Introduction
Phosphodiesterases (PDEs) are a family of enzymes
that are able to lyse phosphodiester bonds, are expressed widely,
and have demonstrable clinical significance (1,2).
PDE10A has been demonstrated to be a potential target for the
treatment of central nervous system (CNS) disorders (3,4).
Previous studies have confirmed that PDE10A inhibitors have
important biological activity in the treatment of psychosis
(5–7). Therefore, screening for PDE10A
inhibitors is an effective strategy for the treatment of CNS
disorders.
The roots of Sophora flavescens (Sophorae
radix) have been used as an herbal medicine for thousands of years
in East Asian countries. They have been demonstrated to possess
diverse pharmacological properties, including antitumor,
antioxidant, anti-inflammatory, antiapoptotic, vasodilatory and CNS
regulatory functions (8–15). Different types of compounds,
including triterpenoids, flavonoids and quinolizidine alkaloids,
have been isolated from the roots of S. flavescens (16–19).
In order to identify natural PDE10A inhibitors, virtual screening
based on a pharmacophore model and molecular docking was performed
to identify the chemical molecule database of S. flavescens.
In addition, literature mining was performed to validate the
biological activity of the top-ranking hits from the virtual
screening. A total of two hits were reported to have inhibitory
activity against cyclic adenosine monophosphate (cAMP)
phosphodiesterase, while one exhibited protective effects against
glutamate-induced oxidative stress in the CNS. The computational
methods used in the present study efficiently identified PDE10A
inhibitors from the roots of S. flavescens. Therefore, this
screening method and workflow may be applied to other traditional
Chinese medicines in the search for potential bioactive
compounds.
Materials and methods
Chemical molecule database of S
flavescens
In order to establish the chemical molecule database
of S. flavescens, the traditional Chinese medicine database
(Chinese Academy of Sciences, Beijing, China; 2009) was searched
using ‘Sophora flavescens’ as the key term from the plant
sources. A total of 78 molecules that were retrieved from S.
flavescens, including kushenol B, kushenol E, kosamol A,
kosamol Q and kushenol X, were downloaded and saved in Mol2 format.
The two-dimensional structures were individually converted to
three-dimensional molecular conformations using the CONCORD module
in SYBYL X-1.2 (Tripos Inc., St. Louis, MO, USA). All the hydrogen
atoms were subsequently added, and the energy optimization was
performed using the Tripos force field in SYBYL X-1.2. The chemical
molecule database of S. flavescens was built as a UNITY hit
list file in SYBYL X-1.2.
Pharmacophore-based virtual
screening
A diverse dataset of 30 experimentally identified
