Introduction
Hereditary elliptocytosis (HE) disease refers to a
type of familial inherited hemolytic disease that is characterized
by a broad spectrum of clinical severities, ranging from an
asymptomatic carrier to life-threatening anemia with
transfusion-dependency (1). The
main inheritance mode of HE is autosomal dominant; the other modes
are associated with true recessive inheritance and novel mutations.
The global incidence of HE disease is high, particularly in the
malaria-endemic areas of West Africa where the prevalence of HE is
2% (2). The typical clinical
characteristics of HE include, splenomegaly, jaundice and anemia,
with the presence of elliptically shaped erythrocytes in peripheral
blood smears. However, due to high levels of heterogeneity,
patients with HE can easily be misdiagnosed and there are ~1/3,000
heterozygous mutation carriers in the world with no obvious signs
of jaundice or anemia (3). A
common molecular biological basis of all types of HE is either a
deficiency or dysfunction of one or more of the following 3
proteins: Protein 4.1, and α- and β-spectrin proteins encoded by
the genes, erythrocyte membrane protein band 4.1 (EPB41), spectrin
α chain erythrocytic 1 (SPTA1) and spectrin β chain erythrocytic
(SPTB), respectively (4). In
particular, α- and β-spectrin proteins alter the morphology of
erythrocytes, from the normal biconcave disc structure toan
elliptical shape, there by causing the irreversible deformity of
red blood cells (RBCs) (5).
Heteromorphic erythrocytes are susceptible to being trapped within
the splenic cord and are removed by the splenic reticuloendothelial
system, which can lead to hemocytolysis and increased free
bilirubin levels.
The SPTA1 gene is 80 k Bin length, contains 52 exons
and is located on chromosome 1q22-q23. The gene encodes a 2,429
amino acid-long protein, α-spectrin, which accounts for 21% of
total erythrocyte membrane proteins. It constitutes the lateral
linkage of the erythrocyte membrane skeleton protein network with
β-spectrin and protein 4.1 (6). In
the present study, a novel molecular defect in α-spectrin,
His54Pro, was detected, which resulted in a heterozygous point
substitution in exon II of the SPTA1 gene. In addition, clinical
characteristics and laboratory results of the proband and their
family are summarized to provide a reference for prenatal check-ups
and diagnosis of HE.
Materials and methods
Routine examination
Written informed consent was obtained from all
participants enrolled in the present study. The study protocol was
approved by the Ethics Committee of The First Affiliated Hospital
at the Guangxi Medical University (Nanning, China). A total of 7 ml
of blood was collected from the proband and 95 healthy volunteers
(2 ml of blood was used for EDTA anticoagulation and 5 ml was used
for separation of coagulation factors). The proband (3 year old
female) was recruited following routine surgery at the Department
of Clinical Laboratory, The First Affiliated Hospital of Guangxi
Medical University, and all participants (57 males and 38 females;
mean age of 23.15±11.59 years; age range of 7–51 years) in the
present study were recruited between January 2016 and December 2016
at The First Affiliated Hospital of Guangxi Medical University.
Blood tests, including RBC count, hemoglobin (HGB)
levels, mean corpuscular volume (MCV), mean corpuscular hemoglobin
(MCH), mean sphered blood cell volume (MSCV), mean reticulocyte
volume, reticulocyte (RET) levels and mean platelet volume (MPV),
were performed using the LH 780 automated hematology analyzer
(Beckman Coulter, Inc., Brea, CA, USA). For assessment of liver
function, levels of total bilirubin (TBiL) and direct bilirubin
(DBiL) were determined using the 7600 Fully Automated Biochemical
Analyzer (Hitachi, Ltd., Tokyo, Japan).
Preparation of RBC membrane protein
extract
A low-osmotic hemolysis method was used to extract
the erythrocyte membrane protein content as previously described
(7). Briefly, the packed RBCs were
separated by centrifugation at 3,000 × g for 5 min at room
temperature. Subsequently, 50 µl of packed RBCs and 15 µl of
phenylmethylsulfonyl fluoride (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) were added to 750 µl
erythrocyte lysis buffer (Solarbio Science & Technology Co.,
Ltd.) and incubated for 30 min at 4°C. Totalerythrocyte membrane
proteins were then harvested by centrifugation at 12,000 × g for 20
min at 4°C. The precipitate was washed three times with 0.1 mol/l
PBS at 4°C. Following the third PBS wash, the precipitate was
stored at −20°C until further use.
Separation and evaluation of RBC
membrane protein content
RBC membrane proteins were analyzed within 15 days
of extraction using 3.5–17% SDS-PAGE according to Fairbanks et
al (8). SDS loading buffer,
containing β-mercaptoethanol, was added to membrane proteins, and
the mixture was incubated at 37°C for 30 min to allow for the
reduction of disulfide bonds, followed by a brief centrifugation at
3,000 × g for 10 sec at room temperature. The gel was stained with
Coomassie brilliant Blue R-250, and then destained at room
temperature overnight in double-distilled water. A Gel Doc 2000
electrophoresis gel imaging system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA) was used to scan the gel and Quantity One
software (version 4.6; Bio-Rad Laboratories, Inc.) was used to
analyze the relative quantitative values of the proteins.
Eosin-5-maleimide (EMA)-labeling of
RBCs and flow cytometry
This method was conducted according to King et
al (9). EMA reagent
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) was diluted in 0.01
mol/l PBS to a final concentration of 0.5 mg/ml, stored at −20°C
and then thawed at 4°Cprior to usage. Freshly anti coagulated blood
was washed three times with PBS and centrifuged at 1,500 × g for 5
min at room temperature following each wash. Subsequently, 25 µl
EMA solution and 5 µl washed RBCs were mixed and incubated at room
temperature for 1 h in the dark. The precipitate was washed three
times with PBS containing 0.5% bovine serum albumin (Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) and centrifuged
at 1,500 × g for 5 min at room temperature following each wash. The
supernatant was discarded and the cells were resuspended in 500 µl
PBS for subsequent analysis using a flow cytometer (Beckman
Coulter, Inc.). A total of 15,000 cells were acquired using the
FL-1 channel and data analysis was conducted using Flow Jo software
(version 10.2; Flow Jo LLC, Ashland, OR, USA). The results were
expressed as the ratio of the mean channel florescence (MCF) of the
study subject to the healthy controls. The formula used was as
follows:
Patients%=ControlMCF–PatientsMCFControlMCFx100%
Where, Control
MCF=(MFC1+MFC2+MFC3+MFC4+MFC5+MFC6+MFC7+MFC8+MFC9+MFC10)/10.
