Introduction
Acute kidney injury (AKI) is one of the most common
and serious complications of sepsis (1). The morbidity of AKI secondary to
sepsis in the intensive care unit may reach 70% (2). The mortality rate for septic patients
with AKI is increased compared with patients with sepsis alone
(3). However, there are no
effective therapeutic strategies to treat this disease. Therefore,
a better understanding of the molecular basis of the disease may
contribute to more targeted therapies of septic AKI.
Autophagy is a protective mechanism of the body
against injury, which may maintain the homeostasis of the body
(4). Recent studies have
demonstrated that autophagy is activated during AKI (5,6).
Leventhal et al (7),
reported that autophagy may be induced by lipopolysaccharide (LPS)
in renal tubular epithelial cells, and that the induction of
autophagy has a protective effect in AKI. As a selective autophagy
signaling adaptor, sequestosome-1 (p62) is one of the indicators of
the level of autophagy (8,9). The ablation of important autophagy
proteins has been observed to increase p62 levels and to damage
renal function. (6) p62 possesses
a number of important domains that facilitate interactions with
numerous signaling activators (10,11).
It has been demonstrated that p62-mediated selective autophagy is
regulated by its PB1 domain, light chain (LC) 3-interacting region
and ubiquitin-associated domain (12). The N-terminal PB1 domain enables
p62 to self-associate and polymerize in the cytoplasm to form
aggregates and cytoplasmic inclusion bodies, and these
self-aggregates are degraded by autophagy (13). The C-terminal ubiquitin-binding
domain enables p62 to recognize and bind to polyubiquitinated
proteins (11). The
LC3-interacting region (LIR) facilitates p62 binding to LC3-II (a
marker of autophagy) and p62-bound polyubiquitinated cargo to the
autophagosomes for degradation (14). The selective turnover of p62 by
autophagy serves an important role in the formation of cytoplasmic
proteinaceous aggregates, identified as a common hallmark of
autophagy-deficient post-mitotic cells (8,15).
Since p62 is implicated in a number of signaling pathways, its
expression level is important. Recently, p62 has been demonstrated
to serve essential roles in various diseases, including liver and
breast cancer (16,17), Paget's disease of bone (18), obesity and insulin resistance
(19,20). However, little is known about the
expression pattern and roles of p62 in septic AKI.
In the present study, a mouse model of endotoxemia
was developed via intraperitoneal injection of LPS, the most common
toxemia model. Kidney tissue was used to detect the expression and
location of p62. In addition, at the cellular level, the present
study overexpressed and knocked down p62 in renal tubular
epithelial cells by transfection of p62 overexpression plasmids and
siRNA, respectively, to analyze the possible roles.
Materials and methods
Animals and treatment
Male C57BL/6 mice at 8 weeks of age, 20~25 g, were
purchased from the Experimental Animal Center of Central South
University (Changsha, China), and housed in a temperature (25°C)
and humidity (40~60%) controlled facility on a 12-h light/dark
cycle with free access to food and water. A total of 30 mice were
divided into two groups: The LPS group (n=20), injected with LPS
(10 mg/kg; intraperitoneal); and the control group (n=10), injected
with saline. Subsequently, the 72-h survival rate was observed. An
additional 40 mice were divided into five groups (n=8) and injected
with LPS (10 mg/kg; intraperitoneal) for 0 (control), 4, 8, 12 and
24 h individually. The mice were anesthetized and bled by heart
puncture. Subsequently, the mice were sacrificed, upper and lower
parts of the left kidneys were collected in liquid nitrogen and
then stored at −80°C for reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis and western blotting.
Middle parts of the left kidneys were collected and fixed in 4%
paraformaldehyde at 4°C overnight for paraffin embedded and 3 µm
sections were cut and stained with immunohistochemistry. The cortex
and medulla of the right kidneys were separated and collected in
liquid nitrogen and then stored at −80°C until for western blotting
analysis. All animal experimental protocols were approved by the
institutional ethics committee for animal experiments of Central
South University.
Assessment of renal function
Renal functional parameters including blood urine
nitrogen (BUN) and creatinine were detected using a Synchron CX7
autoanalyzer (Beckman Coulter, Inc., Brea, CA, USA).
Reverse-transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The isolation of total RNA and the production of
cDNA was performed as previously described (21). qPCR was performed using a 7500 Real
Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) with a Two Step SYBR®
PrimeScript™ RT-qPCR kit (Takara Bio, Inc., Otsu, Japan). The
amplification was performed over 40 cycles with conditions of 95°C
for 30 sec, 95°C for 5 sec and 60°C for 34 sec. The relative
quantitation of mRNA was analyzed using the 2−ΔΔCq
method (22) and normalized by
β-actin. The primers were as follows: β-actin sense,
5′-CATTGCTGACAGGATGCAGAAGG-3′ and antisense,
5′-TGCTGGAAGGTGGACAGTGAGG-3′; p62 sense,
5′-GCTCTTCGGAAGTCAGCAAACC-3′ and antisense,
5′-GCAGTTTCCCGACTCCATCTGT-3′.
Western blotting
Total protein from renal tissues was extracted using
radio immunoprecipitation assay (RIPA) lysis buffer (1% Triton
X-100, 150 mM NaCl, 5 mM EDTA, and 10 mM Tris-HCl, pH 7.0)
containing a protease inhibitor cocktail (cat. no. WB-0071, Beijing
Dingguo Changsheng Biotechnology Co. Ltd, Beijing, China). Proteins
were quantified using a bicinchoninic acid kit. Equal amounts of
protein from each sample (20–80 µg per lane) were mixed with SDS
sample buffer, heated to 100°C for 10 min to expose epitopes and
subjected to electrophoresis on a 10% SDS-PAGE gel. Proteins were
transferred to a polyvinylidene fluoride membrane (EMD Millipore,
Billerica, MA, USA). Following blocking in 2% bovine serum albumin
(Gen-view Scientific Inc., USA) in TBS with Tween 20 for 1 h at
room temperature, membranes were incubated with primary antibodies
against p62 (cat. no. 5114, 1:1,000 dilution, Cell Signaling
Technology, Inc., Danvers, MA, USA), LC-3B (cat. no. 3868, 1:1,000
dilution; Cell Signaling Technology, Inc.) and β-actin (cat. no.