PDE10A inhibitors was retrieved from the published literature
(20–24). The molecules were drawn using the
ISIS-Draw (version 2.5; MDL Informations Systems, Inc., San Ramon,
CA, USA) software and energy-optimized using Tripos force field.
The hydrogen atoms were added and Merck Molecular Force Field 94
charges were assigned using SYBYL X-1.2. All the molecules were
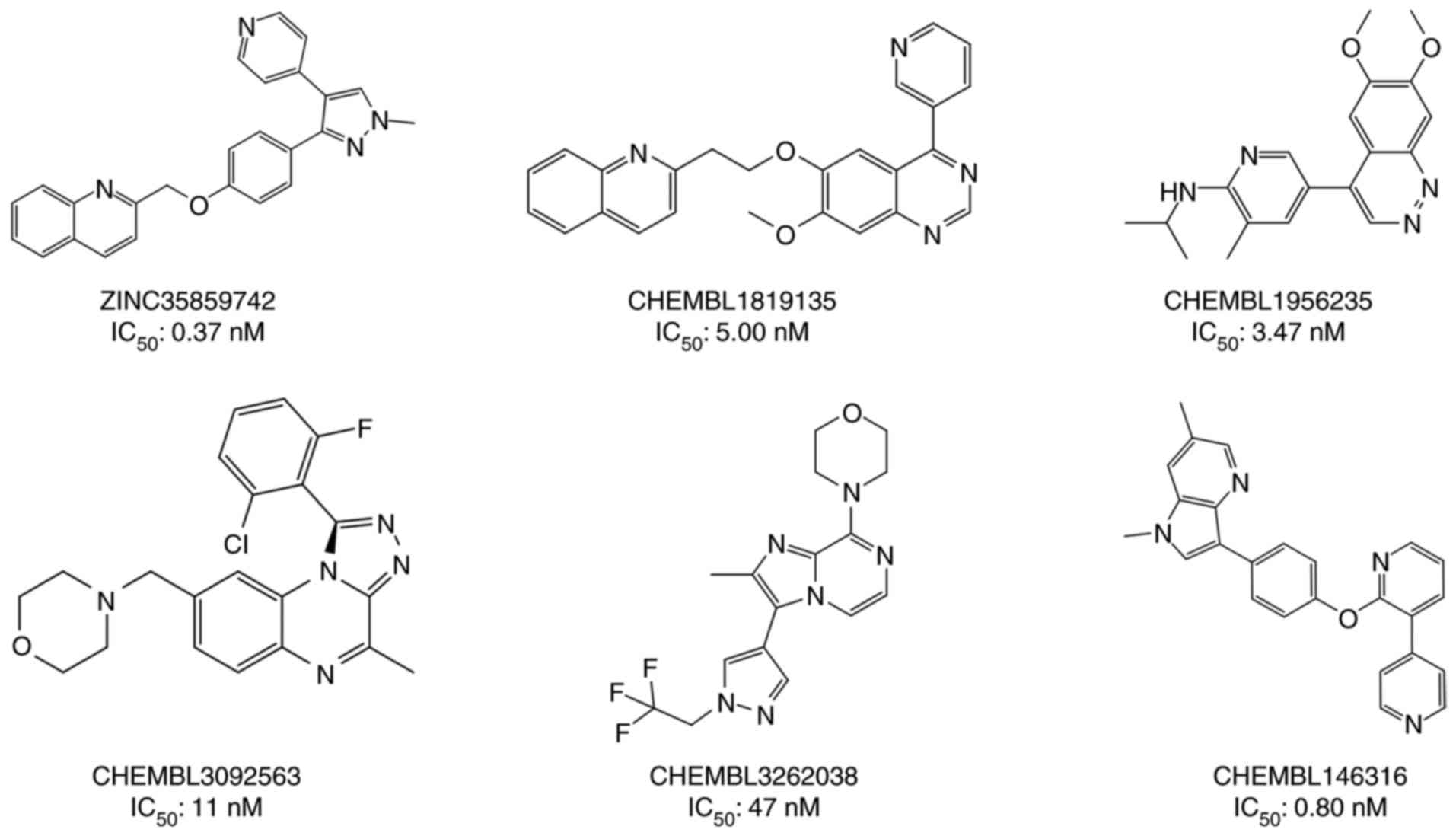
saved in the mol2 format. From the optimized molecules, six
(Fig. 1) were selected to generate
pharmacophore models of PDE10A inhibitors, and they all met the
following criteria: i) Structural diversity; ii) high inhibitory
effect against the PDE10A target; and iii) similar binding mode of
inhibition. The other 24 molecules were used to access the
generated pharmacophore models.
All molecular modeling studies were performed on a
Dell Red Hat Linux workstation using the Common Feature
Pharmacophore Generation protocol in Discovery Studio (version 3.5;
Accelrys, San Diego, CA, USA). The pharmacophore models were
generated using the HIPHOP module. All compounds were energy
minimized using the CHARMM force field. A principal value of 2 and
a maximum omit feature value of 0 was assigned to the six compounds
in the training set. Hydrogen-bond donor (HBD), hydrogen-bond
acceptor (HBA) and hydrophobic (HY) features were selected during
pharmacophore generation. In order to access the generated
pharmacophore models, 100 known PDE10A inhibitors obtained from the
published literature and 300 non-PDE10A inhibiting compounds were
used as a validation set (25–27).
A total of four evaluation parameters [A%, Y%, identified effective
index (N), and comprehensive appraisal index (CAI)] proposed from
previous work were calculated to access the generated pharmacophore
models, according to the following formulae (28): A%=Ha/Ax100; Y%=Ha/Htx100;
N=HaxD/HtxA; CAI=NxA%.
Where D and A represent the total number of
molecules used (n=400) and the total number of known PDE10A
inhibitors (n=100) in the validation set, respectively. Ht and Ha
represent the number of hits obtained using the pharmacophore-based
virtual screening and those from the 100 known PDE10A inhibitors,
respectively. N indicates the ability to recognize the known PDE10A
inhibitors compared with non-PDE10A inhibiting compounds. CAI
expresses the comprehensive ability to discover PDE10A inhibitors
from a specific database (28).
The pharmacophore model with the highest CAI value
was used to screen the chemical molecule database of S.
flavescens using the Search 3D Database module in Discovery
Studio version 3.5 with the default setting. The fit value was
calculated to indicate the pharmacophoric match between the query
and the hits, with a higher value implying that a better alignment
was obtained between the hit and pharmacophore model (29).
