Introduction
The study and development of a novel drug is not
only costly and time-consuming, but the safety and efficacy must be
seriously considered, and determine the success of the drug. Thus,
toxicity is one of the main reasons leading to the failure of drug
development. The liver is considered as the most important organ in
drug toxicity for as it is functionally interposed between the site
of absorption and the systemic circulation, and is a major site of
metabolism and elimination of foreign substances. However, these
features also render it a preferred target for drug toxicity
(1). Drug-induced liver injury is
one of the most common reasons that accounts for the attrition of
candidate drugs during the later stages of drug development
(2). Consequently, early detection
of drug-induced hepatotoxicity is essential before compounds are
tested in animals and enter clinical trials, to save time and
resources (3).
Natural product extracts have long been implemented
as therapeutics all over the world (4). Since the discovery of penicillin,
which was the first pure antibiotic isolated from the fungus
Penicillium rubens, the industry for natural products
developed in 1928 (5). The ocean
covers >70% of the earth's surface, and the marine environment
is home to a taxonomically diverse ecosystem full of bioactive
natural products from diverse sources as microorganisms,
invertebrates, plants and animals (6). Organisms such as algae, mollusks,
sponges, corals and tunicates have evolved to survive the high
concentrations of infectious and surface-fouling bacteria that are
indigenous to ocean waters. Marine microorganism has been
recognized as a rich source of biological macromolecules and the
search for bioactive compounds from marine organisms in recent
decades has produced an abundance of extracts with pharmaceutical
and industrial applications, such as food, cosmetics and
pharmacology (7).
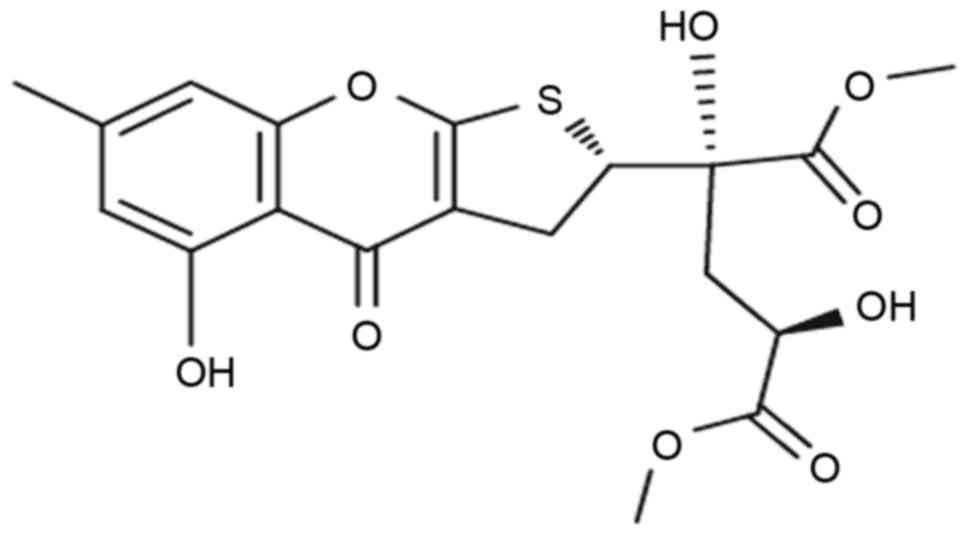
Oxalicumone A (POA) is a constituent isolated from a
culture broth of the South China Sea fungus Penicillium
oxalicum SCSGAF 0023 (8). Its
chemical structure was first identified by Zhang et al
(9) (Fig. 1). POA demonstrates significant
cytotoxicity against several human carcinoma cell lines with
IC50 ≤10 µM (8);
therefore, it represents a potent anticancer bioactive agent.
However, to the best of our knowledge, the influence of POA on
healthy human cells remains to be investigated.
Therefore, the present study aimed to investigate
the cytotoxic effects of POA on L-02 healthy human liver cells, and
the underlying mechanisms, including apoptosis pathways, oxidative
stress and mitochondrial function.
Materials and methods
Chemicals
RPMI 1640 medium and fetal bovine serum (FBS) were
purchased from Biological Industries USA (Cromwell, CT, USA) and
Biological Industries Israel Beit-Haemek (Kibbutz
Beit-Haemek, Israel), respectively. Cell Counting kit-8 (CCK-8) dye
was purchased from Dojindo Molecular Technologies, Inc. (Kumamoto,
Japan). Trypsin, dimethyl sulfoxide (DMSO) and Hoechst 33258 were
purchased from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). The
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI)
double staining kit, cell cycle kit,
5,5,6,6-tetra-chloro-1,1,3,3-tetraethylbenzimidazolyl-carbocyanine
iodide (JC-1) dye and caspase-3 activity assay kits were purchased
from Nanjing KeyGen Biotech Co., Ltd. (Nanjing, China). Glutathione
(GSH; cat. no. CEA294Ge) was purchased from Uscn Life Sciences,
Inc. (Wuhan, China). Radioimmunoprecipitation assay (RIPA) lysis
buffer and an Enhanced Chemiluminescence substrate kit were
purchased from the Biomiga (San Diego, CA, USA) and Beyotime
Institute of Biotechnology (Jiangsu, China), respectively. The
bicinchoninic acid (BCA) protein assay kit was purchased from
Bioteke Corporation (Beijing, China). Fas cell surface death
receptor (Fas; dilution 1:4,000; cat. no. ab133619), B-cell
lymphoma 2 (Bcl-2; dilution 1:4,000; cat. no. ab182858), Bcl-2
X-associated protein (Bax; dilution, 1:4,000; cat. no. ab32503),
cytochrome c (cyt c; dilution, 1:4,000; cat. no. ab76237)
and β-actin (dilution, 1:4,000; cat. no. ab16039) primary
antibodies were purchased from Abcam (Cambridge, England). A
horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
secondary antibody (dilution 1:80,000; cat. no. IH-0011) was
obtained from Wuhan Boster Biological Technology, Ltd. (Wuhan,
China). All other chemicals were obtained from Nanjing Jiancheng
Bio Institute (Nanjing, China).
POA was provided by the South China Sea Institute of
Oceanology (Guangzhou, China). The structure of POA was elucidated
by infrared (IR), nuclear magnetic resonance and mass spectrometry
(MS) analyses, and its >98% purity was determined by high
performance liquid chromatography (HPLC) (8). POA was dissolved in DMSO and during
the experiments, the DMSO content in the medium never exceeded 0.5%
(v/v).
Cell culture
L-02 cells were derived from healthy adult human
livers and obtained from the Guangzhou Jennio Biotech Co., Ltd.
(Guangzhou, China). Cells were maintained in RPMI 1640 media
supplemented with 10% heat-inactivated FBS at 37°C in 5%
CO2. The cells were cultured for 3 days and culture
medium was changed every 2 days. Cells for assay were detached by a
solution of 0.25% trypsin and 0.02% EDTA.