A1978, 1:2,000 dilution, Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) at 4°C overnight. Following washing three times, the
membranes were subsequently incubated with the HRP-conjugated goat
anti-rabbit IgG (cat. no. BA1054, 1:3,000, Boster Biological
Technology, Pleasanton, CA, USA) or goat anti-mouse IgG antibody
(cat. no. BA1050, 1:3,000, Boster Biological Technology) for 1 h at
room temperature. Bands were determined using the enhanced
chemiluminescence method (cat. no. 170-5061, Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The levels of proteins were
quantitatively analyzed using ImageJ software, version 1.48
(National Institutes of Health, Bethesda, MD, USA) and normalized
to the β-actin band density.
Immunohistochemistry
Immunohistochemistry was performed using EnVision
(TM) FLEX Mini kit, according to the manufacturer's protocol (cat.
no. K8024, Dako; Agilent Technologies, Inc., Santa Clara, CA, USA).
Briefly, slides were deparaffinized and hydrated in xylene and a
graded series of alcohol. Heat-induced antigen retrieval was
performed and endogenous peroxidase activity was blocked using a
peroxidase-blocking reagent (as part of the EnVision FLEX Mini kit;
cat. no. K8024-DM821, Dako; Agilent Technologies, Inc., Santa
Clara, CA, USA). Sequentially, the slides were incubated with
primary antibodies, HRP-conjugated secondary antibody (as part of
the EnVision FLEX Mini kit; cat. no. K8024-DM822, Dako; Agilent
Technologies, Inc.), diaminobenzidine and hematoxylin for 30 sec,
at room temperature. Primary antibodies included p62 (cat. no.
5114, 1:50 dilution, Cell Signaling Technology, Inc.), Na-K-2Cl
cotransporter (NKCC-2; cat. no. AB3562P, 1:200 dilution; EMD
Millipore), aquaporin-2 (AQP2, cat. no. 3487S, 1:200 dilution, Cell
Signaling Technology, Inc.), and aquaporin-1 (AQP1, cat. no.
M00865, 1:50 dilution; Boster Biological Technology). Finally, the
slides were analyzed using an optical microscope (Olympus
Corporation, Tokyo, Japan).
Cell culture
The HK-2 cell line from the American Type Culture
Collection (Manassas, VA, USA) was maintained in Dulbecco's
modified Eagle's medium/F12 (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C in an incubator supplemented with 5%
CO2. Medium was renewed every 2 days.
Transfection of plasmids and siRNA
into HK-2 cells
pENTER eukaryotic plasmids expressing p62 and small
interfering RNA (siRNA) of p62 were purchased from Vigene
Biosciences, Inc. (Rockville, MD, USA) and Shanghai GenePharma Co.,
Ltd. (Shanghai, China), respectively. The plasmid was extracted
using Plasmid Maxprep kit (Vigorous Biotechnology, Beijing, China;
www.vigorousbiol.com/), according to the
manufacturer's protocol. Plasmid (2 µg/µl) and siRNA (5 µl)
transfections into HK-2 cells (50,000 cells/cm2) were
performed using Lipofectamine 3000™ (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. After
48 h, protein was extracted for western blot analysis. The siRNA
sequences were as follows: p62-siRNA sense,
5′-GAUCUGCGAUGGCUGCAAUTT-3′ and antisence,
5′-AUUGCAGCCAUCGCAGAUCTT-3′; control-siRNA sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and antisence,
5′-ACGUGACACGUUCGGAGAATT-3′.
Cell grouping
Cells transfected with p62 overexpression
plasmids/siRNA were divided into four groups: i)
PBS+pENTER/negative; ii) PBS+p62-pENTER/p62-siRNA; iii)
LPS+pENTER/negative; and iv) LPS+p62-pENTER/p62-siRNA groups.
According to the preliminary results (23), cells (50,000 cells/cm2)
were treated with 1,000 ng/ml LPS for 12 h. The cells and
supernatants were collected for subsequent experiments.
Analysis of cell proliferation and
lactate dehydrogenase (LDH) level
The viability of cells cultured in 96-well plates
(4,000 cells/cm2) in each group was measured using a
Cell Counting Kit-8 kit (Bimake, Shanghai, China; www.bimake.com/search?q=CCK-8),
according to the manufacturer's protocol. The culture supernatants
were used to measure the LDH level using an LDH cytotoxicity test
kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China),
according to the manufacturer's protocol.
Apoptosis detection using flow
cytometry
The cells in each group were washed, digested and
subjected to apoptosis analysis using an Annexin V-Fluorescein
Isothiocyanate/Propidium Iodide Apoptosis Detection kit (BD
Pharmingen; BD Biosciences, Franklin Lakes, NJ, USA), according to
the manufacturer's protocol. Subsequently, flow cytometric analysis
was performed using a flow cytometer (BD Biosciences). Data were
analyzed using FlowJo software (version 7.6.5; FlowJo, LLC,
Ashland, OR, USA).
Statistical analysis
All experiments were repeated at least three times.
The quantitative data are presented as the mean ± standard
deviation. Comparisons among groups were analyzed using one-way
analysis of variance with Tukey's post hoc test. Survival analysis
was performed using the Kaplan-Meier method and the log-rank test.