Molecular docking-based virtual
screening
The molecular docking-based virtual screening was
performed using a Surflex-Dock module, which has been successfully
utilized for molecular docking and binding free energy calculations
(30–33). The x-ray crystal structure of
PDE10A has been resolved (Protein Data Bank no. 2O8H) and,
therefore, it was selected as the docking protein (34). The PDE10A protein model was cleaned
by removing the co-crystallized water molecules and adding hydrogen
atoms. The Gasteiger-Hückel charges were assigned using SYBYL
X-1.2. An energy optimization was performed using Tripos force
field for 1,000 iterations in SYBYL X-1.2 with the default
parameters.
In order to verify the reliability of the docking
protocol established in the present study, the co-crystallized
PDE10A inhibitor (ligand 227;
C24H29N5O4S) was
extracted and re-docked into the active site of PDE10A. The active
site of PDE10A was defined as the ProtoMol generated using the
steric hydrophobic (CH4) and hydrogen bond (C=O) groups,
and the hydrogen acceptor (N-H) within 0.5 Å of the ligand 227
binding site.
All compounds from the chemical molecule database of
S. flavescens were docked into the active site of PDE10A in
turn using the Surflex-Dock module. The total score was calculated
for each compound following running of the Surflex-Dock module. A
higher total score implied an increased binding affinity between
the protein and the ligand, based on an empirically derived scoring
function.
Results
Pharmacophore-based virtual
screening
Ten HIPHOP models were generated and model
assessment studies (Table I)
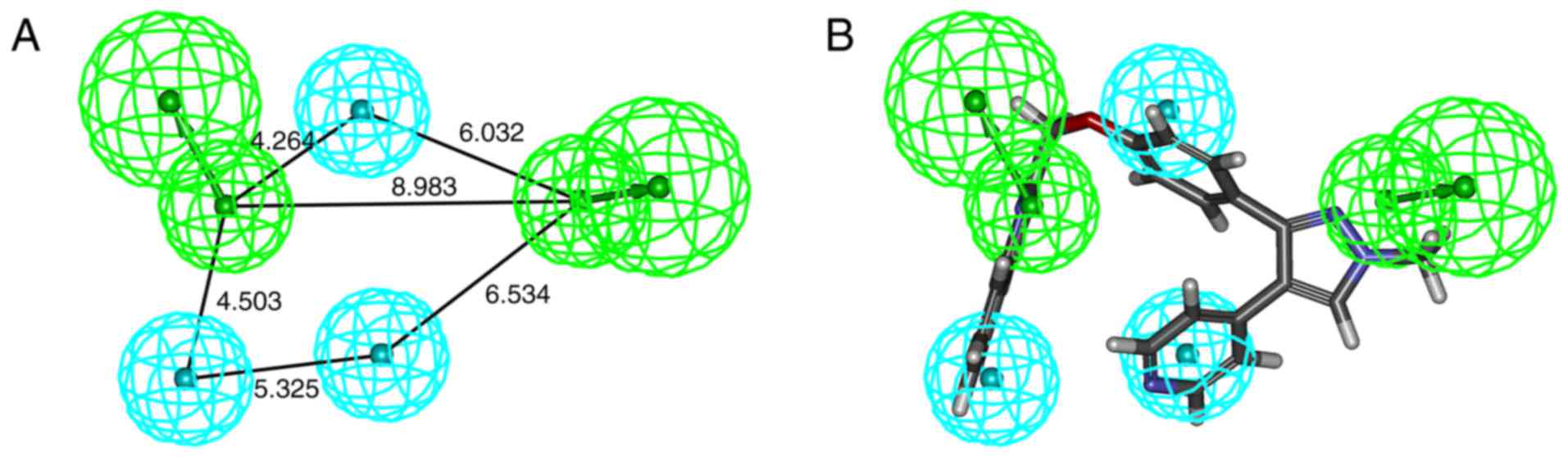
indicated that Model_2 (Fig. 2A)
had the highest CAI and N values (Table I). Therefore, Model_2 was used to
screen the chemical molecular database of S. flavescens.
Model_2 contained two HBA (marked in green) and three HY (marked in
cyan). The best active compound (ZINC35859742) was able to map all
features of Model_2 with a fit value of 2.23 (Fig. 2B). A total of 32 hits were obtained
following the pharmacophore-based virtual screening against the
chemical molecule database of S. flavescens. The top 20 hits
ranked by fit value are presented in Table II.
 | Table I.Assessment of generated pharmacophore
models. |
Table I.