Assessment of cell viability
L-02 cells (1×104 cells/well) were seeded
into 96-well microplates and exposed to various concentrations of
POA (10, 20, 30, 40, 50, 60, 70, 80, 90 or 100 µM) for 24, 48 or 72
h. Cells treated without POA (0 µM) served as a control in each
experiment throughout the study. Subsequently, cells were incubated
with 10 µl CCK-8 for 2 h, which provided effective and reproducible
determination of the proliferative activity of L-02, as the
dehydrogenases in surviving cells can convert CCK-8 to a colored
formazan product. Finally, the optical density was measured at a
wavelength of 450 nm using a microplate reader (PerkinElmer, Inc.,
Waltham, MA, USA) with a reference wavelength of 650 nm. Three
independent experiments were conducted in triplicate.
Assessment of morphological changes in
the cell and nucleus
The morphologies of the L-02 cells after exposure to
20 or 40 µM POA for 24 h were evaluated under a phase contrast
optical microscope (Leica Microsystems GmbH, Wetzlar, Germany). The
morphological changes in the L-02 cells induced by POA were
examined by fluorescent visualization under a fluorescence
microscope (Leica, Microsystems GmbH). Briefly, cells were treated
similarly as described above, then washed twice with PBS, fixed
with 4% paraformaldehyde for 10 min and incubated with Hoechst
33258 fluorescent dye (5 mg/ml) for 5 min. Following this, cells
were washed with PBS, dried, observed and imaged under a
fluorescence microscope.
Assessment of apoptosis by the Annexin
V/PI staining assay
POA-induced apoptosis was measured by Annexin
V-FITC/PI double staining using a FACSCalibur flow cytometer (BD
Biosciences, San Jose, CA, USA). In brief, subsequent to either
sham (0 µM) or POA exposure (10, 20 or 40 µM) for 6 h, L-02 cells
were harvested and washed twice with pre-cooled PBS, and
resuspended with 500 µl binding buffer, 5 µl Annexin V-FITC
conjugate and 5 µl PI buffer, according to manufacturer's protocol.
Cells were incubated in the dark for 15 min at room temperature,
following which they were analyzed by flow cytometry, with each
determination based on the acquisition of 10,000 events. The
percentage of early apoptotic cells was calculated with the ratio
of the total L-02 cell number.
For PI staining, cells were treated as described
above, collected and centrifuged (800 × g for 5 min at 4°C), washed
twice with ice-cold PBS, then fixed in 70% chilled ethanol for 12
h. After fixation, L-02 cells were washed and incubated in PBS
containing 50 mg/ml PI, 1 mg/ml RNase A and Triton X-100 (0.5%) at
4°C for 30 min in the dark according to the instructions provided
by the cell-cycle kit. Finally, the cells were assessed using a
FACScan flow cytometer, and the percentage of nuclei with sub-G1
content was considered as apoptotic cells. The results were counted
by a blind observer.
Activity of caspase 3
The activity of caspase 3 was measured by a caspase
3 activity assay kit. In brief, the cells were treated either sham
or POA (10, 20 or 40 µM) for 24 h, following which cells were
harvested, washed twice with ice-cold PBS and resuspended in cold
lysis buffer for 60 min at 4°C. The lysate was centrifuged (12,000
× g, 4°C, 5 min) and the supernatant was incubated with the enzyme
specific colorimetric substrate Ac-DEVD-pNA in assay buffer for 2 h
at 37°C. The colorimetric release of pNA from the Ac-DEVD-pNA
substrate was examined using a microplate reader (PerkinElmer,
Inc.) with the optical absorbance at 405 nm.
Protein expression levels of Fas, Bax
and Bcl-2
The cytosolic fractions of Fas, Bax and Bcl-2 were
extracted from the cell with RIPA lysis buffer containing 1%
phenylmethanesulfonyl fluoride (PMSF), and the expression levels
were detected by western blotting. Briefly, subsequent to either
sham or POA exposure (20 or 40 µM for 24 h), 5×106 cells
were trypsinized and harvested, then the treated cells were lysed
in precooled lysis buffer and incubated on ice for 30 min. The
homogenate was centrifuged at 8,000 × g for 8 min at 4°C, and the
supernatant was transferred to an ice-cold 1.5 ml tube and stored
at −20°C until further use. The concentration of total protein in
the supernatant was determined by the BCA protein assay kit.
Proteins (60 µg/lane) were separated by 12% SDS-PAGE for 90 min,
and then were electrotransferred to a polyvinylidene difluoride
membrane. Membranes were subsequently incubated with Fas, Bax and
Bcl-2 primary antibodies, followed by incubation with corresponding
HRP-conjugated secondary antibodies. Fas, Bax and Bcl-2 expressions
were then detected using an ECL kit (Beyotime Institute of
Biotechnology, Jiangsu, China) and analyzed by Gel. Doc2000 Gel
imager (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with Image
Lab Software version 3.0 (Bio-Rad Laboratories, Inc.).
Assessment of oxidation state
To detect intracellular oxidation state, L-02 cells
were treated as described above, but in 6-well plates.
Subsequently, the harvested cell suspension was washed with PBS (pH
7.4), sonicated (20 sec) in ice, and then centrifuged at 5,000 × g
for 10 min at 4°C. The supernatant was transferred to fresh tubes
and used for the enzymatic assays.
GSH content
The antioxidant enzyme GSH was measured by a GSH
ELISA kit. The samples were prepared similarly as described above,
according to the manufacturer's protocol. The assay is based on the
depth of yellow, which is inversely associated with the content of
GSH. The content of GSH was determined by measuring the optical
absorbance at a wavelength 450 nm using a microplate reader
(PerkinElmer, Inc.) Activity of GSH is expressed as µg/ml
protein.
Superoxide dismutase (SOD)
activity
The activity of SOD was measured by the reduction of
the formation of reddish violet crystals using a microplate reader
at a wavelength of 550 nm, then the precise result was obtained
through the formula calculation of the manufacturer's protocol
based on cellular protein concentration. Protein concentrations
were determined by using a formula calculation based on absorbance
values at 550 nm (10). Activity
of SOD is expressed as U/mg protein.
Determination of lipid
peroxidation
The content of malondialdehyde (MDA) was measured by
a MDA detection kit (Nanjing Jiancheng Bio Institute) according to
the manufacturer's protocol. MDA reacts with thiobarbituric acid
(TBA) at 95–100°C in acidic conditions and produces a pink colored
MDA-TBA conjugate, which has an absorbance at a wavelength of 532
nm using a microplate reader. MDA was calculated based on cellular
protein concentration. Cellular MDA is expressed nmol/mg cellular
protein.