Statistical analysis was performed using GraphPad Prism 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
Effects of LPS on the survival rate
and renal function of mice
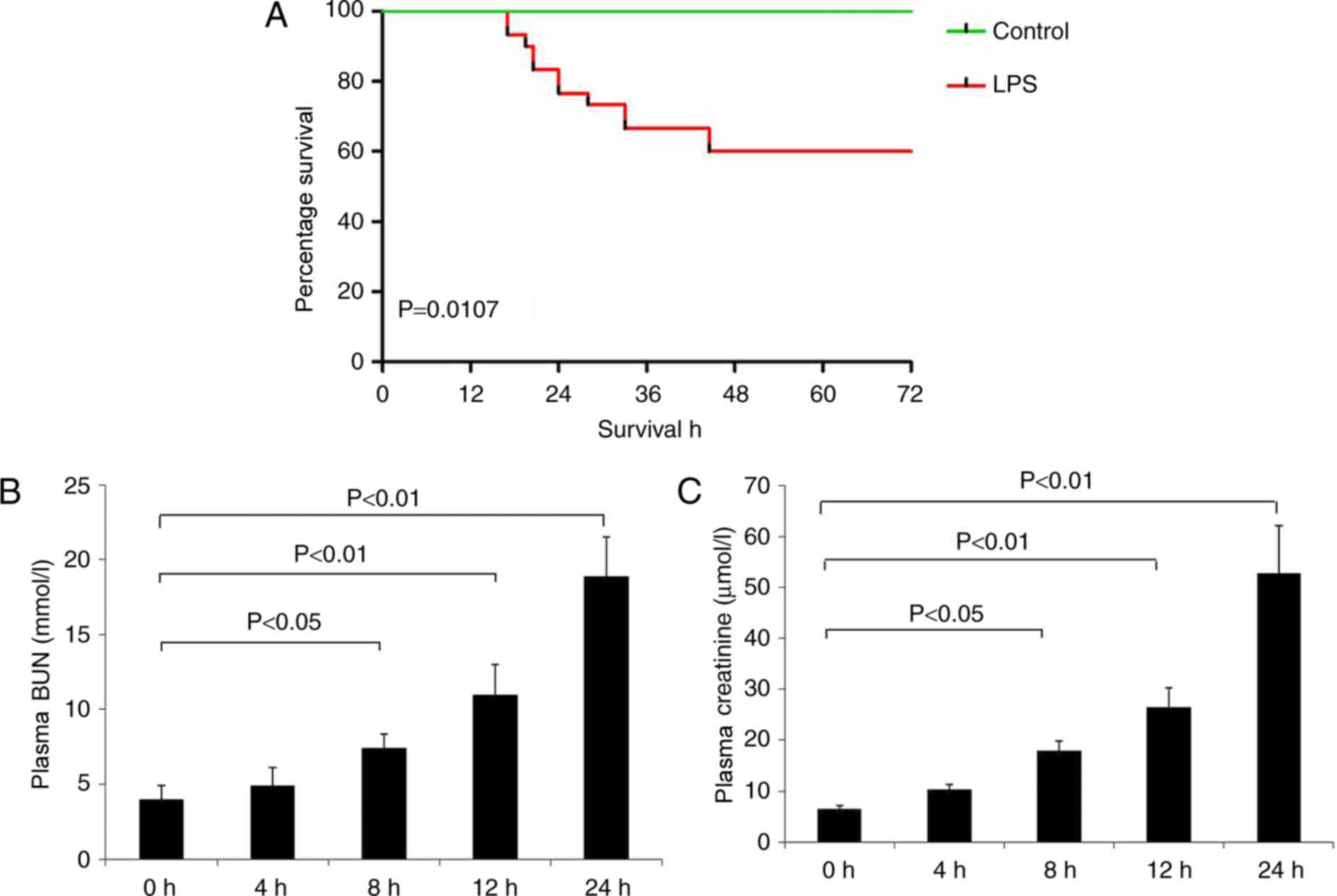
As presented in Fig.
1A, the survival rate of the LPS group was significantly
decreased compared with the control group (P<0.05). LPS
administration additionally decreased renal function at 8–24 h, as
demonstrated by a significant increase in BUN and creatinine levels
compared with the control group (Fig.
1B and C).
p62 and LC-3B expression in the
kidneys of mice during endotoxemia
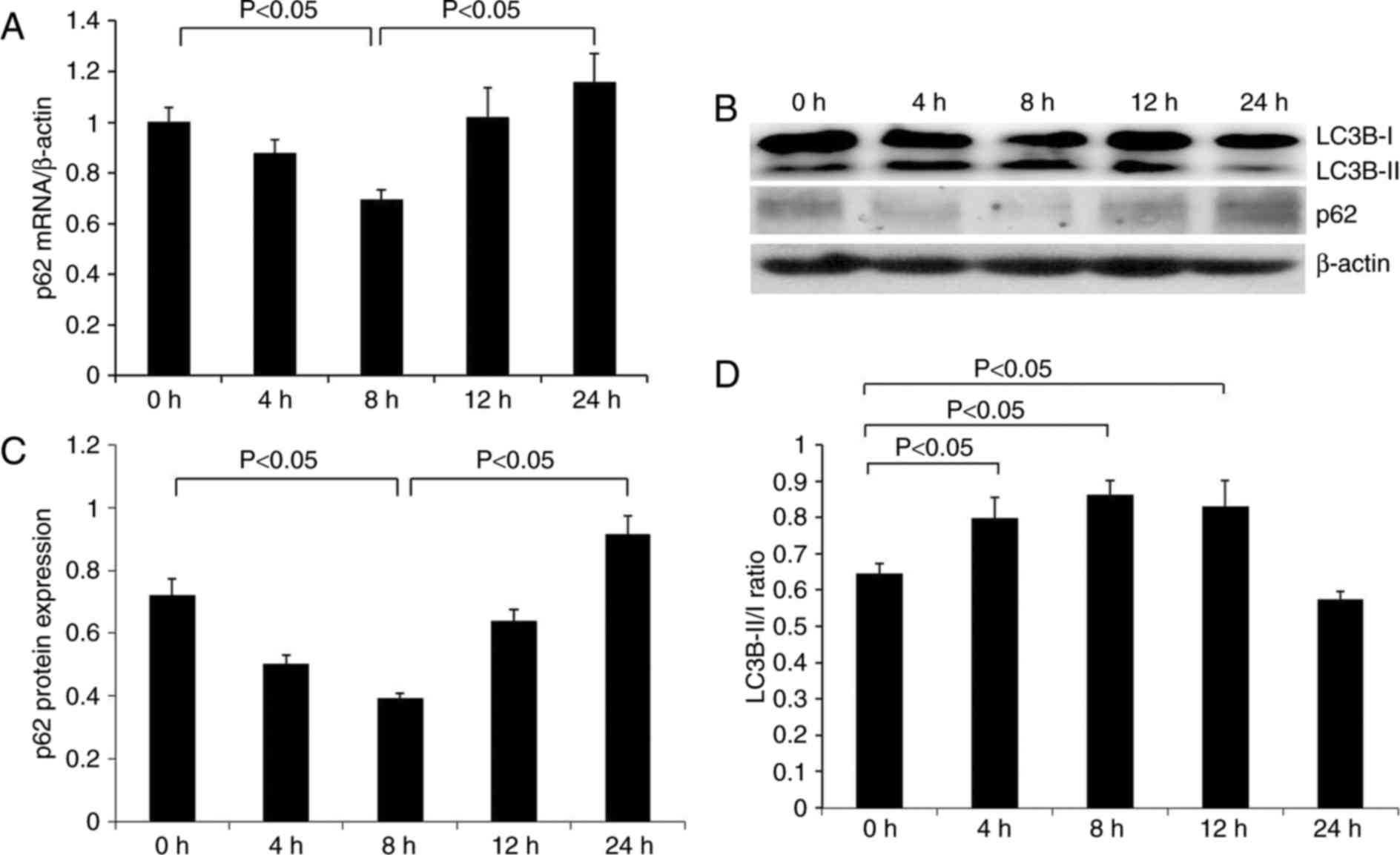
As indicated in Fig.
