
electrophoresis. Br J Haematol. 140:468–470. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
1000 Genomes Project Consortium, .
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA,
Hurles ME and McVean GA: A map of human genome variation from
population-scale sequencing. Nature. 467:1061–1073. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hassoun H, Vassiliadis JN, Murray J,
Njolstad PR, Rogus JJ, Ballas SK, Schaffer F, Jarolim P, Brabec V
and Palek J: Characterization of the underlying molecular defect in
hereditary spherocytosis associated with spectrin deficiency.
Blood. 90:398–406. 1997.PubMed/NCBI
|
|
12
|
Maciag M, Płochocka D, Adamowicz-Salach A
and Burzyńska B: Novel beta-spectrin mutations in hereditary
spherocytosis associated with decreased levels of mRNA. Br J
Haematol. 146:326–332. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sugatani J, Yamakawa K, Yoshinari K,
Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M and
Miwa M: Identification of a defect in the UGT1A1 gene promoter and
its association with hyperbilirubinemia. Biochem Biophys Res
Commun. 292:492–497. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delaunay J, Alloisio N, Morle L, Baklouti
F, Dalla Venezia N, Maillet P and Wilmotte R: Molecular genetics of
hereditary elliptocytosis and hereditary spherocytosis. Ann Genet.
39:209–221. 1996.PubMed/NCBI
|
|
15
|
van Es HH, Bout A, Liu J, Anderson L,
Duncan AM, Bosma P, Oude Elferink R, Jansen PL, Chowdhury JR and
Schurr E: Assignment of the human UDP glucuronosyltransferase gene
(UGT1A1) to chromosome region 2q37. Cytogenet Cell Genet.
63:114–116. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yu Z, Zhu K, Wang L, Liu Y and Sun J:
Association of neonatal hyperbilirubinemia with UGT1A1 gene
polymorphisms: A meta-analysis. Med Sci Monit. 21:3104–3114. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yamamoto K, Sato H, Fujiyama Y, Doida Y
and Bamba T: Contribution of two missense mutations (G71R and
Y486D) of the bilirubin UDP glycosyltransferase (UGT1A1) gene to
phenotypes of Gilbert's syndrome and Crigler-Najjar syndrome type
II. Biochim Biophys Acta. 1406:267–273. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
del Giudice EM, Perrotta S, Nobili B,
Specchia C, d'Urzo G and Iolascon A: Coinheritance of Gilbert
syndrome increases the risk for developing gallstones in patients
with hereditary spherocytosis. Blood. 94:2259–2262. 1999.PubMed/NCBI
|