Assessment of nitric oxide (NO)
The content of NO was measured by a NO detection kit
(Nanjing Jiancheng Bio Institute), which conducted following the
manufacturer's protocol. NO reacts with oxygen and water is
converted into nitrate and nitrite, which can change to a pink azo
compound in a nitrate chromogenic agent. NO levels were determined
by measuring the optical density of colored formation at 550 nm
with a microplate reader. NO content was calculated based on
cellular protein concentration, with sodium nitrite to generate the
standard curve. Cellular NO content is expressed µmol/g cellular
protein.
Measurement of alanine
aminotransferase (ALT) and aspartate aminotransferase (AST)
The levels of ALT and AST were calculated using ALT
and AST detection kits (Nanjing Jiancheng Bio Institute), according
to the manufacturers' protocol. The absorbance was measured at a
wavelength of 510 nm with a microplate reader. Cellular ALT and AST
contents are expressed U/mg cellular protein with pyruvic acid
sodium to generate the standard curve.
Lactate dehydrogenase (LDH) leakage
assay
The LDH leakage assay was assessed by a LDH
cytotoxicity detection ELISA kit (cat. no. CSB-E11720h; CUSABIO
Biotech. Co., Ltd., Wuhan, China) according to the manufacturer's
protocol. The concentration of LDH was determined by measuring the
optical density at 450 nm with a microplate reader according to the
standard curve, and the calculation was based on cellular protein
concentration.
Measurement of reactive oxygen species
(ROS) production
ROS accumulation in cells was measured by a ROS
detection kit (Nanjing Jiancheng Bio Institute) using the cell
permeable fluorescent dye 2,7-dichlorofluorescin diacetate
(DCFH-DA). The intracellular esterases hydrolyze DCFH-DA to DCFH,
which is oxidized to DCF by the oxidants; its fluorescence is a
measure of ROS production in the cell. A BD flow cytometer with BD
CFlow software v.264.15 (BD Biosciences) was used to measure the
fluorescence intensity at excitation and emission wavelengths of
485 and 530 nm, respectively.
Assessment of mitochondrial membrane
potential (MMP)
To monitor the loss of MMP, L-02 cells
(5×105 cells/ml) in 6-well culture plates were treated
without or with POA (10, 20 or 40 µM) for 24 h. Reaction medium (pH
7.4) containing 5 µg/ml JC-1 cationic dye were preincubated at
37°C. Cells were trypsinized and harvested, washed twice with
ice-cold PBS, then incubated with reaction medium for 30 min in
darkness at 37°C. Finally, cells were washed twice with PBS then
analyzed by flow cytometry as described above, within 1 h.
Assessment of adenosine triphosphate
(ATP)
The ATP level was determined using an ATP detection
kit (Nanjing Jiancheng Bio Institute) according to the
manufacturer's protocol. In brief, L-02 cells were treated without
or with POA (10, 20 or 40 µM) for 24 h, then cells were harvested
and centrifuged (2,400 × g, 5 min, 4°C), and the cell pellet was
ultrasonicated for 5 sec. The optical density was detected at a
wavelength of 660 nm with a microplate reader and the concentration
was calculated according to the standard curve, based on cellular
protein concentration. Cellular ATP content is expressed as U/mg
cellular protein.
Assessment of mitochondrial
permeability transition pore (MPTP)
The MPTP was determined with a MPTP kit (Nanjing
Jiancheng Bio Institute) according to the manufacturer's protocol.
Mitochondrial relative fluorescence intensity as an indicator of
MPTP was estimated from the decrease using an excitation wavelength
of 480 nm and an emission wavelength of 530 nm. Data are reported
as fold in fluorescence intensity relative to the control.
Assessment of the release of cyt c
from mitochondria
The cytosolic fraction of cyt c was extracted from
the cell with RIPA lysis buffer containing 1% PMSF and detected by
western blotting. Briefly, L-02 cells were treated without or with
POA (20 or 40 µM for 24 h), 5×106 cells were trypsinized
and harvested, and the treated cells were lysed in precooled lysis
buffer on ice for 30 min. The homogenate was centrifuged at 8,000 ×
g for 8 min at 4°C, and the supernatant was transferred to an
ice-cold 1.5 ml tube and stored at −20°C until further use. The
protein concentration in the supernatant was determined by the BCA
protein assay kit. Equal amounts of protein (60 µg/lane) were
separated by 12% SDS-PAGE and were electrotransferred to a
polyvinylidene difluoride membrane. The membrane was then incubated
in 5% non-fat milk in TBS with Tween-20 (TBST) for 2 h followed by
overnight incubation with the cytochrome c antibody at 4°C.
The incubated membrane was washed with TBST before incubation for 1
h with a HRP-conjugated goat anti-rabbit IgG secondary antibody at
25°C. Membranes were washed with TBST, and proteins were detected
by an ECL kit (Beyotime Institute of Biotechnology, Jiangsu, China)
and analyzed by Gel. Doc2000 Gel imager (Bio-Rad Laboratories,
Inc.) with Image Lab Software version 3.0 (Bio-Rad Laboratories,
Inc.).
Electron microscopy of
mitochondria
The morphological changes of mitochondria were
observed by electron microscopy assay. In brief, L-02 cells were
exposed to POA (0, 10, 20 or 40 µM for 24 h), then trypsinized and
harvested for transmission electron microscopy. Cells samples of a
maximum size of 1 mm3 were collected and prefixed in PBS
(pH 7.2) supplemented with 2.5% glutaraldehyde for 4 h, according
to a method described previously (11). Finally, the sections were observed
under a transmission electron microscope.
Statistical analysis
Values are presented as the mean ± standard
deviation of at least three independent experiments. Statistical
differences were analyzed by one way analysis of using SPSS version
17.0 software (SPSS, Inc., Chicago, IL, USA). Student's t-test was
used for comparison between two groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
Cytotoxicity induced by POA in L-02
cells
The cytotoxicity of POA in L-02 cells was determined
using the CCK-8 assay, which is a sensitive, quantitative and
reliable method to analyze cell viability and cell growth. The cell
growth inhibition after treatments with various concentrations of
POA for 24, 48 or 72 h is presented in Fig. 2. As expected, in comparison with
the control cells, POA significantly decreased L-02 cell viability
in a typical dose- and time-dependent manner (P<0.01 vs.
control). DMSO alone did not show any inhibitory effect on the cell
viability.
 | Figure 2.Concentration- and time-dependent
inhibition effect of POA in L-02 cells. L-02 cells were treated
with 0, 10, 20, 30, 40, 50, 60, 70, 80, 90 or 100 µM POA for 24, 48
or 72 h. Data are presented as the mean ± standard deviation from
three independent experiments. *P<0.01 vs. 24 h at each
concentration. POA, oxalicumone A. |
Morphological changes induced by POA
in L-02 cells
As presented in Fig.