Gene sequence analysis
Genomic DNA from peripheral blood samples was
extracted using the Gene JET Whole Blood Genomic DNA Purification
Mini kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Primers were designed based on exon
sequences and adjacent intron sequences of SPTA1, SPTB and EPB41,
and were synthesized by BGI Sequencing (Cambridge, MA, USA), and
were as follows: Exon 2, forward 5′-ACACATATAAGCGGGGCAAC-3′ and
reverse, 5′-TTGTACCCACACATACCCATTAAC-3′; exon 40, forward
5′-TGAGTGAATATAGATTTTCCGGC-3′ and reverse
5′-TTCTACATTTGGGCCAGTCC-3′; and intron 45, forward
5′-TGGACAGATTCATGTTTTGTGG-3′ and reverse
5′-TGGAATGAAAATGTCTCAGCAC-3′. The polymerase chain reaction (PCR)
mixture consisted of 25 µl of 2X Taq PCR Master Mix (Takara
Biotechnology Co., Ltd.), 2 µl of genomic DNA, 1 µl each of the
forward and reverse primers, and ddH2O to a final volume
of 50 µl. The reaction conditions were as follows: Pre-denaturation
at 95°C for 5 min, followed by 35 cycles of 95°C for 30 sec,
annealing (at the optimal annealing temperature per primer set) for
30 sec 72°C for 40 sec. The final extension was at 72°C for 8 min
and 4°C for storage. The PCR products were sent to BGI Sequencing
for purification, recovery and bi-directional sequencing. The
sequencing results were compared with the standard sequences stored
in the NCBI database (https://www.ncbi.nlm.nih.gov/gene/). Sequencing
results were confirmed by twice repeated PCR amplification and
sequencing analysis via comparison with the published sequences of
homologous species obtained from the NCBI database (https://www.ncbi.nlm.nih.gov/gene/), which was
performed using Chromas 2.6.4 software (Technelysium Pty Ltd.,
Brisbane, Queensland, Australia).
Matrix-assisted laser
desorption-ionization time of flight mass spectrometry (MALDI-TOF
MS)
In-gel enzymolysis was performed according to the
methods described by Gharahdaghi et al (10) Protein samples of the proband and
the healthy controls were separated by SDS-PAGE and stained with
Coomassie brilliant blue according to the aforementioned protocol.
Distinct target protein bands were removed from the gel and
collected. Following de-staining and washing, the proteins were
dehydrated for 10 min using pure acetonitrile (ACN) at room
temperature and vacuum dried. The proteins then underwent overnight
enzymatic digestion in 1U/µl trypsin in 30 µl enzymolysis buffer
(40 mM NH4HCO3 and 10% ACN) at room
temperature. Proteins were extracted and centrifuged at 10,000 × g
at room temperature for 5 min. The top-layer solution was
extracted, concentrated and dried in a vacuum dryer, then
re-dissolved in 0.1% trifluoroacetic acid. The proteins were
analyzed using a 4800 Plus System Mass spectrometer (SCIEX,
Framingham, MA, USA).
High resolution melting (HRM)
PCR amplification and HRM curve analysis were
conducted using a Light Cycler 480 II Fluorescence Quantitative PCR
Instrument (Roche Applied Science, Penzberg, Germany) and the Light
Cycler 480 High Resolution Melting Master kit (Roche Applied
Science) in accordance with the manufacturer's protocol. The total
reaction volume was 20 µl, containing 10 µl of Master Mix, 3.6 µl
of Mg2+, 2 µl of DNA template, and 0.5 µl each of the
forward and reverse primers. Based on the mutations of the proband
and their family members, HRM primers were designed using Premier
5.0 software (Premier Biosoft International, Palo Alto, CA, USA).
The primer sequences were as follows: Forward,
5′-GAGGCGTCAGGAAGTGTT-3′ and reverse, 5′-CTGCATCTCGCTTGAAAA-3′. HRM
detection and data analysis were conducted using the Light Cycler
480 II instrument. There action conditions were as follows: Enzyme
activation at 95°C for 5 min, followed by PCR amplification at 95°C
for 10 sec, 49°C for 15 sec 72°C for 25 sec (fluorescence
detection) for a total of 40 cycles. Melting was performed at 95°C
for 1 min, 40°C for 1 min, 70°C for 1 sec 95°C
(fluorescencedetection at 40 times/sec) for 1 min, followed by cool
down at 40°C for 10 sec. Following the completion of the reaction,
HRM curve analysis was performed using the Light Cycler 480 Gene
Scanning software (version 1.5; Roche Applied Science).
Results
Patient and blood routine tests
The proband, a 3 year old female, was hospitalized
due to recurrent xanthochromia over a period of 3 years and
exhibited brown-colored urine. Immediately following birth,
neonatal pathologic jaundice and ABO hemolytic disease of the
newborn were observed in this patient. In the local hospital, this
patient received exchange transfusion, phototherapy to treat
jaundice and additional treatments, and was discharged following
signs of improvement. The mother had a history of hepatitis B (Hb)
and tested positive for Hb surface antigen, Hb e-antigenand Hb core
antibody. Due to an infection, the patient was examined at the
local hospital, where test results revealed 33.3×109/l
white blood cells (WBCs), 72 g/l HGB and 383 µmol/l TBiL. Following
anti-infection and liver protection treatments, the levels of WBC,
HGB and TBiL were reduced to 6.99×109/l,93 g/l and 166.7
µmol/l, respectively. The patient was then transferred to The First
Affiliated Hospital of Guangxi Medical University on July 15th,
2016.
Physical and auxiliary examinations performed
following hospitalization revealed that the patient was conscious
with no signs of fever or bleeding under the skin. The proband
presented with xanthochromia, scleral icterus in both eyes and
slight redness in the pharynx. The liver and spleen were palpable
at 2 cmunder the ribs, with a medium hardness, and were slightly
obtuse on the edge. Ultrasound examination revealed hepatomegaly
and splenomegaly, with multiple masses near the abdominal aorta. No
echo abnormalities were observed in gallbladder or spleen.
Laboratory tests demonstrated 1+urobilinogen, 0.19 µmol/24 h urine
copper, increased activated partial thromboplastin time (53.20
sec), 580.2 mg/l ceruloplasmin, MSCV<MCV and increased osmotic
fragility of erythrocytes. Analysis of peripheral erythrocyte
morphology revealed anisocytosisthat was dominated by smaller
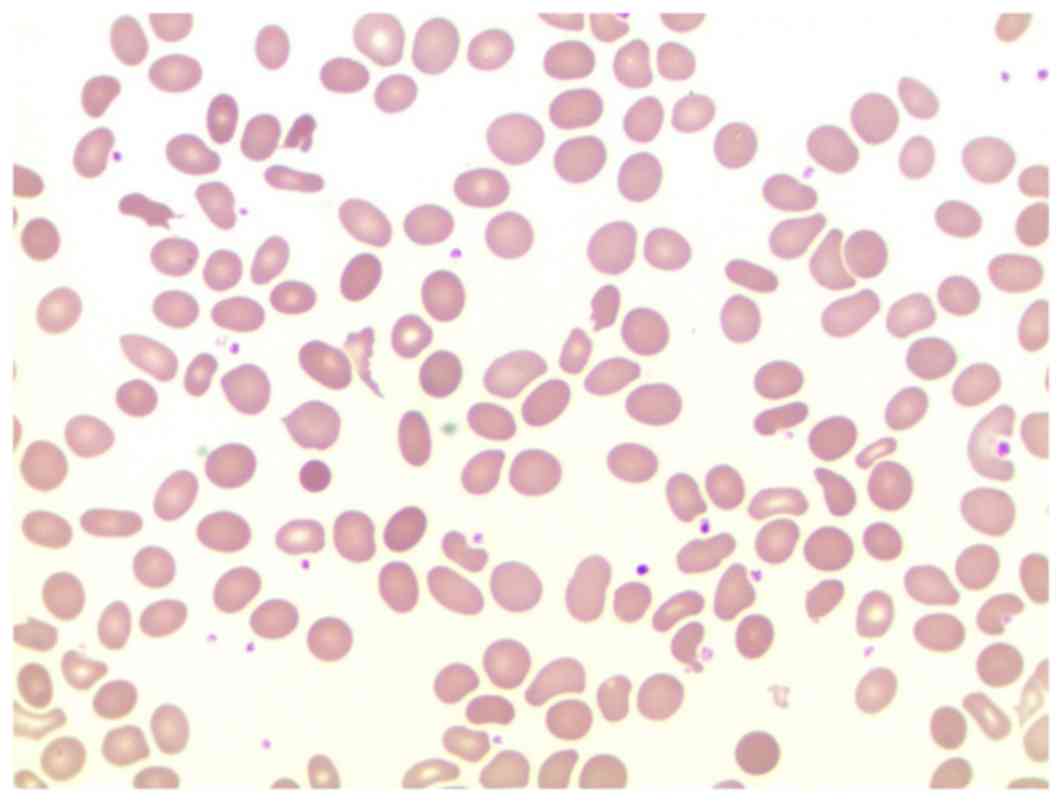
erythrocytes. Elliptocytes, characterized by helmet-shaped and
spherical RBCs, were markedly identifiable and accounted for ~16%
of the total erythrocyte population (Fig. 1). Glucose-6-phosphate dehydrogenase
enzymatic activity was normal, and no abnormalities were detected
in hemoglobin electrophoresis analysis and thalassemia-gene
analysis. The patient tested negative for the Coombs test; the
clinical manifestations and the laboratory test results led to the
diagnosis of the patient with HE. Subsequently, routine blood and
biochemical tests were conducted on the patient's family members.