2A, renal p62 mRNA expression was apparent in the control group
(0 h) compared with β-actin mRNA. The p62 signal was gradually
decreased at 4 h subsequent to LPS injection, reaching the lowest
level at 8 h and thereafter increasing at 12–24 h. The protein
expression pattern of p62 was similar to that of p62 mRNA (Fig. 2B and C). p62 protein was expressed
at a relatively high level in normal renal tissues, gradually
decreasing to the lowest level at 8 h following treatment with LPS,
and subsequently increased over time. p62 protein expression was
increased compared with the control group at 24 h.
In order to clarify the effect of LPS on renal
autophagy, the expression of the autophagy-associated protein LC3B
was additionally examined. The ratio of LC3-II/LC3-I has been
previously used to represent the level of autophagy (24). It was identified that the ratio of
LC3B-II/LC3B-I was significantly increased at 4 h following
treatment with LPS, reaching a peak at 8 h, and was slightly
decreased at 12 h, although significantly higher than the control
group. At 24 h, the ratio was significantly reduced below the
normal level (Fig. 2B and D).
Cellular localization of p62 protein
in the renal cortex of mice during endotoxemia
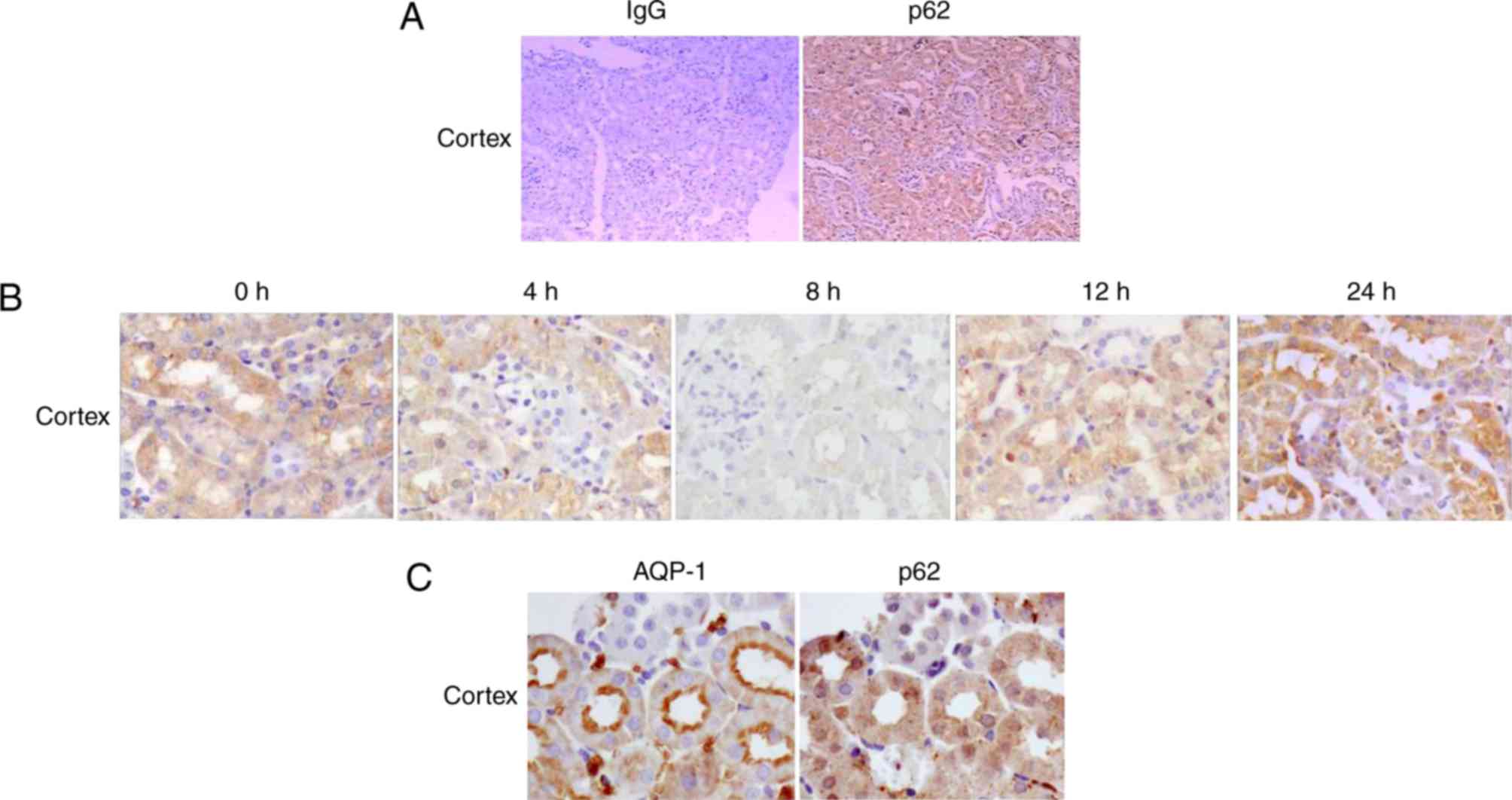
The renal cellular localization of p62 in each group
was determined by immunohistochemical staining at various time
points following LPS injection. The results demonstrated that the
expression pattern of p62 in mouse kidneys during endotoxemia was
dynamic. In the control group (0 h), p62 protein was predominantly
localized in the renal cortex (Fig.
3A). In addition, p62 was expressed primarily in renal cortical
tubules with a small amount in the glomeruli (Fig. 3A).
In mice treated with LPS, the pattern of p62 protein
expression in the renal cortex was consistent with findings from
RT-qPCR analysis and western blotting, demonstrating minimal
expression in mice treated with LPS for 8 h and increasing in mice
treated with LPS for 24 h (Fig.
3B). Fig. 3C illustrates that
AQP-1 was expressed primarily in proximal tubular segments in the
renal cortex, and it was used as a marker of proximal tubules.
Co-staining of kidney sections for p62 and AQP-1 confirmed the
localization of p62 in the proximal tubules of the renal cortex. In
addition, p62 protein was primarily detected in the cytoplasm of
the cortical tubules, and little was detected in the nucleus.