3A, in the absence of POA, L-02 cells had a common spindle
shape, whereas POA treatment (20 and 40 µM) resulted in increased
shrinkage and detachment from the substrate as the exposure dose
increased (Fig. 3A). After the
treatments, the L-02 cells were stained with Hoechst 33258 and
visualized to investigate the incidence of apoptosis. As presented
in Fig. 3B, in the absence of POA,
the L-02 cells had almost uniform Hoechst 33258 (blue) staining
with intact nuclei, and appeared to proliferate in spindle-like
shapes in the wells. While in the presence of POA (20 and 40 µM),
the apoptotic cells were characterized by brighter blue with
nuclear condensation and chromatin margination in a dose-dependent
manner, indicating an increased apoptosis in these cells (Fig. 3B).
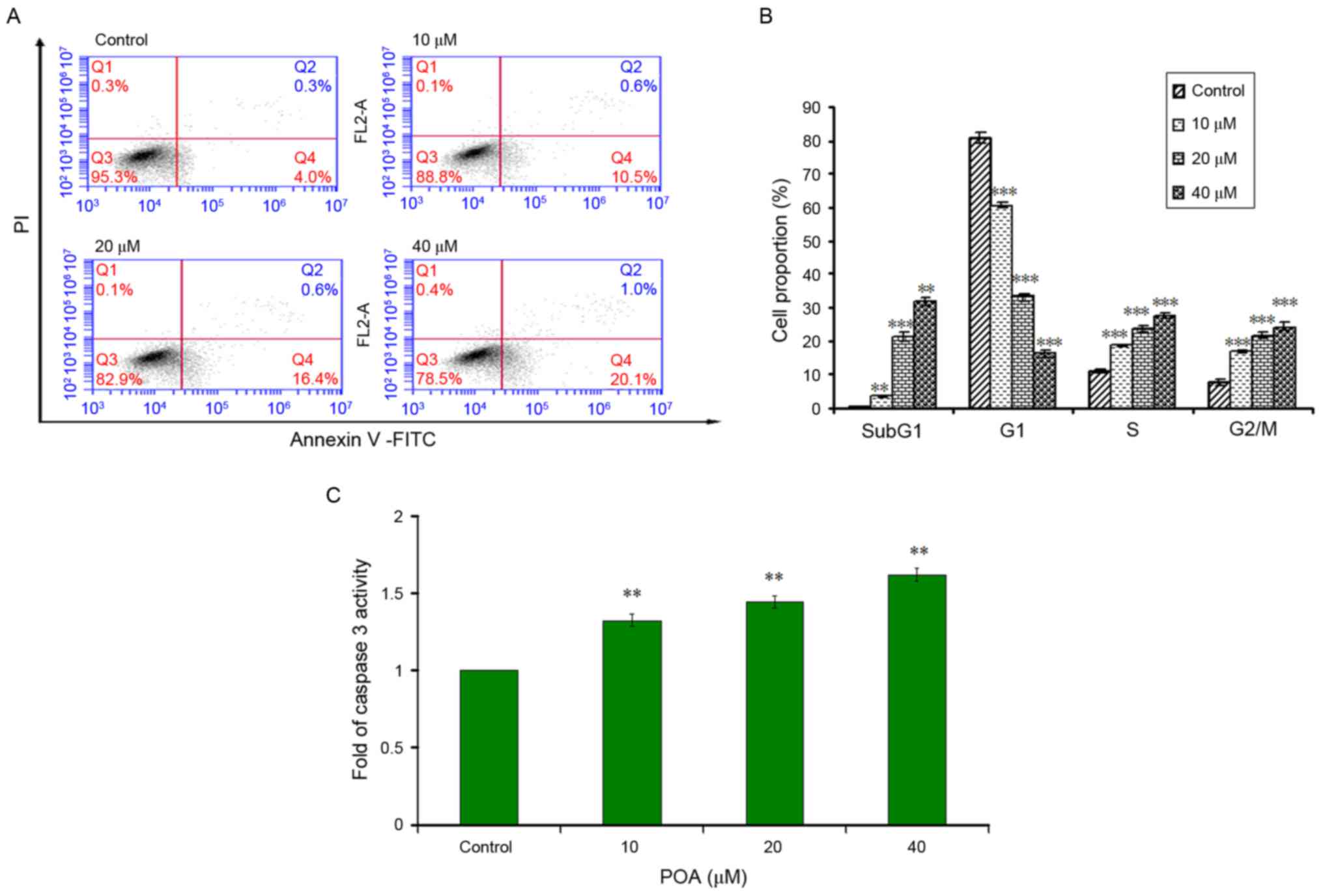
Effects of POA on early apoptosis and
cell cycle in L-02 cells
As presented in Fig.
4A, when L-02 cells were treated with 0, 10, 20 or 40 µM POA
for 6 h, significant increases in the percentage of early apoptotic
cells (Annexin V+/PI− staining) were
observed, with increases from 4.0% at 0 µM, to 10.5% at 10 µM,
16.4% at 20 µM and 20.1% at 40 µM. The inhibitory effect of POA on
L-02 cell growth was further investigated using flow cytometric
analysis. The amounts of apoptotic cells were determined by the
sub-G1 apoptotic peaks, which may be attributed to their lower
cellular DNA content in case of DNA crazing. Apoptotic peaks and
the distributions of the cells in various phases of the cell cycle
were observed after 24 h treatment with POA (10, 20 and 40 µM). As
presented in Fig. 4B, POA induced
an increase in sub-G1 apoptotic peaks and S-phase cell populations
in a dose-dependent manner, indicating an accelerated cell cycle.
The result was accompanied by a marked increase of the percentage
of cells in the G2/M phase, while the percentage of cells in the G1
phase exhibited a significant reduction. The results of Annexin
V-PI double-labeling assay and cell-cycle analysis indicated that
growth inhibition was accompanied with an increase in apoptotic
cells in a dose-dependent manner.
Effect of POA on caspase 3 activity in
L-02 cells
Caspase 3 mainly exists in the cytoplasm in normal
state; during apoptosis, it is energized into two large and two
small subunit compositions, which can break the cytoplasm and
nucleus, eventually leading to apoptosis. As presented in Fig. 4C, when L-02 cells were exposed to
POA (10, 20 and 40 µM) for 24 h, the activities of caspase 3 were
significantly increased (P<0.01) in a concentration-dependent
manner, compared with cells treated without POA.