The results are presented in Table
I.
 | Table I.Laboratory test results of the
hereditary elliptocytosis proband and their family members. |
Table I.
Laboratory test results of the
hereditary elliptocytosis proband and their family members.
| Test | Proband | Father | Mother | Grandmother | Normal range |
|---|
| TBiL (µmol/l) | 73.50 | 15.90 | 19.30 | 8.30 | 3.40–20.50 |
| DBiL (µmol/l) | 10.20 | 7.50 | 4.20 | 2.90 | 0.00–6.80 |
| RBC
(×1012/l) | 3.23 | 4.85 | 4.90 | 5.42 | 4.00–5.50 |
| HGB (g/l) | 90.10 | 137.40 | 134.20 | 109.00 | 120.00–160.00 |
| PLT
(×109/l) | 452.20 | 204.80 | 253.60 | 311.70 | 125.00–350.00 |
| MCV (fl) | 85.85 | 85.56 | 82.87 | 69.20 | 82.00–100.00 |
| MCH (pg) | 27.92 | 28.33 | 27.40 | 18.72 | 27.00–34.00 |
| MCHC (g/l) | 325.20 | 331.10 | 330.60 | 316.20 | 316.00–354.00 |
| MSCV (f/l) | 79.50 | 84.00 | 85.74 | 65.98 | 84.00–104.00 |
| MRV (f/l) | 101.93 | 107.05 | 118.75 | 82.94 | 101.00–119.00 |
| RET (%) | 0.109 | 0.020 | 0.009 | 0.019 | 0.000–0.020 |
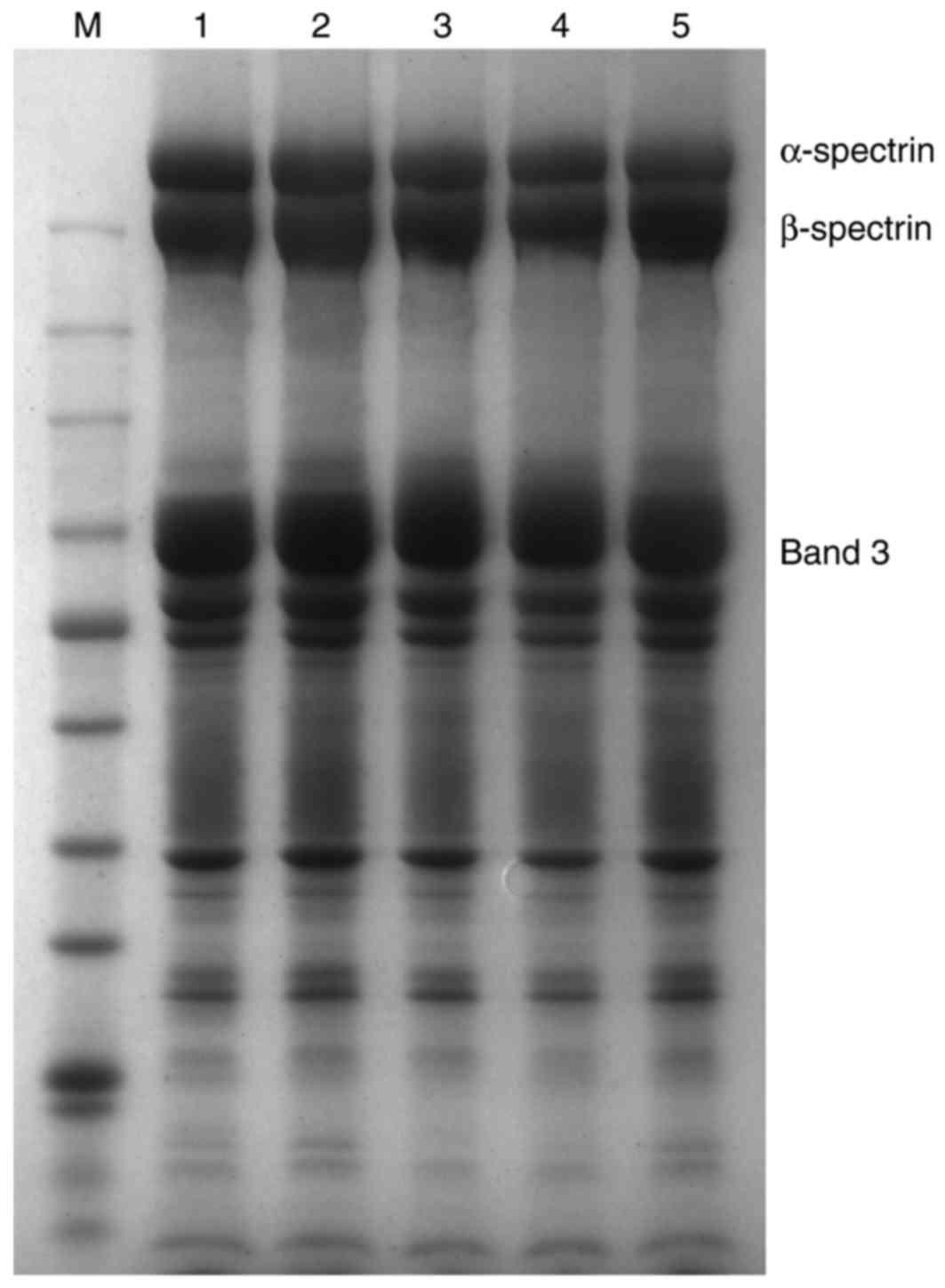
Analysis of the erythrocyte membrane
proteins
Erythrocyte membrane proteins of the proband and
their family members were analyzed by SDS-PAGE. Coomassie brilliant
blue staining was quantified using Quantity One analysis software,
and the level of α-spectrin expression was expressed as
α-spectrin/b and 3 (Fig. 2). The
results of the present study indicated that there were no marked
differences in the amount of membrane proteins between the HE
patient or their family members when compared with healthy
individuals.