Expression and cellular localization
of p62 protein in the renal medullas of mice during
endotoxemia
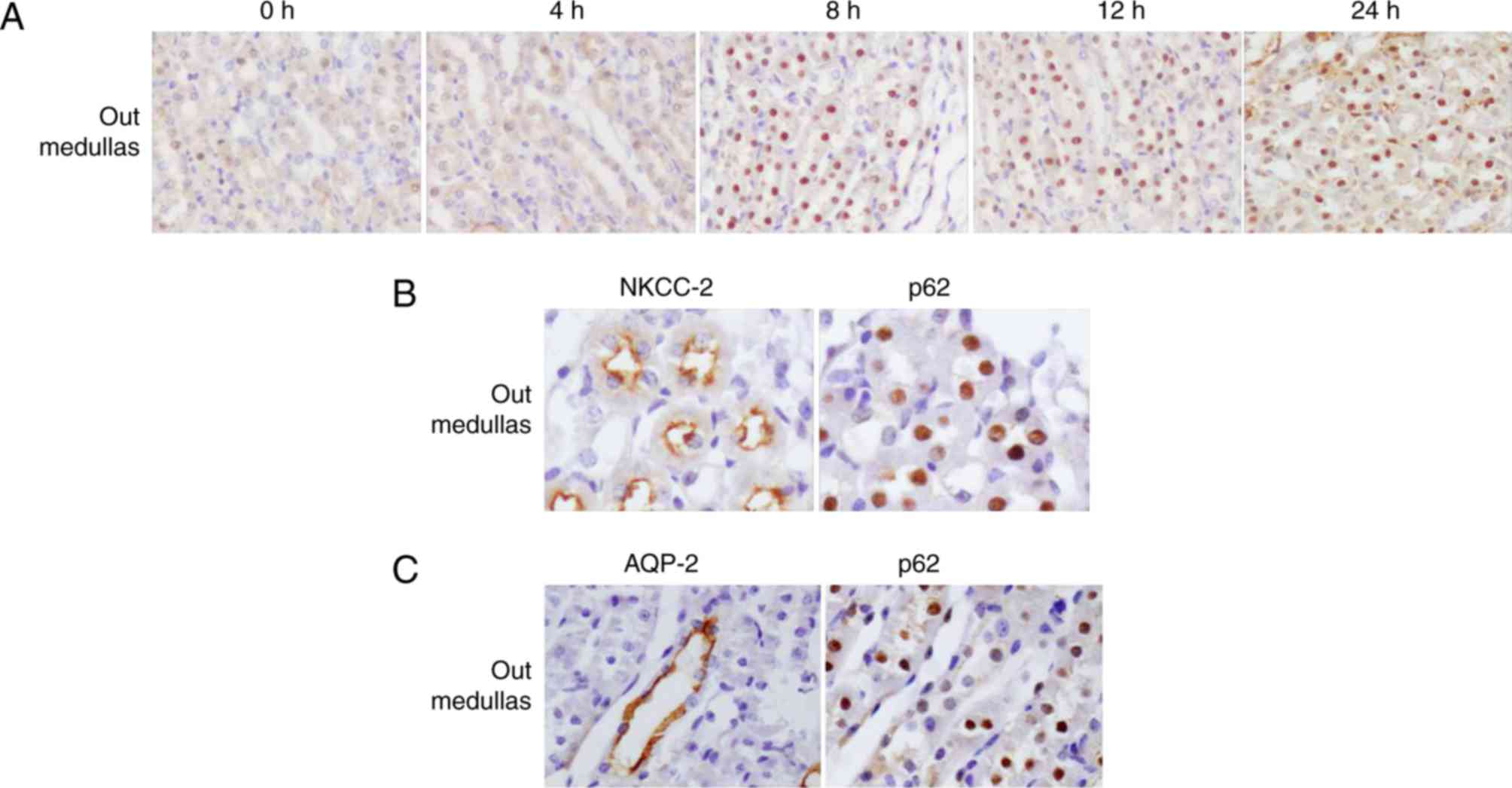
The expression and cellular localization of p62
protein in renal medullas was LPS-dependent. From 8 h following
treatment with LPS, it was observed that p62 was redistributed from
the cortex to the outer medullas (Fig.
4A). In order to determine the exact location of p62, the
present study detected the expression of p62 and NKCC-2 (a marker
of the thick ascending limb) using consecutive kidney sections at 8
h following LPS administration. The results demonstrated that p62
and NKCC-2 were co-localized, confirming that p62 was located in
the thick ascending limb (Fig.
4B). In addition, as a marker of collecting ducts, AQP2 was
additionally detected using consecutive sections. By contrast, the
AQP2-positive regions exhibited a weak p62 signal (Fig. 4C), indicating substantially
decreased p62 expression in the collecting ducts in the outer
medullas. It was unexpected that p62 protein was primarily detected
in the nuclei of tubular cells in outer medulla.