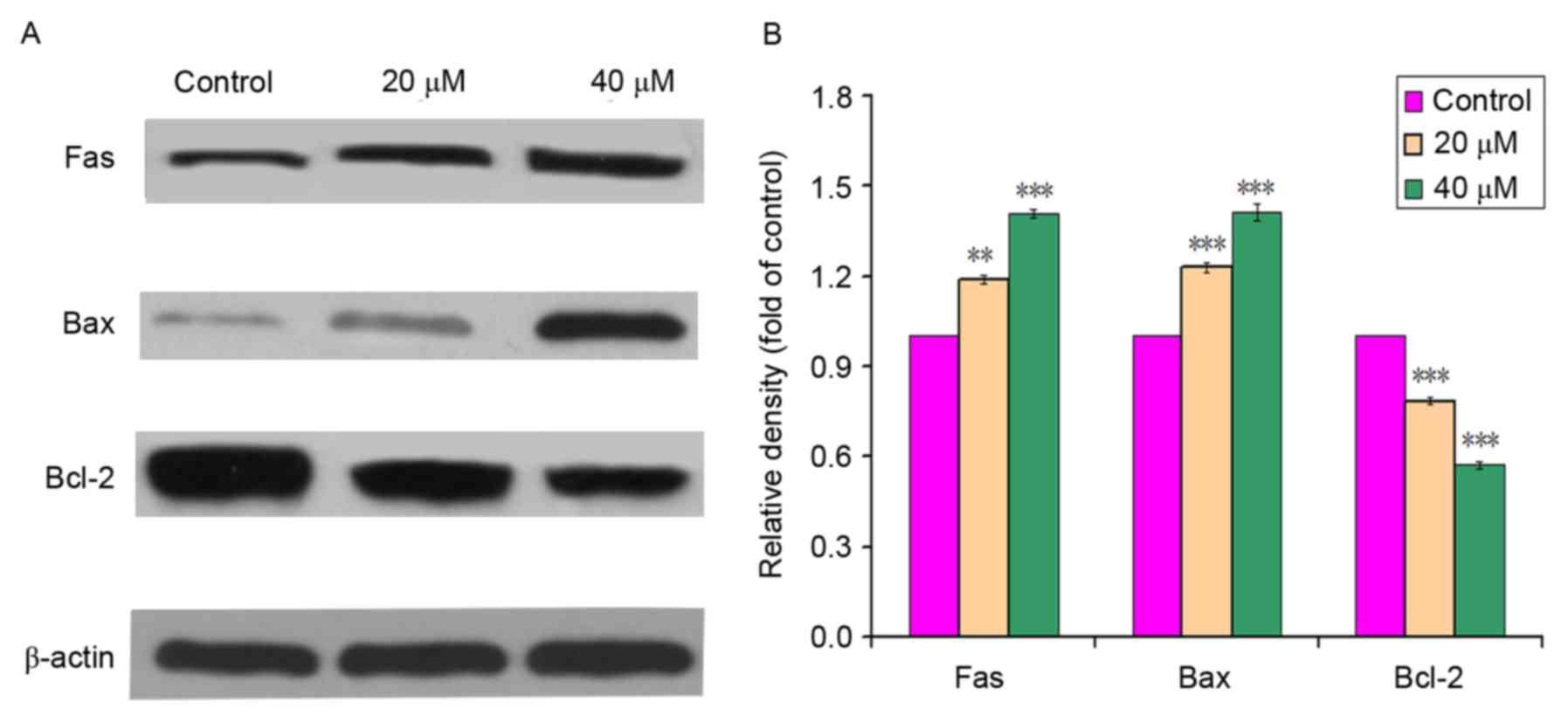
Effect of POA on levels of apoptotic
proteins in L-02 cells
As presented in Fig. 5A
and B, POA (0, 20 or 40 µM) treatment for 24 h increased the
protein expression of Fas compared with the control (0 µM).
Additionally, the protein expression of Bcl-2 decreased with
exposure to POA at increasing concentrations (0, 20 and 40 µM),
while that of Bax increased in POA-treated cells in comparison with
control cells. The results of the expression levels of apoptotic
proteins indicated that POA may upregulate the expression levels of
Fas and Bax, and downregulate Bcl-2 expression, in a
concentration-dependent manner.
Effects of POA on oxidative
stress
GSH is a ubiquitous sulfhydryl-containing molecule
in cells that is responsible for maintaining cellular
oxidation-reduction homeostasis. Changes in GSH homeostasis can be
monitored as an indication of cell damage. As presented in Fig. 6A, POA induced a decrease in the
level of GSH. The levels of GSH were 0.932-fold at 10 µM,
0.821-fold at 20 µM and 0.719-fold at 40 µM of control,
respectively. Compared with the control (44.03 U/ml), POA markedly
decreased SOD activity to 37.31 U/ml at 10 µM, 32.73 U/ml at 20 µM
and 30.72 U/ml at 40 µM (Fig. 6B).
Additionally, POA induced a significant increase in the level of
MDA from 0.57 nmol/mg at 0 µM to 0.71 nmol/mg at 10 µM and 0.79
nmol/mg at 20 µM (P<0.05 vs. control) and 1.36 nmol/mg at 40 µM
(P<0.001 vs control; Fig. 4C).
Furthermore, POA inhibited NO production in a dose-dependent
manner, from 21.43 µmol/l in control cells to 13.41, 8.94 and 3.14
µmol/l after 10, 20 and 40 µM POA, respectively (Fig. 4D).
 | Figure 6.Effect of POA on (A) GSH activity,
(B) SOD activity, (C) MDA formation, (D) NO content, (E) ALT
content, (F) AST content and (G) LDH content in L-02 cells. Data
are presented as the mean ± standard deviation. *P<0.05,
**P<0.01, ***P<0.001. GSH, glutathione; MDA, malondialdehyde;
NO, nitric oxide; ALT, alanine aminotransferase; AST, aspartate
aminotransferase; LDH, lactate dehydrogenase; SOD, superoxide
dismutase; POA, oxalicumone A. |
The results indicated that POA increased the
activities of ALT (Fig. 6E) and
AST (Fig. 6F) in a dose-dependent
manner. The ALT level was increased to 2.38, 3.99, 5.87 U/g protein
compared with the control (0.65 U/g protein), and AST levels were
increased to 2.75, 6.25, 8.43 compared with the control (1.75 U/g
protein) in cells treated with 10, 20 and 40 µM for 24 h,
respectively.
Membrane damage that leads to LDH leakage is
generally considered irreversible. Thus, LDH leakage was applied to
as a biomarker for cellular viability. As presented in Fig. 6G, LDH leakage was identified in
POA-induced L-02 cells in a dose dependent manner: 1.48-fold at 10
µM, 2.08-fold at 20 µM and 3.06-fold at 40 µM compared with control
cells.
Effect of POA on ROS production in
L-02 cells
DCFH-DA is generally used to measure ROS generation
in cells. As presented in Fig. 7,
ROS production following a 24-h exposure to POA increased in a
concentration-dependent manner. The level of ROS in L-02 cells
increased from 4.2% at 0 µM to 14.2% at 10 µM, 19.5% at 20 µM and
23.1% at 40 µM.