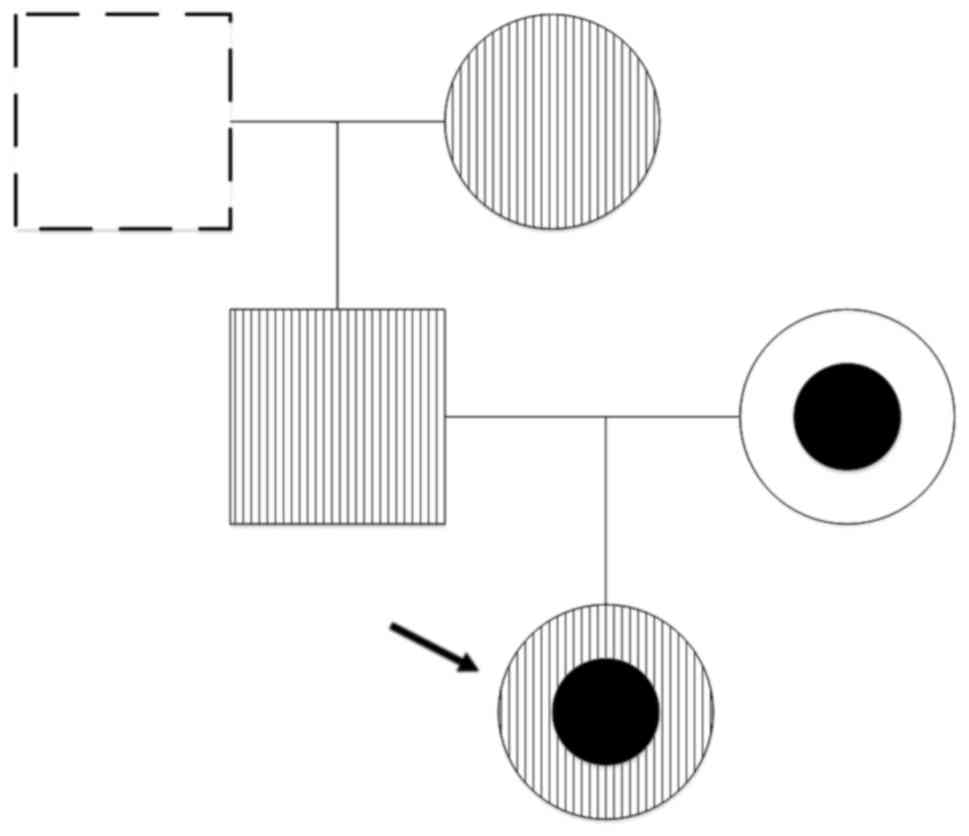
DNA analysis
Compared with the standard sequences that are
available in the NCBI database, PCR amplification and direct
sequencing of the proband's genomic DNA revealed 3 mutations in the
SPTA1 gene within the hereditary elliptocytosis family (Fig. 3). The first mutation was a
heterozygous mutation c.161A>C in exon 2, which resulted in a
His54>Pro54 substitution in α-spectrin.
The second mutation was a heterozygous mis sense mutation
c.5572C>G in exon 40, which resulted in
Leu1858>Val1858. The third mutation was a
heterozygous mutation 6531-12C>T in intron 45. Mutation analysis
of the proband's family members confirmed that their father and
paternal grandmother were carriers of the c.161A>C mutation, and
their mother was a carrier of both the c.5572C>G and 6531-12
C>Tmutations (Fig. 4).
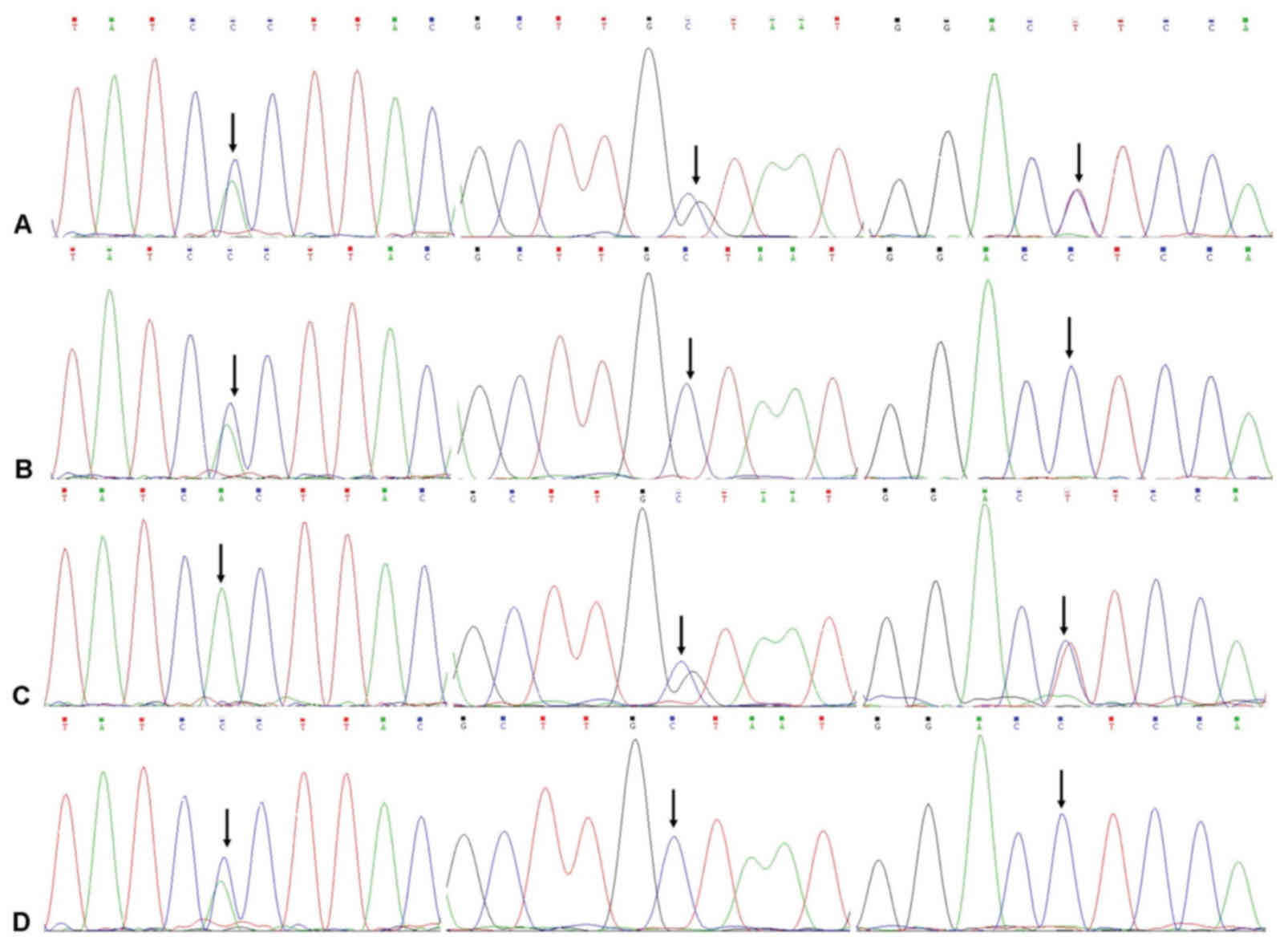 | Figure 4.DNA sequences of, from top to bottom,
the proband, the proband's father, the proband's mother and the
proband's grandmother. (A) Proband heterozygous point mutations
A→C, C→G and C→T were located in exon 2, exon 40 and intron 45,
respectively. (B) Heterozygous point mutation A→C in the father was
detected in exon 2. (C) Heterozygous point mutations C→G and C→T in
the proband and their mother, were detected in exon 40 and intron
45, respectively. (D) Heterozygous point mutation A→C, was found in