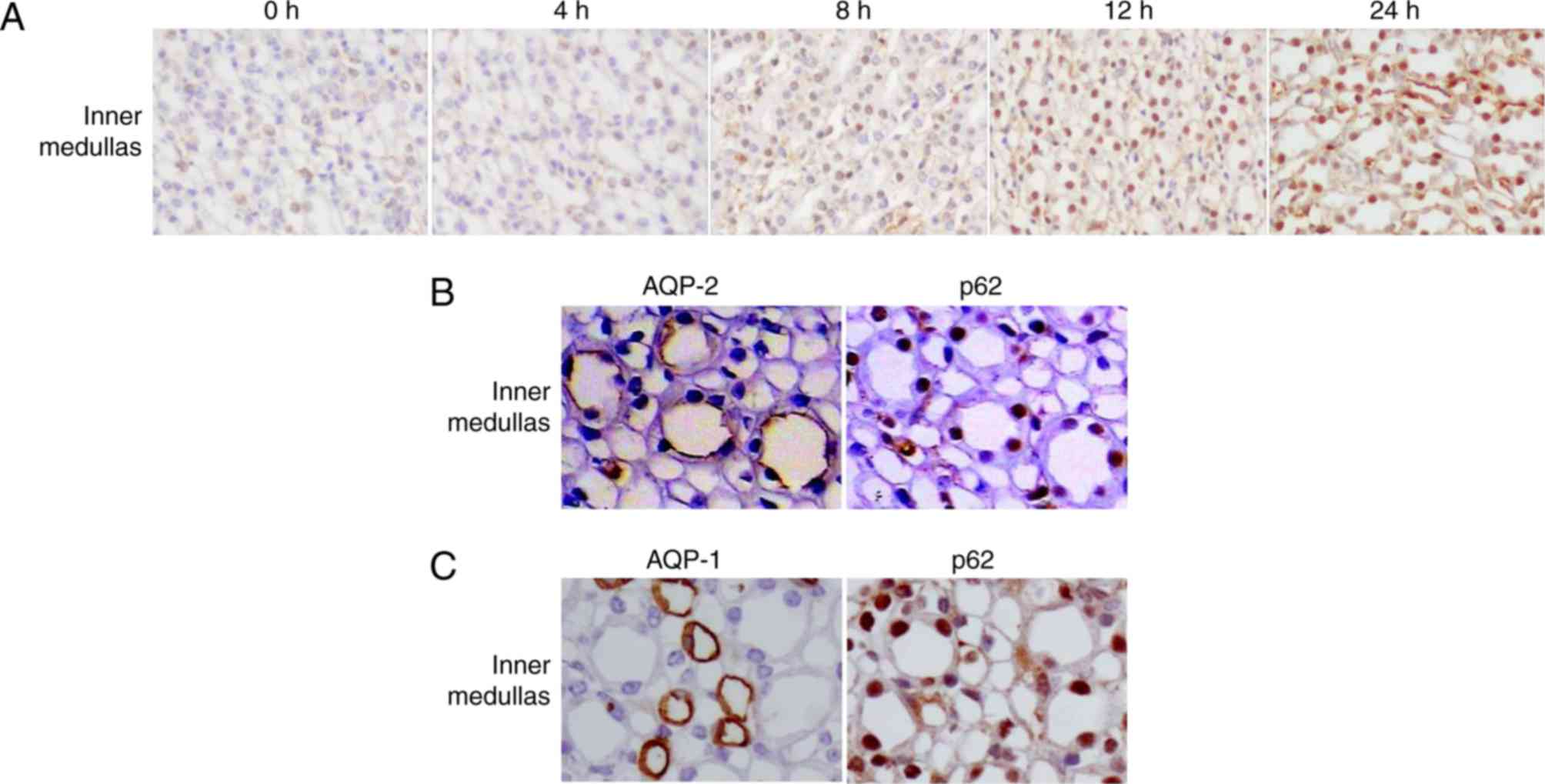
The redistribution of p62 in the inner medulla
(Fig. 5A) was different compared
with that in the outer medulla. In control mice, the expression of
p62 was low in the inner medullas. However, in mice treated with
LPS, high levels of p62 were detected in the nuclei of tubular
cells in the inner medulla (Fig.
5A) from 12 h. Co-staining of kidney sections for p62 and AQP-2
confirmed the accumulation of p62 in collecting ducts (Fig. 5B). The low level of p62 in AQP-1
(additionally a marker of the thin limb of the loops of
Henle)-positive cells (Fig. 5C)
indicated that little p62 protein was accumulated in the thin limb
of the loops of Henle.
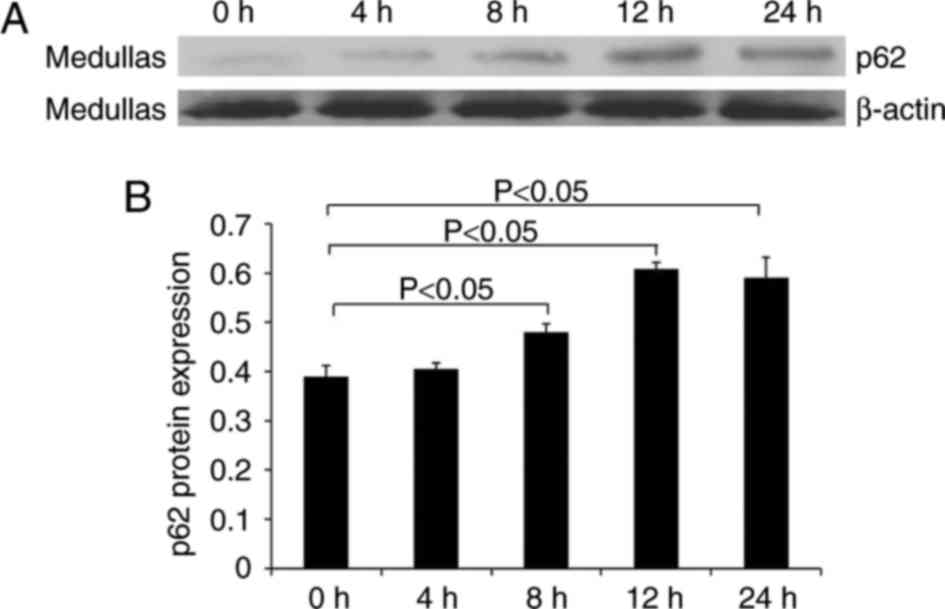
In addition, the protein level of p62 in medullary
tissue was examined using western blotting. Fig. 6 illustrates the increased level of
p62 in LPS-treated mice compared with control mice. The level of
p62 reached a maximum level at 12 h, which was maintained until 24
h.