Effects of POA on mitochondrial
function in L-02 cells
MMP was determined by the uptake of JC-1 (a
molecular probe) using flow cytometric assay. As presented in
Fig. 8A, MMP depolarization
following a 24-h exposure to POA (10, 20 and 40 µM) significantly
increased in a dose-dependent manner. The percentage of depolarized
cells were 14.2, 19.5 and 23.1% in the samples treated with 10, 20
and 40 µM, respectively, while the value in control cells was 4.2%.
Therefore, POA-induced mitochondrial damage resulted in disruption
of MMP.
 | Figure 8.Effects of POA on (A) MMP, (B) ATP
and (C) MPTP in L-02 cells. Data are presented as the mean ±
standard deviation. ***P<0.001. MMP, mitochondrial membrane
potential; ATP, adenosine triphosphate; MPTP, mitochondrial
permeability transition pore; POA, oxalicumone A; JC-1,
5,5,6,6-tetra-chloro-1,1,3,3-tetraethylbenzimidazolyl-carbocyanine
iodide. |
Concentrations of ATP in L-02 cells treated with POA
are presented in Fig. 8B. The ATP
level decreased from 1.85 U/mg at 0 µM to 1.39 U/mg at 10 µM, 0.81
U/mg at 20 µM and 0.50 U/mg at 40 µM, which indicated that POA
induced the decrease of ATP in a dose-dependent manner.
The results of the MPTP assay are presented in
Fig. 8C. The relative fluorescence
intensity decreased compared with the control in a dose-dependent
manner, and was 0.78 at 10 µM, 0.60 at 20 µM and 0.41 at 40 µM.
This suggested an increasing number of cells with mitochondrial
membrane depolarization or breakdown.
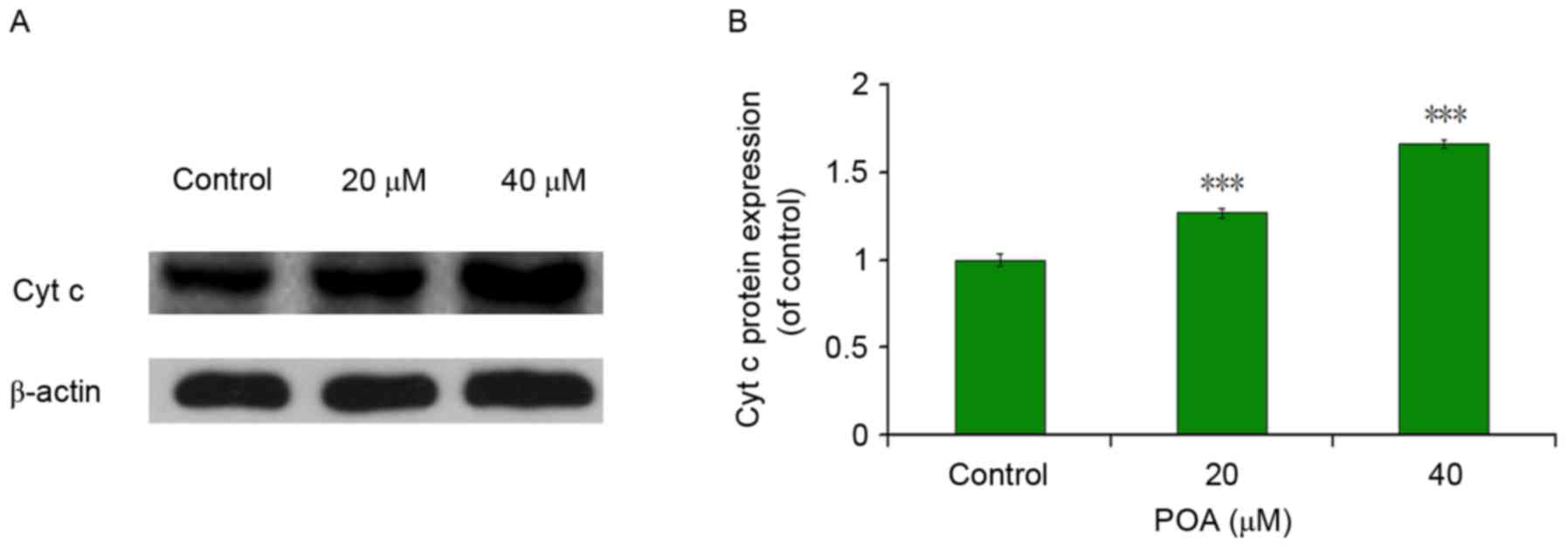
Effects of POA on cyt c
As presented in Fig.
9, POA treatment significantly increased the protein expression
levels of cyt c compared with control cells (P<0.001 vs.
control), which in parallel with mitochondrial membrane breakdown,
indicated that POA increases the release of the cyt c
protein from the mitochondria into the cytosol.
Electron microscopy findings
A transmission electron microscope was used to
observe the cell mitochondrial internal structure. In L-02 cells
there were lesions in the mitochondria of POA treated cells
compared to sham treated cells. In the blank control group,
mitochondrial structures had a regular shape, were distributed in
cells regularly and mitochondrial crests were arranged by rule
(Fig. 10A), whereas cells treated
with 10 (Fig. 10B), 20 (Fig. 10C) and 40 µM (Fig. 10D) POA exhibited increased vacuole
sizes, mitochondrial swelling and disappearance of mitochondrial
matrix particles. Furthermore, the areas of vacuoles increased to
more than half than that of the mitochondria, and integrated
mitochondria around the nucleus were rare as the dose
increased.
Discussion
POA was isolated from a culture broth of the South
China Sea fungus Penicillium oxalicum SCSGAF 0023 (8). Although POA has previously been
indicated to induce cytotoxicity in human carcinoma (12), to the best of our knowledge, there
have not been previous studies on the cytotoxicity of POA on L-02
cells human normal liver cells, which has been a major limitation
in the clinical application of POA. The present study investigated
POA-induced cytotoxicity and its toxic mechanisms in L-02 cells. It
was demonstrated that the toxicological mechanisms of POA on human
normal liver cells mainly included oxidative stress injury and
mitochondrial dysfunction.
In the CCK-8 assay of cell viability, POA inhibited
the proliferation of L-02 cells in a concentration- and
time-dependent manner, which demonstrated that POA induced
cytotoxicity in L-02 cells. POA inhibited L-02 cell proliferation,
potentially via apoptosis-associated processes. Apoptosis, a highly
structured and ordered process, eliminates superfluous, harmful and
metabolically perturbed cells, and is a fundamental form of cell
death (13). In the present study,
apoptosis was induced by POA in L-02 cells, as evidenced by typical
morphological changes in the cell and nucleus assayed by Hoechest
33258 staining, such as karyopyknosis, deepened stains and
karyorrhexis, along with cellular shrinkage, chromatin
condensation, nuclear fragmentation shrinkage and nuclear
condensation or fragmentation. Furthermore, Annexin V/PI double
staining and cell cycle arrest assays further confirmed apoptosis
by flow cytometry; they demonstrated that POA markedly and
progressively increased the percentage of early apoptosis of the
cells as the concentration increased, compared with the control.