exon 2 of the proband's grandmother. Arrows indicate point
mutations. |
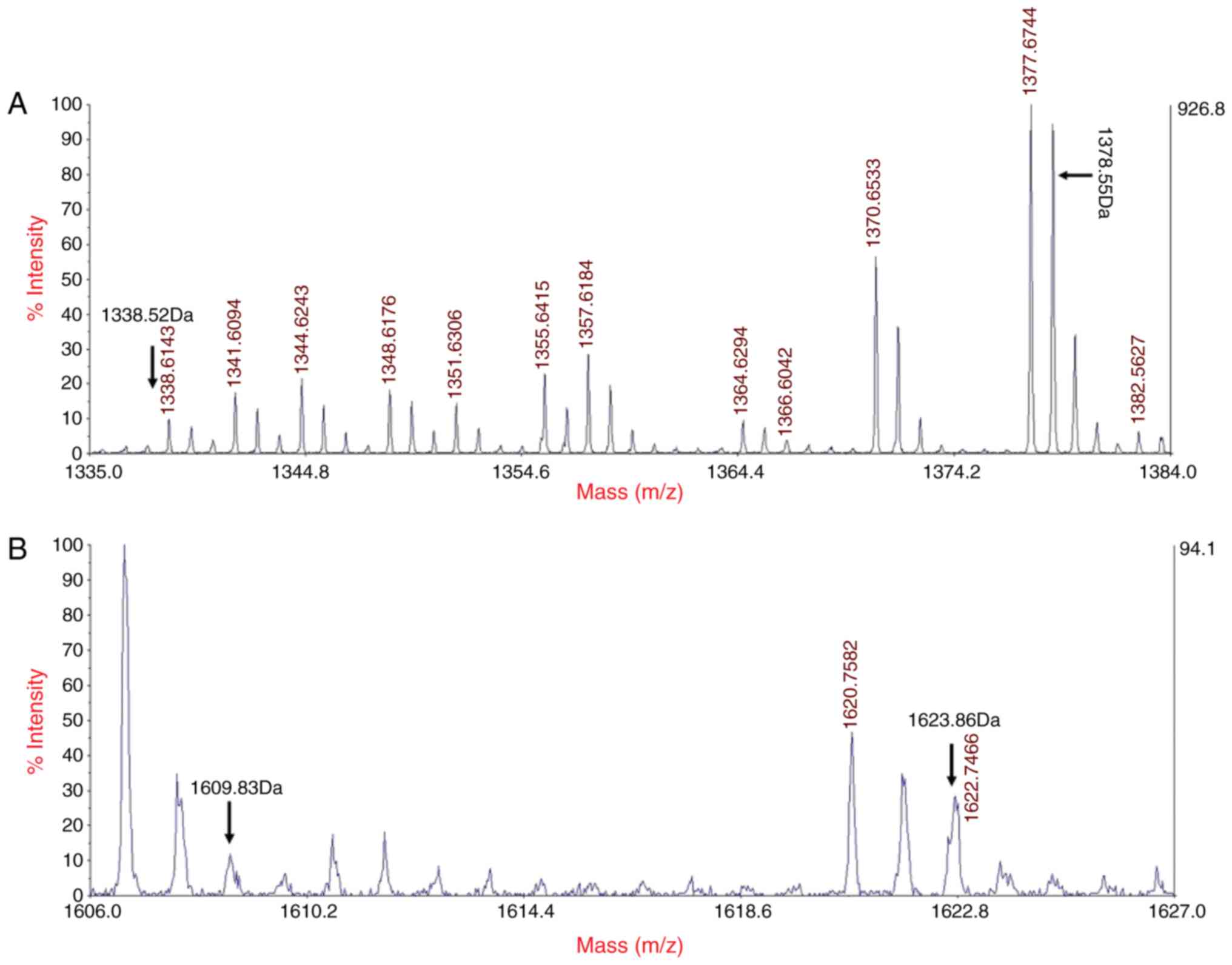
MALDI-TOF MS analysis
MALDI-TOF MS analysis of the proband's α-spectrin
peptide chain indicated that the digested fragments of the mutant
peptides were LEDSYPLQVFK and GDCGDTLAATQSLVMK rather than
LEDSYHLQVFK and GDCGDTLAATQSLLMK, which are present in the normal
peptide. These mutations were heterozygous and identifiable by 2
molecular ion peaksat 1,378.55–1,338.52 and 1,623.86–1,609.83 kDa,
respectively; these corresponded to the mass numbers of the normal
peptide and the mutant peptide of the 2 mutations detected in the
proband (Fig. 5).
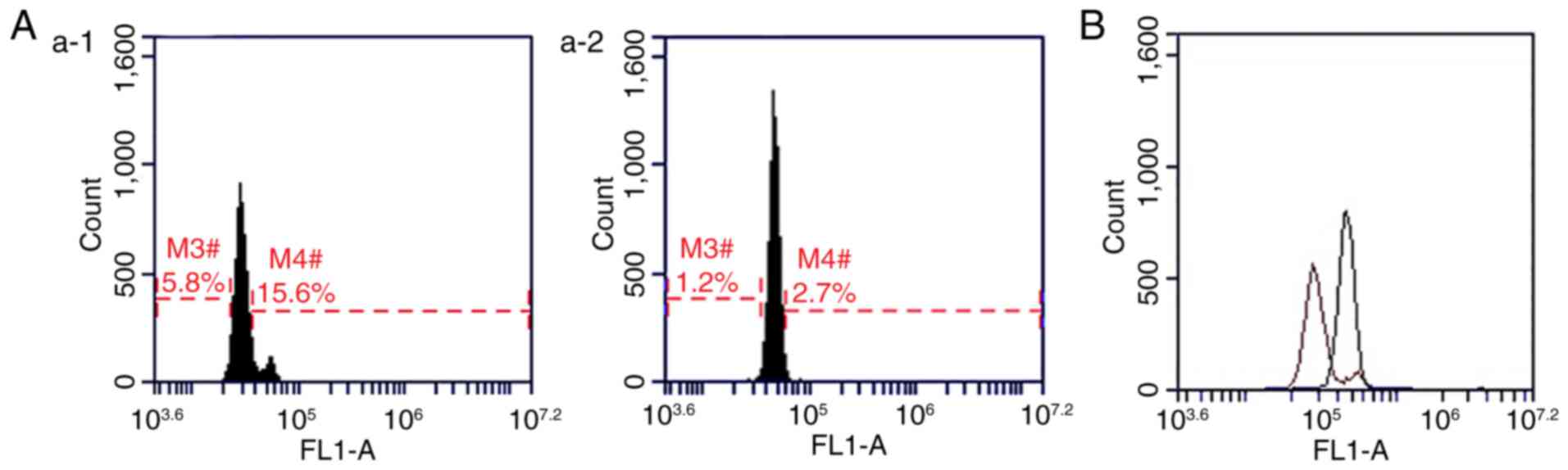
Typical fluorescence profiles for
EMA-labeled RBCs
In order to reduce the level of error, blood samples
collected from 10 healthy individuals (5 males and 5 females; mean
age of 7±2.09 years; age range of 5–12 years) were used as negative
controls for EMA-labeled RBC analysis (11) The results of the present study
indicated that, when compared with the healthy controls, the
fluorescence peaks of the proband, and their father and grandmother
were shifted to the left, and the MCF readings were reduced by
30.81, 12.50 and 17.53%, respectively. In addition, no significant
differences were observed in the fluorescence peaks between the
proband's mother and healthy controls (Fig. 6).