Assessment of generated pharmacophore
models.
| Model | Ht | Ha | A, % | Y, % | N | CAI |
|---|
| Model_1 | 139 | 69 |
69.00 | 49.64 | 1.99 | 1.37 |
| Model_2 | 171 | 101 | 101.00 | 59.06 | 2.36 | 2.39 |
| Model_3 | 141 | 83 |
83.00 | 58.87 | 2.36 | 1.95 |
| Model_4 | 174 | 92 |
92.00 | 52.87 | 2.12 | 1.95 |
| Model_5 | 154 | 80 |
80.00 | 51.95 | 2.08 | 1.66 |
| Model_6 | 160 | 76 |
76.00 | 47.50 | 1.90 | 1.44 |
| Model_7 | 161 | 84 |
84.00 | 52.17 | 2.09 | 1.75 |
| Model_8 | 153 | 81 |
81.00 | 52.94 | 2.12 | 1.72 |
| Model_9 | 160 | 94 |
94.00 | 58.75 | 2.35 | 2.21 |
| Model_10 | 176 | 88 |
88.00 | 50.00 | 2.00 | 1.76 |
| Table II.Hits obtained using
pharmacophore-based virtual screening. |
Table II.
Hits obtained using
pharmacophore-based virtual screening.
| ID | Name | QFIT |
|---|
| 1 | Isokurarinone | 4.03 |
| 2 | Leachianone A | 4.03 |
| 3 | Kushenol X | 3.80 |
| 4 | Kosamol Q | 3.73 |
| 5 | Kushenol C | 3.67 |
| 6 | Kuraridinol | 3.57 |
| 7 |
2′-Methoxykurarinone | 3.51 |
| 8 | Kuraridin | 3.50 |
| 9 | Kushenol P | 3.48 |
| 10 | Norkurarinol | 3.36 |
| 11 | Kurarinone | 3.32 |
| 12 | Kushenol G | 3.31 |
| 13 | Neokurarinol | 3.09 |
| 14 | Sophoraflavanone
G | 3.05 |
| 15 | Kushenol E | 2.60 |
| 16 | Kosamol V | 2.47 |
| 17 | Kosamol A | 2.45 |
| 18 | Kushenol B | 2.42 |
| 19 | Kushenol L | 2.35 |
| 20 | Kushenol K | 2.11 |
Molecular docking-based virtual
screening
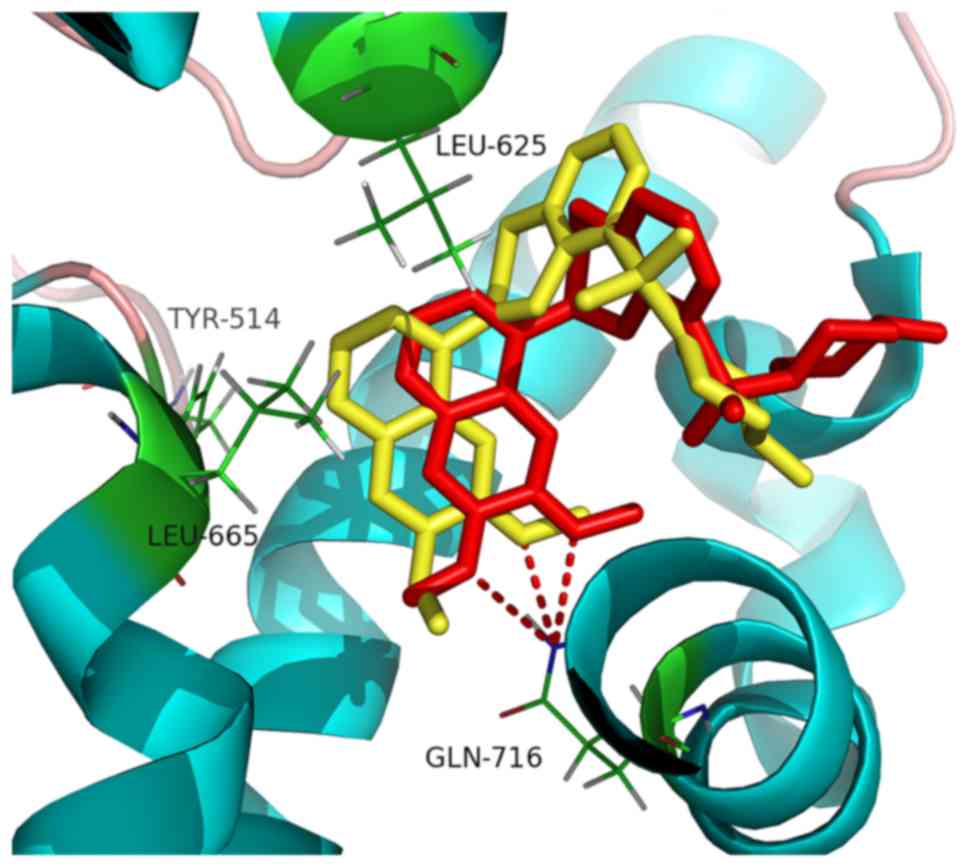
The root-mean-square deviation (RMSD) of the
conformations between the re-docked and co-crystallized ligand 227
(Fig. 3) was calculated as 2.6 Å,
which indicated that the difference between the co-crystal and
re-docked conformation of ligand 227 was very small. The docking
protocol established in the present study has a strong ability to
reproduce the co-crystal conformation and binding mode of PDE10A
inhibitors. The molecular docking analysis of the chemical molecule
database of S. flavescens and the PDE10A protein resulted in
a hit list of 14 molecules with total scores >6.0 (Table III).
| Table III.Hits obtained through docking-based
virtual screening. |
Table III.