Meanwhile, POA largely increased the sub-G1 cell population in a
whole cell cycle, which is a hallmark of apoptosis (14), along with causing S-phase arrest.
Additionally, POA was able to activate caspase 3, which is a key
executioner of apoptosis (15),
thus providing a reasonable explanation for cytotoxic activities in
L-02 cells induced by POA, directly demonstrating that the
mechanism of cytotoxicity is associated with the apoptosis.
To elucidate the molecular toxic mechanism of the
POA, the expression of apoptosis proteins were detected, including
Fas, Bax and Bcl-2. It has previously been reported that the
extrinsic apoptotic signaling pathway is initiated by binding of
the Fas ligand to the ectodomain of the surface death receptor Fas
protein, which triggers activation of caspases that induce cell
death (16,17). Thus, Fas serves an important role
in the initiation of the cell death signaling pathway (18). The present study identified that
POA promoted the expression of Fas protein, which further
demonstrated that POA induced apoptosis in L-02 cells in accordance
with the results described above, potentially via the intrinsic
signaling apoptotic pathway.
The Bcl-2 family, which serves a key role in
deciding the fate of cells, can be divided into either
anti-apoptotic or pro-apoptotic members (19). Apoptosis is also understood to be
critically dependent on the balance between the Bax and the Bcl-2
proteins (20,21). Therefore, the expression levels of
Bax and Bcl-2 were detected by western blotting. The results
indicated that POA increased the expression of the pro-apoptotic
protein Bax and decreased the expression of anti-apoptotic protein
Bcl-2, suggesting that POA may alter the balance between anti- and
pro-apoptotic protein members, ultimately inducing apoptosis in
L-02 cells involving the Bcl-2 family protein.
Oxidative stress serves a role in the mechanisms of
toxicity in a number of compounds, whether by production of free
radicals or by depletion of cellular antioxidant capacity. Cellular
integrity is affected by oxidative stress when the production of
reactive oxidants overwhelms antioxidant defense mechanisms. The
disruption of cellular redox balance by exogenous substances may
lead to cell damage. GSH, non-enzymatic substance, is an
omnipresent sulfhydryl-containing molecule in cells, which is
responsible for maintaining cellular oxidation-reduction
homeostasis and changes in the homeostasis that are as a sign of
cell damage (22). SOD is a
ubiquitous component of cellular antioxidant systems, which can
convert superoxide anions to hydrogen peroxide and fight the damage
resulting from the oxygen free radical to cells, rapidly repairing
damaged cells. The present study demonstrated that POA reduced the
content of GSH and the activity of SOD. These results indicated
that the GSH and SOD antioxidant system is involved in POA-induced
oxidative stress injury in L-02 cells. It has been reported that
the promotion of MDA, which is the main end production of lipid
peroxidation, is an indication of membrane lipid damage, as
membrane lipids are major targets of free radicals (23). On the other hand, NO is a free
radical, which takes part in many pathological processes; a small
amount of NO serves a physiological role, whereas a large amount of
NO induces cell damages. The present study revealed that POA
elevated MDA and NO levels, which suggested that POA induced lipid
peroxidation injury in L-02 cells, and the oxidative stress served
an important role in POA-induced cytotoxicity.
ALT and AST mainly exist in liver cells, and
translocate to the outer membrane from the interior once necrosis
or liver damage occur. They are sensitive indicators for the
examination of liver function in clinical treatment, and are used
to assess the damage of liver cells. The present study demonstrated
an increase in levels of ALT and AST in comparison with the
control, which revealed that POA induced damage in L-02 cells.
Similarly, LDH is a glycolytic enzyme that exists in the cytoplasm
of cells; when cells are damaged, it leaks from the interior to the
outer of cells. The LDH leakage test indicated that the amount of
LDH in treatments was increased compared with the sham, which
indicated that POA induced damage in L-02 cells, consistent with
ALT and AST results.
ROS are by-products of biological redox reactions
and are associated with various pathological conditions. They
increase following exposure to harmful substances and rely on the
balance between oxidative and antioxidant cellular system (24). In turn, excessive harmful
substances can disturb this balance by creating increased formation
of ROS through mitochondria dysfunction or diminishing and/or
inhibiting antioxidant system, and the overproduction of ROS may
induce apoptosis and cell death (25). The present study demonstrated that
POA significantly increased ROS production in a dose-dependent
manner, which is consistent with the above results of cytotoxicity
and oxidative stress injury induced by POA in L-02 cells.
Mitochondria serve a key role in apoptosis, and the
mitochondrial apoptotic pathway has been suggested as a pivotal
signaling pathway of apoptosis (26). The most crucial effect of oxidant
stress is the increased opening of the MPTP, resulting in the
collapse of membrane potential and interruption of ATP synthesis
(27). Certain drugs can lead
swollen mitochondria, abnormal or cracked cristae and reduced
matrix density in liver cells. The MMP formed by electrons within
the mitochondrial respiratory chain couples with the extrusion of
protons from the mitochondrial matrix to the intermembrane space,
which is the driving force for phosphorylation of ADP in the
process of oxidative phosphorylation (28). A previous study confirmed that the
depolarization of MMP is an irreversible cause of early apoptosis
(29). In the present study,
incubation of activated mitochondria with POA resulted in an abrupt
depletion of ATP synthesis, MPTP opening and mitochondria swelling,
vacuolation, and reduced matrix density. Furthermore, POA induced
the depletion of MMP; as it is reported that alternations in the
mitochondrial functions via the increase of ROS generation and
disruption of MMP may lead to cell death (30), these results indicated that POA
induced damage in mitochondria, thus causing cytotoxicity in L-02
cells. Furthermore, the exposure of L-02 cells to POA resulted in
an abrupt release of cyt c in comparison to control,
indicating an impairment of mitochondrial function.
In conclusion, the present study demonstrated that
POA may lead to persistent toxicity in L-02 cells. The mechanisms
of POA-mediated hepatocellular toxicity potentially involve
apoptosis via oxidative stress injury and impairment of
mitochondrial function in L-02 cells. The alteration of
mitochondrial function and oxidative stress may be the major cause
for the activation of the caspase cascade that results in apoptosis
in response to POA-induced hepatocellular toxicity. However,
whether other mechanisms are associated with POA-induced
cytotoxicity remain to be further investigated.