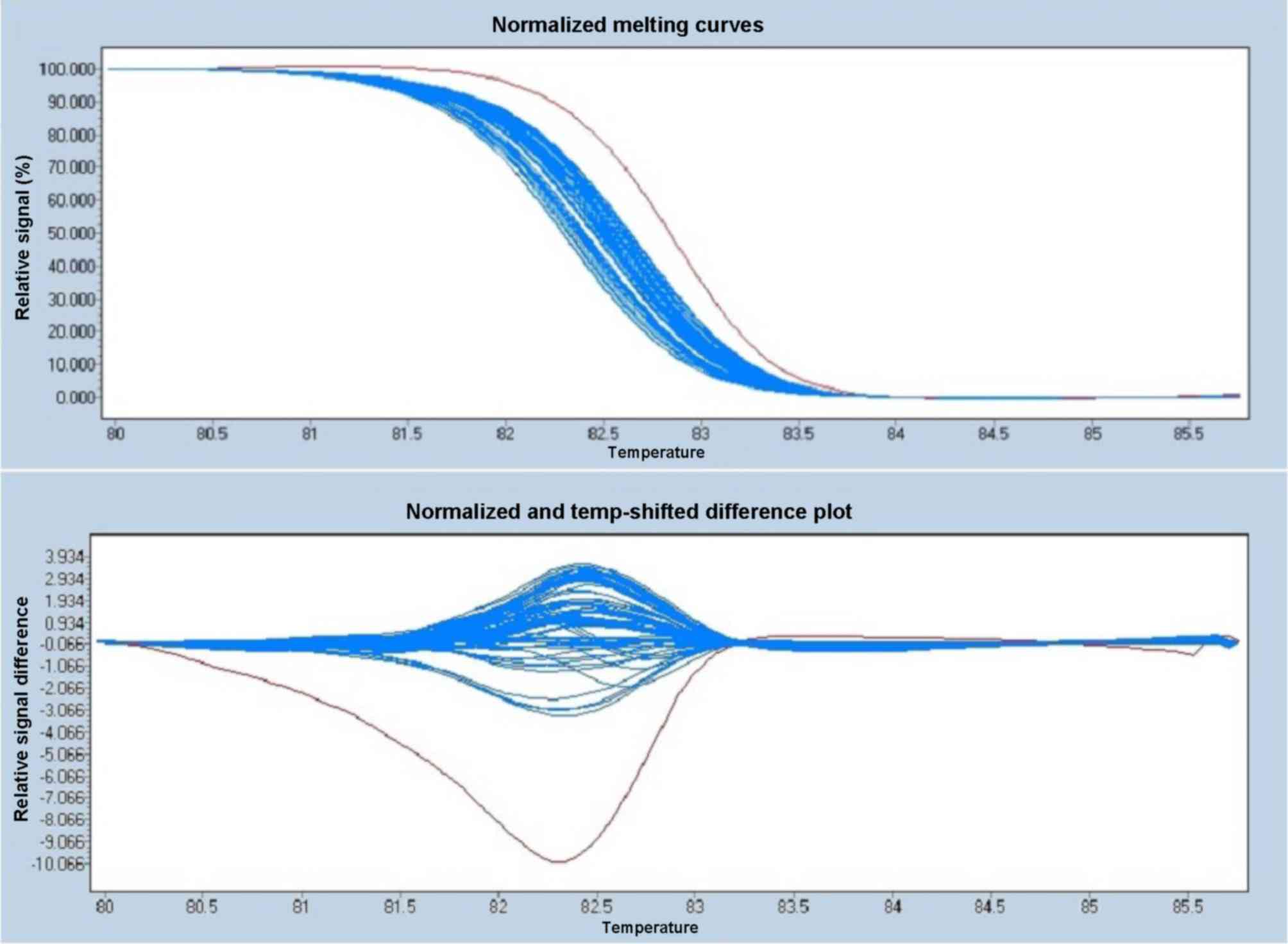
Polymorphism analysis
HRM analysis of the proband (known genotype) and 95
controls (unknown genotype) demonstrated that the proband had an
A/C genotype, while all 95 healthy controls hadan A/A genotype. The
allele frequency in the proband was 1.0% and the melting curves are
presented in Fig. 7.
Discussion
HE is a type of extra vascular hemolytic anemia,
characterized by an increased number of elliptocytes in the
peripheral blood (12). HE has
been reported globally, and its incidence rate greatly varies among
a variety of ethnicities and geographical regions (13) The primary difference between
hereditary spherocytosis (HS) and HE, is that HE is caused by
mutations in the genes encoding transverse linkages of cytoskeleton
proteins in the erythrocyte membrane, resulting in functional
changes of the cell membrane and decreased morphotropism. The most
common causes of HE are structural defects in contractile proteins,
which have been reported in 65% of patients with HE (14). As the number of α-and β-spectrin
dimmers that form into tetramers decreases, certain segments of the
normal peptide chain are replaced by abnormal peptides, resulting
in abnormal RBC function, reduced erythrocyte membrane surface area
and increased membrane osmotic fragility (15) These abnormal RBCs are detained and
broken into RBC debris in the spleen, leading to hemolytic anemia.
Notably, a significant portion of RBC debris can be misclassified
by blood cell analyzers as platelets, resulting in a false increase
in the level of platelets (16).
In this case, it is recommended to perform manual counting.
As HE can result from various genetic defects,
clinical symptoms can vary greatly among patients, ranging from
asymptomatic to chronic hemolytic anemia to a plastic crisis.
Therefore, based on the patient's RBC count, hemoglobin, RET, level
of jaundice and splenomegaly, and other indicators, HE may be
categorized as asymptomatic, compensated hemolytic anemiaor
hemolytic anemia. Asymptomatic and compensated hemolytic anemic
patients exhibit no apparent clinical symptoms, including anemia,
jaundice and splenomegaly; therefore, without genetic testing, HE
may remain unidentified. Generally, patients with a single
heterozygous mutation are often considered asymptomatic, such as
the absence of anemia and hemolytic tendency in the proband's
father and grandmother. Homozygous mutations or compound
heterozygous mutations may account for ~10% of cases, and patients
often exhibit moderate to severe hemolysis and require a blood
transfusion (17).
In the present study, 3 heterozygous mutations were
identified within the proband; laboratory tests indicated that the
proband exhibited symptoms of anemia, jaundice and splenomegaly,
and an increased level of RET. Only families carrying mutations in
the SPTA1 gene may be asymptomatic and symptomatic, while families
carrying mutations in the SPTB gene or EPB41 gene may not exhibit
such diverse clinical manifestations. For the proband, the defect
in the self-assembly of spectrin was one of the major causes of
abnormal morphology of RBCs (18).
DNA analysis of the probandrevealed no mutations within the SPTB
and EPB41 geneshowever, 3 mutations were identified in the SPTA1
gene. In particular, the heterozygous mutationc. 161A>C in exon
2 resulted in a histidine to proline substitution at amino acid 54,
a novel mutation site; its role in disease pathogenesis has not yet
been identified, however, this mutation may affect the mRNA
expression levels or structural stability of proteins, or block the
self-assembly of spectrin dimers to tetramers, resulting in
abnormal transverse linkage with actin proteins. In addition, DNA
mutation analysis of the proband's family members revealed that the
patient's father and grandmother carried the c.161A>C point
mutation. HRM-based genotyping demonstrated that the A/A form was
predominant among healthy controls. At present, The Single
Nucleotide Polymorphism database of the 1,000 Genomes Project
(www.ncbi.nlm.nih.gov/projects/SNP/), does not have a
record of mutations at this site; thus, the frequency among the
general population is unknown. Therefore, based on the associations
between clinical phenotype and genotype inheritance, this locus may
be associated with the manifestation of HE in the proband.