Hits obtained through docking-based
virtual screening.
| Compound | Name | Total score |
|---|
| 1 | Kosamol Q | 8.82 |
| 2 | Kosamol A | 8.39 |
| 3 | Kushenol X | 8.08 |
| 4 | Kurarinol | 7.34 |
| 5 | Neokurarinol | 7.21 |
| 6 | Kurarinone | 7.21 |
| 7 | Norkurarinone | 7.09 |
| 8 | Kushenol E | 7.02 |
| 9 | Kosamol S | 6.92 |
| 10 | Sophoraflavanone
G | 6.91 |
| 11 | Kuraridinol | 6.90 |
| 12 | Kushenol O | 6.89 |
| 13 | Kushenol B | 6.72 |
| 14 | 5-O-Methyl kushenol
C | 6.69 |
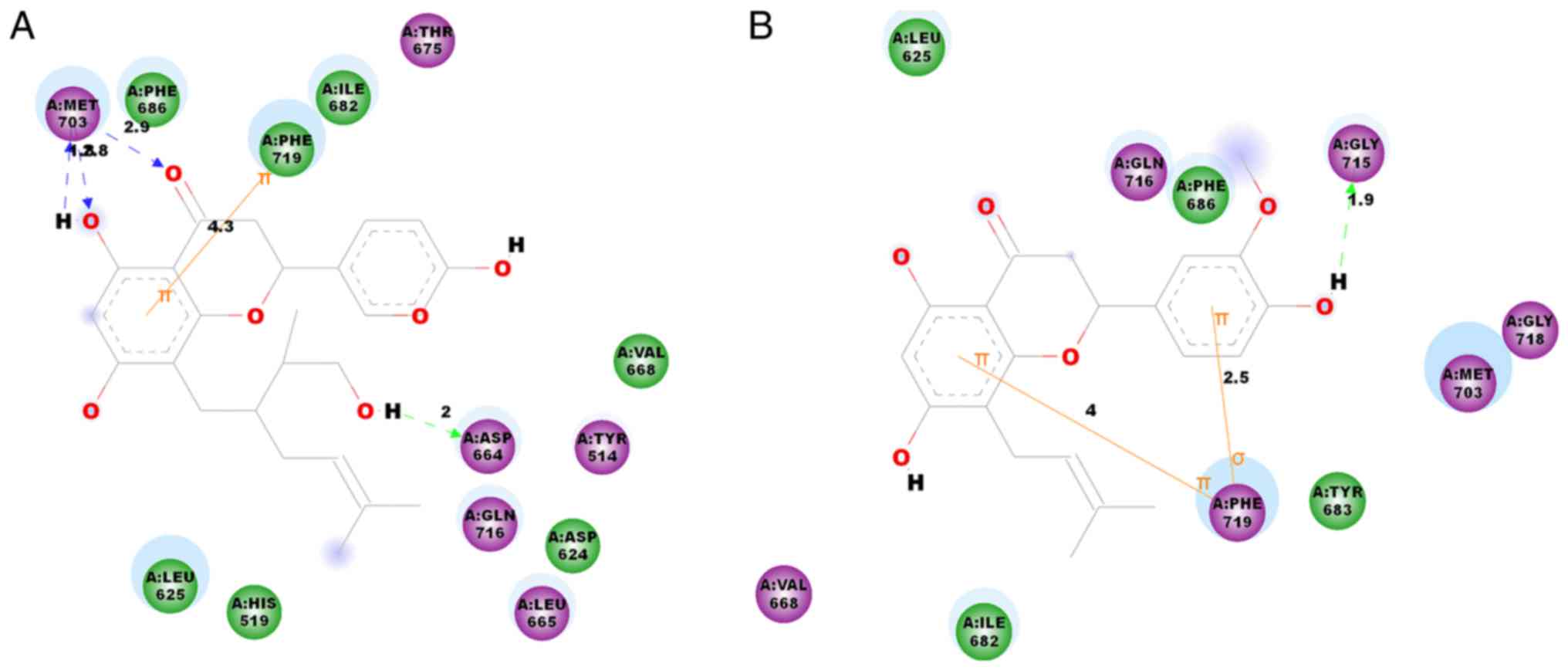
Ligand-protein binding mode analysis is important
for the study of molecular interactions, binding affinity and
active ingredient identification from traditional Chinese medicine
(35,36). The binding mode between PDE10A and
kosamol Q and kosamol A (Fig. 4A and
B, respectively) was investigated.