Acknowledgements
The present study was supported by the National
Marine Public Welfare Research Project of China (grant no.
201305017), the National Natural Science Foundation of China (grant
no. 81573638), the Natural Science Foundation of Guangdong Province
(grant nos. 2016A030313859 and 2017A030313666) and the Science
Program for Overseas Scholar of Guangzhou University of Chinese
Medicine (Torch Program; grant no. XH20150107).
References
|
1
|
Russmann S, Kullak-Ublick GA and
Grattagliano I: Current concepts of mechanisms in drug-induced
hepatotoxicity. Curr Med Chem. 16:3041–3053. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gerets HH, Tilmant K, Gerin B, Chanteux H,
Depelchin BO, Dhalluin S and Atienzar FA: Characterization of
primary human hepatocytes, HepG2 cells and HepaRG cells at the mRNA
level and CYP activity in response to inducers and their
predictivity for the detection of human hepatotoxins. Cell Biol
Toxicol. 28:69–87. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
O'Brien PJ, Irwin W, Diaz D,
Howard-Cofield E, Krejsa CM, Slaughter MR, Gao B, Kaludercic N,
Angeline A, Bernardi P, et al: High concordance of drug-induced
human hepatotoxicity with in vitro cytotoxicity measured in a novel
cell-Based model using high content screening. Arch Toxicol.
80:580–604. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aggarwal BB, Sundaram C, Malani N and
Ichikawa H: Curcumin: The Indian solid gold. Adv Exp Med Biol.
595:1–75. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Houbraken J, Frisvad JC and Samson RA:
Fleming's penicillin producing strain is not Penicillium
chrysogenum but P. rubens. IMA Fungus. 2:87–95. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cragg GM and Newman DJ: Natural products:
A continuing source of novel drug leads. Biochim Biophys Acta.
1830:3670–3695. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Senni K, Pereira J, Gueniche F,
Delbarre-Ladrat C, Sinquin C, Ratiskol J, Godeau G, Fischer AM,
Helley D and Colliec-Jouault S: Marine polysaccharides: A source of
bioactive molecules for cell therapy and tissue engineering. Mar
Drugs. 9:1664–1681. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun YL, Bao J, Liu KS, Zhang XY, He F,
Wang YF, Nong XH and Qi SH: Cytotoxic dihydrothiophene-condensed
chromones from marine-derived fungus Penicillium oxalicum.
Plata Med. 79:1474–1479. 2013. View Article : Google Scholar
|
|
9
|
Zhang XY, Bao J, Wang GH, He F, Xu XY and
Qi SH: Diversity and antimicrobial activity of culturable fungi
isolated from six species of the South China Sea gorgonians. Microb
Ecol. 64:617–627. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai Q, Wei J, Zhao W, Shi S, Zhang Y, Wei
R, Zhang Y, Li W and Wang Q: Toxicity of Evodiae fructus on
rat liver mitochondria: The role of oxidative stress and
mitochondrial permeability transition. Molecules. 19:21168–21182.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang JQ, Shen M, Zhu CC, Yu FX, Liu ZQ,
Ally N, Sun SC, Li K and Liu HL: 3-Nitropropionic acid induces
ovarian oxidative stress and impairs follicle in mouse. PLoS One.
9:e865892014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang XY, Bao J, Zhong J, Xu XY, Nong XH
and Qi SH: Enhanced production of a novel cytotoxic chromone
oxalicumone a by marine-derived mutant Penicillium oxalicum
SCSIO 24-2. Appl Microbiol Biotechnol. 97:9657–9663. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jacobson MD, Weil M and Raff MC:
Programmed cell death in the animal development. Cell. 88:347–354.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pirocanac EC, Nassirpour R, Yang M, Wang
J, Nardin SR, Gu J, Fang B, Moossa AR, Hoffman RM and Bouvet M:
Bax-induction gene therapy of pancreatic cancer. J Surg Res.
106:346–351. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wilson MR: Apoptotic signal transduction:
Emerging pathways. Biochem Cell Biol. 76:573–782. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Daniel PT, Wieder T, Sturm I and
Schulze-Osthoff K: The kiss of death: Promises and failures of
death receptors and ligands in cancer therapy. Leukemia.
15:1022–1032. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Itoh N, Yonehara S, Ishii A, Yonehara M,
Mizushima S, Sameshima M, Hase A, Seto Y and Nagata S: The
polypeptide encoded by the cDNA for human cell surface antigen fas
can mediate apoptosis. Cell. 66:233–243. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Waring P and Müllbacher A: Cell death
induced by the Fas/Fas ligand pathway and its role in pathology.
Immunol Cell Biol. 77:312–317. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Certo M, Del Gaizo V, Nishino M, Wei G,
Korsmeyer S, Armstrong SA and Letai A: Mitochondria primed by death
signals determine cellular addiction to antiapoptotic BCL-2 family
members. Cancer Cell. 9:351–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lanave C, Santamaria M and Saccone C:
Comparative genomics: The evolutionary history of the Bcl-2 family.
Gene. 333:71–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hussain SM and Frazier JM: Cellular
toxicity of hydrazine in primary rat hepatocytes. Toxicol Sci.
69:424–432. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Valko M, Jomova K, Rhodes CJ, Kuča K and
Musílek K: Redox- and non-redox-metal-induced formation of free
radicals and their role in human disease. Arch Toxicol. 90:1–37.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Farber JL, Kyle ME and Coleman JB:
Mechanisms of cell injury by activated oxygen species. Lab Invest.
62:670–679. 1990.PubMed/NCBI
|
|
25
|
Sinha K, Das J, Pal PB and Sil PC:
Oxidative stress: The mitochondria-dependent and
mitochondria-independent pathways of apoptosis. Arch Toxicol.
87:1157–1180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao J, Jiang Z, Duan W, Huang J, Zhang L,
Hu L, He L, Li F, Xiao Y, Shu B and Liu C: Involvement of
mitochondrial pathway in triptolide-induced cytotoxicity in human
normal liver L-02 cells. Biol Pharm Bull. 31:592–597. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramachandran A, Lebofsky M, Baines CP,
Lemasters JJ and Jaeschke H: Cyclophilin d deficiency protects
against acetaminophen-induced oxidant stress and liver injury. Free
Radic Res. 45:156–164. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Haasio K, Koponen A, Penttilä KE and
Nissinen E: Effects of entacapone and tolcapone on mitochondrial
membrane potential. Eur J Pharmacol. 453:21–26. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Urra FA, Cordova-Delgado M, Pessoa-Mahana
H, Ramirez-Rodriguez O, Weiss-Lopez B, Ferreira J and
Araya-Maturana R: Mitochondria: A promising target for anticancer
alkaloids. Curr Top Med Chem. 13:2171–2183. 2013. View Article : Google Scholar : PubMed/NCBI
|