Effects of p62 overexpression on
LPS-induced injury in the renal tubular epithelial cell line
HK-2
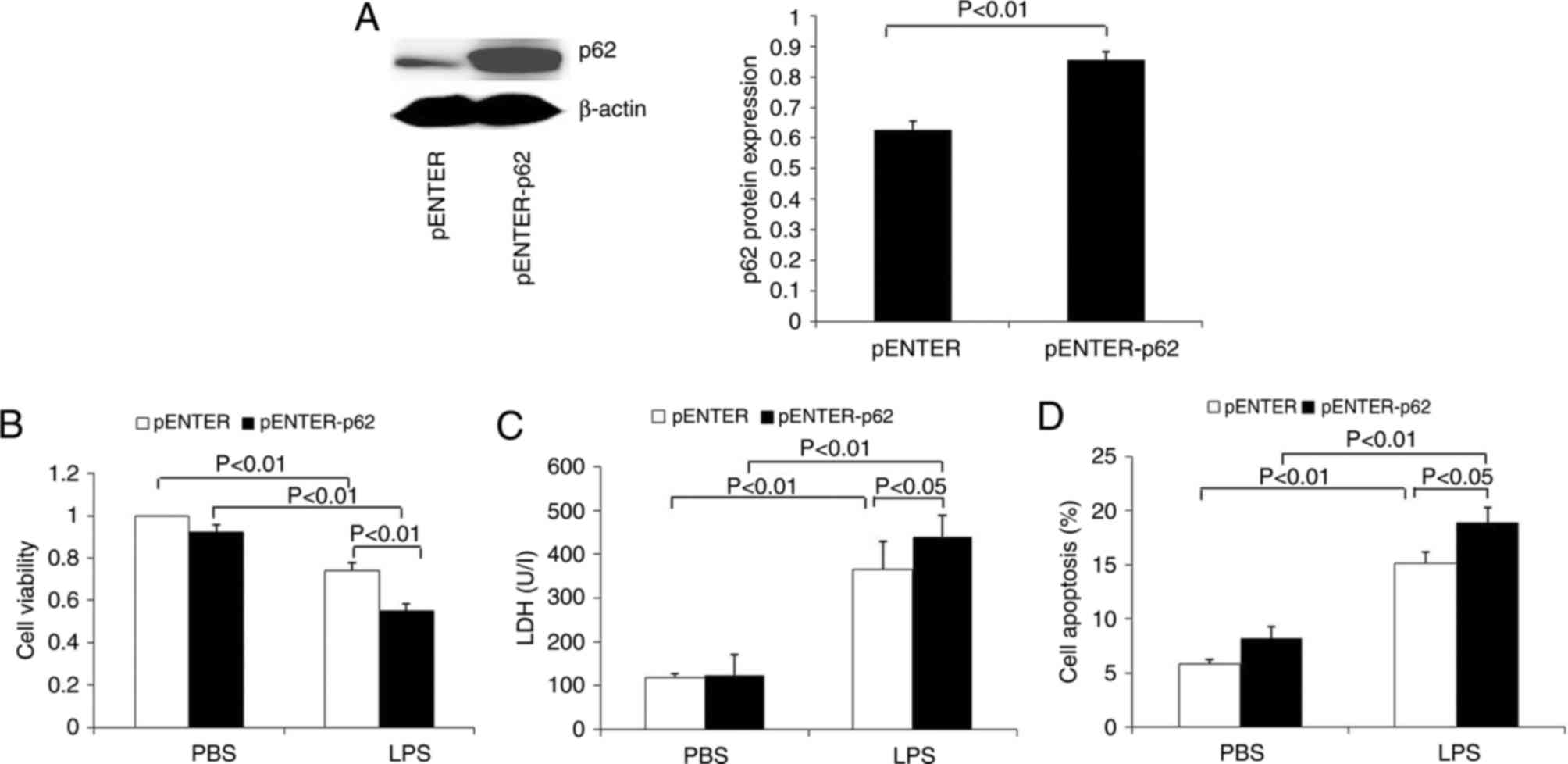
Western blotting results demonstrated that the
levels of p62 protein were significantly increased in HK-2 cells
transfected with p62 plasmid (Fig.
7A). In addition, these cells were treated with LPS to explore
the effects of p62 on LPS-induced viability, LDH level and
apoptosis. The results demonstrated that p62 overexpression
significantly decreased cell viability and increased LDH release
(Fig. 7B and C). Flow cytometry
analysis demonstrated that p62 overexpression significantly
increased the apoptosis of HK-2 cells (Fig. 7D).
Effects of interference with p62
expression on LPS-induced injury in the renal tubular epithelial
cell line HK-2
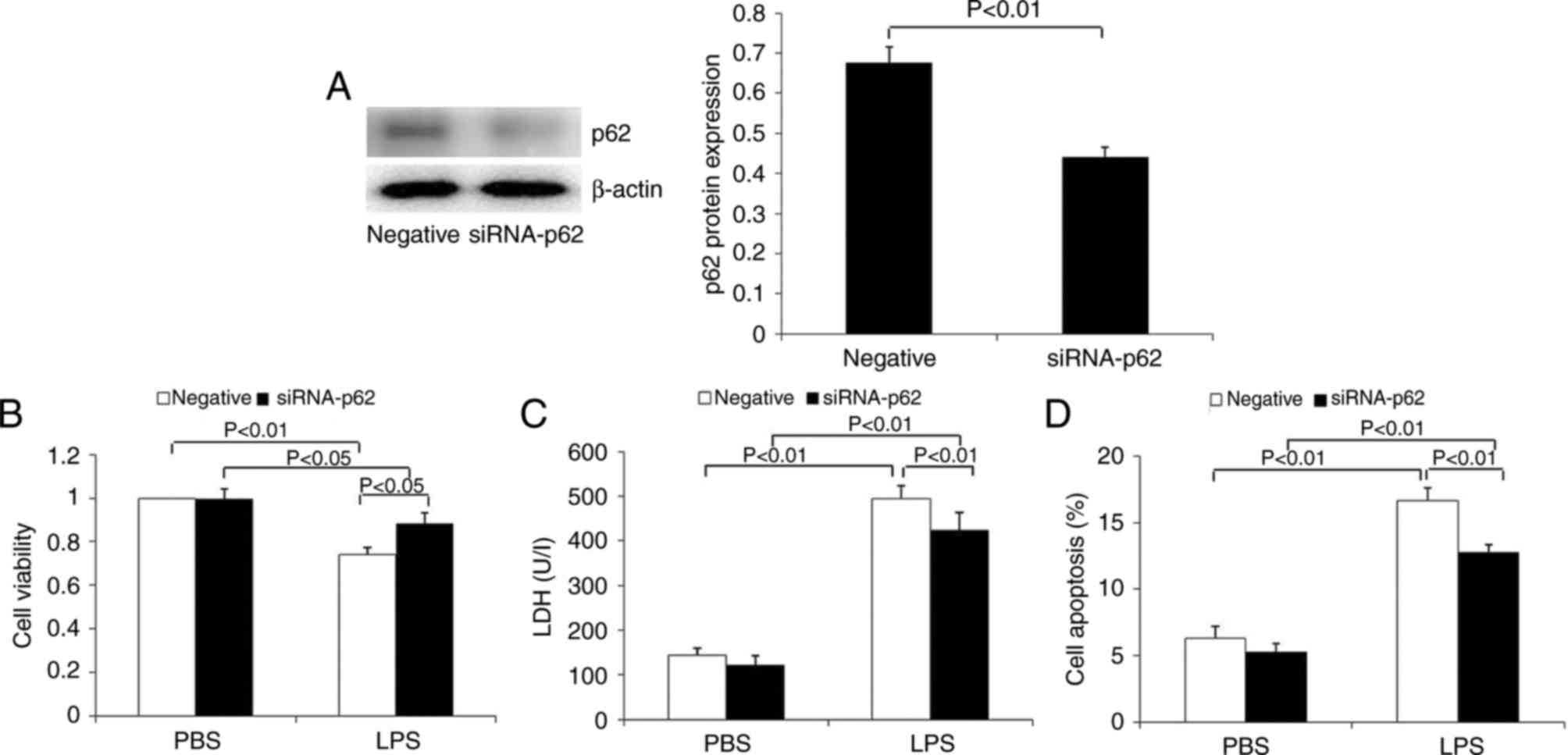
The expression of p62 was significantly inhibited in
HK-2 cells transfected with p62 siRNA at the protein level
(Fig. 8A). In contrast to the
overexpression of p62 in HK-2 cells, p62 downregulation
significantly increased cell viability, and decreased LDH release
(Fig. 8B and C). Flow cytometry
demonstrated that p62 downregulation significantly decreased the
apoptosis of HK-2 cells induced by LPS (Fig. 8D). These results demonstrated that
p62 is able to promote apoptosis and the release of the toxic
substance LDH, and to inhibit the proliferation of renal tubular
epithelial cells.