Discussion
The combination of three-dimensional pharmacophore
modeling and molecular docking in the present study demonstrated
marked advantages for identifying direct PDE10A inhibitors from the
chemical molecule database of S. flavescens. The
three-dimensional pharmacophore model focused on quick generation
of the common characteristics of the known PDE10A inhibitors, while
the molecular docking method was able to rapidly calculate the
binding force between the small-molecule ligands and the target
protein. In addition, molecular docking was able to provide the
binding conformations between the ligands and the PDE10A protein,
which is important for the identification of active compounds from
traditional Chinese herbs, in addition to the modification and
optimization of molecular structures.
According to the virtual screening based on the
pharmacophore model and molecular docking, nine compounds were
obtained as hits. These nine hits were kushenol B, kurarinone,
sophoraflavanone G, kosamol Q, kosamol A, kushenol X, neokurarinol,
kushenol E and kuraridinol. It has been reported that kushenol B
and kurarinone have in vitro inhibitory activity against
cAMP phosphodiesterase, and their half-maximal inhibitory
concentrations were determined as 31 and 25 µmol/l, respectively
(37). The CNS protective effect
of four lavandulyl flavanones isolated from S. flavescens
was examined and sophoraflavanone G was observed to exhibit the
function of protecting HT22 immortalized hippocampal cells against
glutamate-induced oxidative stress (38). Therefore, the published data
confirmed, to a certain extent, the predictions of the
computational model approach used in the present study. The models
additionally elucidated the recognition mode of the intermolecular
actions between the compounds and PDE10A.
In conclusion, pharmacophore- and molecular
docking-based virtual screening provided an effective approach to
identify PDE10A inhibitors from Chinese medical herbs, and nine
molecules were determined to be potential inhibitors. In addition,
molecular docking is a feasible strategy to characterize the
interactions of the natural ingredients from S. flavescens
with the PDE10A target. In particular, kosamol compounds, including
kosamol Q and kosamol A, which exhibit pharmacological effects,
including CNS protectant activities, may be promising PDE10A
inhibitors. These compounds required further investigation,
including additional bioactivity evaluation.
Acknowledgements
The present study was supported by the Beijing
Municipal Natural Science Foundation (grant no. 7164239) and the
National Natural Science Foundation of China (grant no.
81603311).
References
|
1
|
Otero C, Peñaloza JP, Rodas PI,
Fernández-Ramires R, Velasquez L and Jung JE: Temporal and spatial
regulation of cAMP signaling in disease: Role of cyclic nucleotide
phosphodiesterases. Fundam Clin Pharmacol. 28:593–607. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Goo L, Luo L, Ju R, Chen C, Zhu L, Li J,
Yu X, Ye C and Zhang D: Carboxyamidotriazole: A novel inhibitor of
both cAMP-phosphodiesterases and cGMP-phosphodiesterases. Eur J
Pharmacol. 746:14–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chappie T, Humphrey J, Menniti F and
Schmidt C: PDE10A inhibitors: An assessment of the current CNS drug
discovery landscape. Curr Opin Drug Discov Devel. 12:458–467.
2009.PubMed/NCBI
|
|
4
|
Chen H, Lester-Zeiner D, Shi J, Miller S,
Glaus C, Hu E, Chen N, Able J, Biorn C, Wong J, et al: AMG 580: A
novel small molecule phosphodiesterase 10A (PDE10A) positron
emission tomography tracer. J Pharmacol Exp Ther. 352:327–337.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Siuciak JA, McCarthy SA, Chapin DS,
Fujiwara RA, James LC, Williams RD, Stock JL, McNeish JD, Strick
CA, Menniti FS and Schmidt CJ: Genetic deletion of the
striatum-enriched phosphodiesterase PDE10A: Evidence for altered
striatal function. Neuropharmacology. 51:374–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Natesan S, Ashworth S, Nielsen J, Tang SP,
Salinas C, Kealey S, Lauridsen JB, Stensbøl TB, Gunn RN, Rabiner EA
and Kapur S: Effect of chronic antipsychotic treatment on striatal
phosphodiesterase 10A levels: A [(1)(1)C]MP-10 PET rodent imaging
study with ex vivo confirmation. Transl Psychiatry. 4:e3762014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bartolomé-Nebreda JM, Alonso DDS, Artola
M, Delgado F, Delgado Ó, Martín-Martín ML, Martínez-Viturro CM,
Pena MÁ, Tong HM, Van Gool M, et al: Identification of a novel
orally bioavailable phosphodiesterase 10A (PDE10A) inhibitor with
efficacy in animal models of schizophrenia. J Med Chem. 58:978–993.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rahimi R, Ghiasi S, Azimi H, Fakhari S and
Abdollahi M: A review of the herbal phosphodiesterase inhibitors;
future perspective of new drugs. Cytokine. 49:123–129. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bai L, Zhu LY, Yang BS, Shi LJ, Liu Y,
Jiang AM, Zhao LL, Song G and Liu TF: Antitumor and
immunomodulating activity of a polysaccharide from Sophora
flavescens Ait. Int J Biol Macromol. 51:705–709. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang N, Liang B, Srivastava K, Zeng J,
Zhan J, Brown L, Sampson H, Goldfarb J, Emala C and Li XM: The
Sophora flavescens flavonoid compound trifolirhizin inhibits
acetylcholine induced airway smooth muscle contraction.