The c.5572C>G mutation in exon 40 and the
6531-12C>T mutation in intron 45 constituted the polymorphic,
high frequency and low expression αLELY locus. The
former caused an amino acid substitution, while the latter led to a
50% reduction in mRNA transcription levels of exon 46 of
α-spectrin, inhibiting the ability to form stable dimmers (19). Individuals with the
αLELY polymorphism, whether heterozygous or homozygous,
do not exhibit any clinical symptoms as αLELY in
cis maintains an over expression of the α-spectrin protein.
However, when αLELY is present with other mutations in
SPTA1, these mutations will increase the concentration of
α-spectrin αLELY in transon the erythrocyte
membrane, and consequently reduce the concentration of
αLELY in cis (20). As a result, the carrier will
exhibit more severe clinical symptoms (21). In the proband's family, their
paternal line carried the c.161A>C mutation and their maternal
line carried the αLELY polymorphism. As these mutations
have no effect on α-spectrin protein expression levels when they
occur independently, no apparent clinical symptoms were observed in
individuals of the parental lines. However, when the proband
inherited the two mutations, the αLELY polymorphism
exacerbated the pathogenic role of c.161A>C in trans,
resulting in more significant hemolysis in the proband. Iolascon
et al (22) reported a
patient with elliptopoikilocytosis who demonstrated a common
polymorphic SPTA1 αLELY allele in trans to the
SPTA1 αExeter allele. The SPTA1 αExeter
allele was inherited from the patient's father and the
αLELY allele was inherited from their healthy,
asymptomatic mother. Polymorphism αLELY, when in
trans to Exeter mutations, aggravated the symptoms in
proportion to the intrinsic severity of the αExeter
intrinsic severity.
MALDI-TOF MS is a novel type of soft-ionization MS
that has fast, accurate, sensitive and high-throughput properties
(23). In the present study,
MALDI-TOF MS was employed, which demonstrated that the proband
possessed the mutant peptides, including 1,338.52 kDa (LEDSYPLQVFK)
and 1,609.83 kDa (GDCGDTLAATQSLVMK), as well as the normal peptides
1,378.55 kDa (LEDSYHLQVFK) and 1,623.86 kDa (GDCGDTLAATQSLLMK),
indicating the presence of heterozygous mutations.
Laboratory tests are important for assisting the
diagnosis of HE. While the diagnosis of HE mainly relies on the
presence of >25% elliptocytes in the peripheral blood smear
(24), the elliptocyte count of
the proband in the present study was <25%. Therefore, further
investigation into clinical manifestations and accompanying
laboratory tests is required (25). The proband exhibited a typical
clinical manifestation; however, information regarding family
history was unavailable. During the early period of
hospitalization, the proband demonstrated no improvement in
jaundice symptoms following treatment with reducing glutathione
tablets, and was thereby re-admitted to the hospital. Blood cell
parameters of the proband were evaluated and increased MCV levels
were observed when compared with their MSCV levels. In addition,
erythrocyte morphological examination indicated the presence of
elliptocytes. Considering the patient's history, clinical
manifestations and laboratory test results, the patient was
diagnosed with HE. Consistent with the genetic analysis of their
family members, laboratory test results revealed that the patient's
father and grandmother had MCV>MSCV levels, indicating that
MCV>MSCV has a diagnostic value in HS patients (26) and also a diagnostic potency for HE
patients.
In the present study, a defect in the proband's
membrane proteins was not detected by SDS-PAGE analysis. Excessive
RET affected the gradient concentration of the separation gel and
the separation of membrane proteins (27); however, the EMA-binding test
revealed abnormalities in the RBCs of the proband. The EMA-binding
test is primarily used to label band 3 proteins on the erythrocyte
membrane and is recommended as a screening method for the diagnosis
of HS (28). It has been suggested
that the band 3protein had the same diagnostic potency in other RBC
membrane diseases (9,17). The results of the present study
revealed that the proband had reduced levels of EMA-labeled
erythrocytes compared with healthy controls, suggesting that the
EMA-binding test may be promising for the screening of HE.
HRM analysis is an emerging molecular diagnostic
technique based on the monitoring of changes in the melting curves
of nucleic acids, indicated by the saturation of fluorescence by
quantitative PCR. HRM analysis is not limited by nucleotide
positions and types, and does not require sequence-specific probes
(29). In addition, HRM analysis
identifies mutations by analyzing changes in the melting curves of
PCR products. The formation of a heterologous double-stranded DNA
(dsDNA) in a heterozygous mutation that results in a melting curve
that is significantly different compared with that of homozygous
dsDNA. In the present study, HRM was used for gene mutation
screening as it has many advantages, including high sensitivity and
specificity, low cost and high-throughput. HRM analysis was
employed to screen the His54Pro mutation in the SPTA1 gene of the
proband and 95 healthy controls, which revealed that the His54Pro
mutation rate was 1.0%. This indicated that the His54Pro mutation
was a rare mutation site with a low incidence rate among the
healthy population.
Special treatment is generally not required when a
patient with HE exhibits no or only mild signs of anemia (30). During the early stages, infection
in the proband aggravated their anemia and jaundice, and their HGB
levels (72 g/l) fell to a level that required blood transfusion.
Following control of the infection, the proband's HGB
levelsreturned to 93 g/l and their bilirubin levelsmarkedly
reduced. This suggestedthat an infection in a HE patient requires
timely anti-infection treatment and close monitoring rather than an
immediate blood transfusion as the HGB count can eventually recover
independently. Unnecessary transfusion of blood products can induce
systemic allergic reactions and also increase the risk of
infectious diseases (31).
Furthermore, the transfused RBCs may be abnormally damaged in the
recipient's body due to immune or non-immune responses, which may
result in further hemolysis.
In conclusion, a novel SPTA1 gene mutation was
detected in a HE proband and the corresponding mutant peptide
segments was confirmed by MALDI-TOF MS. The HE-associated clinical
manifestations are quite diverse; therefore, HE may be misdiagnosed
or undetected. In the present study, comprehensive analysis using
EMA-labeled flow cytometry and SDS-PAGE was performed to detect
protein defects; blood smear morphologic examinations and routine
laboratory tests for Hb, MCV, MSCV, RET%, TBIL and DBIL may improve
the rate of diagnosis and timely treatment of HE, as well as
reducing the risk of HE-associated complications.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81360263).