Phytochemistry. 95:259–267. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sim KM, Kim KH, Hwang GB, Seo S, Bae GN
and Jung JH: Development and evaluation of antimicrobial activated
carbon fiber filters using Sophora flavescens nanoparticles. Sci
Total Environ. 493:291–297. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zheng K, Li C, Shan X, Liu H, Fan W and
Wang Z: A study on isolation of chemical constituents from Sophora
flavescens Ait. and their anti-glioma effects. Afr J Tradit
Complement Altern Med. 11:156–160. 2013.PubMed/NCBI
|
|
13
|
He X, Fang J, Huang L, Wang J and Huang X:
Sophora flavescens Ait: Traditional usage, phytochemistry and
pharmacology of an important traditional Chinese medicine. J
Ethnopharmacol. 172:10–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, You RL, Qin WJ, Hai LN, Fang MJ,
Huang GH, Kang RX, Li MH, Qiao YF, Li JW and Li AP: Anti-tumor
activities of active ingredients in Compound Kushen Injection. Acta
Pharmacol Sin. 36:676–679. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhu L, Pan QX, Zhang XJ, Xu YM, Chu YJ,
Liu N, Lv P, Zhang GX and Kan QC: Protective effects of matrine on
experimental autoimmune encephalomyelitis via regulation of ProNGF
and NGF signaling. Exp Mol Pathol. 100:337–343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhou Y, Wu Y, Deng L, Chen L, Zhao D, Lv
L, Chen X, Man J, Wang Y, Shan H and Lu Y: The alkaloid matrine of
the root of Sophora flavescens prevents arrhythmogenic effect of
ouabain. Phytomedicine. 21:931–935. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han R, Takahashi H, Nakamura M, Bunsupa S,
Yoshimoto N, Yamamoto H, Suzuki H, Shibata D, Yamazaki M and Saito
K: Transcriptome analysis of nine tissues to discover genes
involved in the biosynthesis of active ingredients in Sophora
flavescens. Biol Pharm Bull. 38:876–883. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu C, Yang N, Song Y, Wang L, Zi J, Zhang
S, Dunkin D, Busse P, Weir D, Tversky J, et al: Ganoderic acid C1
isolated from the anti-asthma formula, ASHMI™ suppresses
TNF-α production by mouse macrophages and peripheral blood
mononuclear cells from asthma patients. Int Immunopharmacol.
27:224–231. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao FC, Li H, Chen LM, Gao HM, Zhang QW,
Wang ZM and Wu PE: Study on quality standard of Sophora flavescens
root extract. Zhongguo Zhong Yao Za Zhi. 40:245–250. 2015.(In
Chinese). PubMed/NCBI
|
|
20
|
Asproni B, Murineddu G, Pau A, Pinna GA,
Langgård M, Christoffersen CT, Nielsen J and Kehler J: Synthesis
and SAR study of new phenylimidazole-pyrazolo [1,5-c]quinazolines
as potent phosphodiesterase 10A inhibitors. Bioorg Med Chem.
19:642–649. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kehler J, Ritzen A, Langgård M, Petersen
SL, Farah MM, Bundgaard C, Christoffersen CT, Nielsen J and Kilburn
JP: Triazoloquinazolines as a novel class of phosphodiesterase 10A
(PDE10A) inhibitors. Bioorg Med Chem Lett. 21:3738–3742. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bauer U, Giordanetto F, Bauer M, O'Mahony
G, Johansson KE, Knecht W, Hartleib-Geschwindner J, Carlsson ET and
Enroth C: Discovery of 4-hydroxy-1,6-naphthyridine-3-carbonitrile
derivatives as novel PDE10A inhibitors. Bioorg Med Chem Lett.