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
HE
|
hereditary elliptocytosis
|
|
MSCV
|
mean sphered corpuscular volume
|
|
EMA
|
eosin-5-maleimide
|
|
MALDI-TOF
|
matrix-assisted laser
desorption-ionization time of flight
|
|
MCF
|
mean channel florescence
|
|
HRM
|
high resolution melting
|
References
|
1
|
An X and Mohandas N: Disorders of red cell
membrane. Br J Haematol. 141:367–375. 2008.PubMed/NCBI
|
|
2
|
Gallagher PG: Hereditary elliptocytosis:
Spectrin and protein 4.1R. Semin Hematol. 41:142–164. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Christensen RD, Nussenzveig RH, Reading
NS, Agarwal AM, Prchal JT and Yaish HM: Variations in both
alpha-spectrin (SPTA1) and beta-spectrin (SPTB) in a neonate with
prolonged jaundice in a family where nine individuals had
hereditary elliptocytosis. Neonatology. 105:1–4. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gaetani M, Mootien S, Harper S, Gallagher
PG and Speicher DW: Structural and functional effects of hereditary
hemolytic anemia-associated point mutations in the alpha spectrin
tetramer site. Blood. 111:5712–5720. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iolascon A, Miraglia del Giudice E and
Camaschella C: Molecular pathology of inherited erythrocyte
membrane disorders: Hereditary spherocytosis and elliptocytosis.
Haematologica. 77:60–72. 1992.PubMed/NCBI
|
|
6
|
Delaunay J, Alloisio N, Morle L, Baklouti
F, Dalla Venezia N, Maillet P and Wilmotte R: Molecular genetics of
hereditary elliptocytosis and hereditary spherocytosis. Ann Genet.
39:209–221. 1996.PubMed/NCBI
|
|
7
|
Rungaldier S, Oberwagner W, Salzer U,
Csaszar E and Prohaska R: Stomatin interacts with GLUT1/SLC2A1,
band 3/SLC4A1, and aquaporin-1 in human erythrocyte membrane
domains. Biochim Biophys Acta. 1828:956–966. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fairbanks G, Steck TL and Wallach DF:
Electrophoretic analysis of the major polypeptides of the human
erythrocyte membrane. Biochemistry. 10:2606–2617. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
King MJ, Jepson MA, Guest A and Mushens R:
Detection of hereditary pyropoikilocytosis by the eosin-5-maleimide
(EMA)-binding test is attributable to a marked reduction in
EMA-reactive transmembrane proteins. Int J Lab Hematol. 33:205–211.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gharahdaghi F, Weinberg CR, Meagher DA,
Imai BS and Mische SM: Mass spectrometric identification of
proteins from silver-stained polyacrylamide gel: A method for the
removal of silver ions to enhance sensitivity. Electrophoresis.
20:601–605. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hunt L, Greenwood D, Heimpel H, Noel N,
Whiteway A and King MJ: Toward the harmonization of result
presentation for the eosin-5′-maleimide binding test in the
diagnosis of hereditary spherocytosis. Cytometry B Clin Cytom.
88:50–57. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Soderquist C and Bagg A: Hereditary
elliptocytosis. Blood. 121:30662013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakanishi H, Wada H, Suemori S and
Sugihara T: Hereditary red cell membrane disorders in Japan:
Comparison with other countries. Rinsho Ketsueki. 56:760–770.
2015.(In Japanese). PubMed/NCBI
|
|
14
|
An X: The red cell membrane, part 2:
Disorders of the red cell membrane. Clin Adv Hematol Oncol.
12:606–608. 2014.PubMed/NCBI
|
|
15
|
Alloisio N, Morlé L, Maréchal J, Roux AF,
Ducluzeau MT, Guetarni D, Pothier B, Baklouti F, Ghanem A, Kastally
R and Delaunay J: Sp alpha V/41: A common spectrin polymorphism at
the alpha IV-alpha V domain junction. Relevance to the expression
level of hereditary elliptocytosis due to alpha-spectrin variants
located in trans. J Clin Invest. 87:2169–2177. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mullier F, Lainey E, Fenneteau O, Da Costa
L, Schillinger F, Bailly N, Cornet Y, Chatelain C, Dogne JM and
Chatelain B: Additional erythrocytic and reticulocytic parameters
helpful for diagnosis of hereditary spherocytosis: Results of a
multicentre study. Ann Hematol. 90:759–768. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Suemori S, Wada H, Nakanishi H, Tsujioka
T, Sugihara T and Tohyama K: Analysis of hereditary elliptocytosis
with decreased binding of Eosin-5-maleimide to red blood cells.
Biomed Res Int. 2015:4518612015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gallagher PG and Forget BG:
Hematologically important mutations: Spectrin variants in
hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood
Cells Mol Dis. 22:254–258. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wilmotte R, Maréchal J, Morlé L, Baklouti
F, Philippe N, Kastally R, Kotula L, Delaunay J and Alloisio N: Low
expression allele alpha LELY of red cell spectrin is associated
with mutations in exon 40 (alpha V/41 polymorphism) and intron 45
and with partial skipping of exon 46. J Clin Invest. 91:2091–2096.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Delaunay J, Nouyrigat V, Proust A,
Schischmanoff PO, Cynober T, Yvart J, Gaillard C, Danos O and
Tchernia G: Different impacts of alleles alphaLEPRA and alphaLELY
as assessed versus a novel, virtually null allele of the SPTA1 gene
in trans. Br J Haematol. 127:118–122. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tolpinrud W, Maksimova YD, Forget BG and
Gallagher PG: Nonsense mutations of the alpha-spectrin gene in
hereditary pyropoikilocytosis. Haematologica. 93:1752–1754. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iolascon A, King MJ, Robertson S, Avvisati
RA, Vitiello F, Asci R, Scoppettuolo MN and Delaunay J: A genomic
deletion causes truncation of alpha-spectrin and
ellipto-poikilocytosis. Blood Cells Mol Dis. 46:195–200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhao G, Xu B, Li X, Tang C, Qin H, Wang H,
Yang S, Wang W, Gao H, He K and Liu X: Detection of serum peptides
in patients with lung squamous cell carcinoma by MALDI-TOF-MS and
analysis of their correlation with chemotherapy efficacy. Zhongguo
Fei Ai Za Zhi. 20:318–325. 2017.PubMed/NCBI
|
|
24
|
Gallagher PG: Red cell membrane disorders.
Hematology Am Soc Hematol Educ Program. 2005:13–18. 2005.
|
|
25
|
Nie N, Shao YQ, Shi J, Ge M and Zhen Y:
Clinical analysis of five patients with hereditary elliptocytosis.
Clin J Hematol. 6:540–541. 2013.(In Chinese).
|
|
26
|
Liao L, Deng ZF, Qiu YL, Chen P, Chen WQ
and Lin FQ: Values of mean cell volume and mean sphered cell volume
can differentiate hereditary spherocytosis and thalassemia.
Hematology. 19:393–396. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
King MJ and Zanella A: Hereditary red cell
membrane disorders and laboratory diagnostic testing. Int J Lab
Hematol. 35:237–243. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
King MJ, Garçon L, Hoyer JD, Iolascon A,
Picard V, Stewart G, Bianchi P, Lee SH and Zanella A: ICSH
guidelines for the laboratory diagnosis of nonimmune hereditary red
cell membrane disorders. Int J Lab Hematol. 37:304–325. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Druml B and Cichna-Markl M: High
resolution melting (HRM) analysis of DNA-its role and potential in
food analysis. Food Chem. 158:245–254. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Barcellini W, Bianchi P, Fermo E,
Imperiali FG, Marcello AP, Vercellati C, Zaninoni A and Zanella A:
Hereditary red cell membrane defects: Diagnostic and clinical
aspects. Blood Trasfus. 9:274–277. 2011.
|
|
31
|
Yan J, Deng D, Luo M, Cheng P, Chi B, Yuan
Y, Liao L and Lin F: Dysfibrinogenemia in a patient undergoing
artificial abortion after misdiagnosis and review of the
literature. Clin Chim Acta. 447:86–89. 2015. View Article : Google Scholar : PubMed/NCBI
|