22:1944–1948. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu E, Kunz RK, Rumfelt S, Chen N, Bürli R,
Li C, Andrews KL, Zhang J, Chmait S, Kogan J, et al: Discovery of
potent, selective, and metabolically stable 4-(pyridin-3-yl)
cinnolines as novel phosphodiesterase 10A (PDE10A) inhibitors.
Bioorg Med Chem Lett. 22:2262–2265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malamas MS, Stange H, Schindler R, Lankau
HJ, Grunwald C, Langen B, Egerland U, Hage T, Ni Y, Erdei J, et al:
Novel triazines as potent and selective phosphodiesterase 10A
inhibitors. Bioorg Med Chem Lett. 22:5876–5884. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Helal CJ, Kang Z, Hou X, Pandit J, Chappie
TA, Humphrey JM, Marr ES, Fennell KF, Chenard LK, Fox C, et al: Use
of structure-based design to discover a potent, selective, in vivo
active phosphodiesterase 10A inhibitor lead series for the
treatment of schizophrenia. J Med Chem. 54:4536–4547. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang Z, Lu X, Xu J, Rothfuss J, Mach RH
and Tu Z: Synthesis and in vitro evaluation of new analogues as
inhibitors for phosphodiesterase 10A. Eur J Med Chem. 46:3986–3995.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ho GD, Yang SW, Smotryski J, Bercovici A,
Nechuta T, Smith EM, McElroy W, Tan Z, Tulshian D, McKittrick B, et
al: The discovery of potent, selective, and orally active
pyrazoloquinolines as PDE10A inhibitors for the treatment of
Schizophrenia. Bioorg Med Chem Lett. 22:1019–1022. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang X, Xiang Y, Ren Z, Zhang Y and Qiao
Y: Rational questing for inhibitors of endothelin converting
enzyme-1 from Salvia miltiorrhiza by combining ligand-and
structure-based virtual screening. Canadian J Chem. 91:448–456.
2013. View Article : Google Scholar
|
|
29
|
Wang X, Zhang Y, Liu Q, Ai Z, Zhang Y,
Xiang Y and Qiao Y: Discovery of dual ETA/ETB receptor antagonists
from traditional chinese herbs through in silico and in vitro
screening. Int J Mol Sci. 17:3892016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang X, Zhang Y, Yang Y, Wu X, Fan H and
Qiao Y: Identification of berberine as a direct thrombin inhibitor
from traditional Chinese medicine through structural, functional
and binding studies. Sci Rep. 7:440402017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo C, Wang X, An C, Hwang CF, Miao W,
Yang L, Xu M, Bai A and Deng S: Molecular inhibition mechanisms of
cell migration and invasion by coix polysaccharides in A549 NSCLC
cells via targeting S100A4. Mol Med Rep. 15:309–316. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen YX, Li GZ, Zhang B, Xia ZY and Zhang
M: Molecular evaluation of herbal compounds as potent inhibitors of
acetylcholinesterase for the treatment of Alzheimer's disease. Mol
Med Rep. 14:446–452. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, Ren Z, He Y, Xiang Y, Zhang Y and
Qiao Y: A combination of pharmacophore modeling, molecular docking
and virtual screening for iNOS inhibitors from Chinese herbs.
Biomed Mater Eng. 24:1315–1322. 2014.PubMed/NCBI
|
|
34
|
Chappie TA, Humphrey JM, Allen MP, Estep
KG, Fox CB, Lebel LA, Liras S, Marr ES, Menniti FS, Pandit J, et
al: Discovery of a series of 6,7-dimethoxy-4-pyrrolidylquinazoline
PDE10A inhibitors. J Med Chem. 50:182–185. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mobley DL and Dill KA: Binding of
small-molecule ligands to proteins: ‘What you see’ is not always
‘what you get’. Structure. 17:489–498. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Laskowski RA and Swindells MB: LigPlot+:
Multiple ligand-protein interaction diagrams for drug discovery. J
Chem Inf Model. 51:2778–2786. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nikaido T, Ohmoto T, Kinoshita T, Sankawa
U, Delle Monache F, Botta B, Tomimori T, Miyaichi Y, Shirataki Y,
Yokoe I, et al: Inhibition of adenosine 3′,5′-cyclic monophosphate
phosphodiesterase by flavonoids. III. Chem Pharm Bull (Tokyo).
37:1392–1395. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jeong GS, Li B, Lee DS, Byun E, An RB, Pae
HO, Chung HT, Youn KH and Kim YC: Lavandulyl flavanones from
Sophora flavescens protect mouse hippocampal cells against
glutamate-induced neurotoxicity via the induction of heme
oxygenase-1. Biol Pharm Bull. 31:1964–1967. 2008. View Article : Google Scholar : PubMed/NCBI
|