Discussion
The overall 72-h survival rate and the renal
functional parameters indicated that the AKI model was successfully
established. In the present study, the renal expression and
location of p62 was examined in a mouse model of endotoxemia
induced via injection of LPS. In mice treated with LPS, the protein
levels of p62 significantly decreased and subsequently increased.
Previous studies have demonstrated that p62 is one of the selective
mammalian autophagy cargo receptors (13). Under normal conditions, p62 is
incorporated into the autophagosome and degraded by autophagy
(25). Certain stimuli, including
hunger and hypoxia, may increase autophagy, which may induce p62
degradation, resulting in decreased intracellular levels of p62
(26). Therefore, the expression
of p62 may indirectly represent the level of autophagy. The
decreased expression of p62 protein and the increased expression of
LC3-II protein in the results of the present study indicated that
autophagy was induced in the kidneys of LPS-treated mice,
suggesting that autophagy may serve a role in the mechanism of
LPS-induced AKI. A significant decrease in the mRNA levels of p62
was additionally observed in the kidneys of mice treated with LPS.
The results of the present study demonstrated the inhibited
transcription and production of p62 in the kidney at the early
stages of endotoxemia, in addition to the degradation of p62, which
contributed to the reduction of p62 in LPS-induced AKI. In
addition, the expression level of p62, whether mRNA or protein, was
demonstrated to increase gradually and was increased compared with
the normal level at 24 h. It was suggested that p62 may serve as an
autophagy substrate and may serve a role in septic AKI. It was
reported that the accumulation of p62 may additionally activate
nuclear factor-κB, increase interleukin-1β production, and promote
the activation of caspase-1 and caspase-8, which aggravate
inflammation and apoptosis (27,28).
However, whether p62 may serve a role by regulating signaling
pathways in LPS-induced AKI remains unknown.
Immunohistochemistry indicated that p62 protein was
predominantly located in the cytoplasm of proximal tubules in the
renal cortex under normal conditions. Consistent with the
expression of p62 in the whole kidney demonstrated by RT-qPCR
analysis and western blotting, p62 was observed to be decreased in
the cytoplasm of proximal tubules, followed by an increase with a
longer treatment with LPS. A previous study demonstrated that the
LIR domain enables p62 to combine with LC3-II, and the UBA domain
enables p62 to bind to ubiquitin; consequently, ubiquitinated
proteins may be degraded by autophagy (8,29).
Therefore, the intracellular level of p62 is regulated by autophagy
through the interaction of LC3-II with p62. Previously, the
LPS-induced expression of LC3-II was detected in mouse kidneys
(30,31). Consistently, the results of the
present study demonstrated that LC3-II in the kidneys increased
during treatment with LPS, reaching a peak at 8–12 h; thereafter,
it decreased at 24 h. Despite the distinct time frame of the
expression of renal p62 and LC3-II, they exhibit the same cellular
localization (32). Following
treatment with LPS, the expression of LC3-II in the proximal
tubules of the renal cortex was significantly increased compared
with normal mice, indicating the formation of autophagosomes
(30). However, the expression of
LC3-II in the renal medulla was not notably increased (30). The results of the present study
demonstrated that autophagy was predominantly induced in the
proximal tubules of the renal cortex during LPS-induced AKI.
AQP-1 is a marker of proximal tubules.
Co-localization analysis of p62 and AQP-1 at proximal tubules using
immunohistochemistry demonstrated that p62 was localized in the
cytoplasm of renal tubular epithelial cells. Therefore, the roles
of p62 were examine using the renal tubular epithelial cell line
HK-2. The results of the present study demonstrated that the
overexpression of p62 aggravated LPS-induced injury to renal
tubular epithelial cells, however, interference with p62 expression
decreased the damage to HK-2 cells. As a multifunctional protein,
p62 may be upregulated via autophagy deficiency or inhibition
(10). The inhibition of autophagy
may upregulate the accumulation of p62 and enhance
caspase-8-dependent apoptotic pathways (33–35).
However, the role of p62 in LPS-induced AKI remains largely
unknown. The results of the present study demonstrated that the
overexpression of p62 promoted the apoptosis of renal tubular
epithelial cells. In combination with the results of the animal
experiments, it was indicated that the degradation of p62 by
activated autophagy at the early stages of endotoxemia contributed
to the inhibition of apoptosis; however, at the late stages of
endotoxemia, inhibition or dysfunction of autophagy resulted in p62
accumulation, which increased the apoptosis of renal tubular
epithelial cells and accelerated renal injury.
In the present study, it was unexpected that p62 was
observed to redistribute to the nucleus of the tubule cells in the
medulla from 8 h subsequent to treatment with LPS. p62 has
generally been considered to be a cytosolic protein, and little
attention has been paid to the possible nuclear roles of this
protein. In eukaryotic cells, molecular trafficking between the
nucleus and the cytoplasm is highly regulated via cellular
homeostasis and signaling (36).
However, such regulation may be disturbed under stress through the
disruption of nucleocytoplasmic transport pathways, resulting in
the nucleocytoplasmic redistribution of a number of functional
proteins (36). Consistent with
the results of the present study, Pankiv et al (37) demonstrated that p62 may be shuttled
between the cytoplasm and nucleus. p62 contains two important
domains, the nuclear localization signal and nuclear export signal
domains, which may facilitate the shuttling of p62 between the
cytoplasm and nucleus. Additionally, Pankiv et al (37) additionally discovered that p62 may
recruit nuclear polyubiquitinated proteins or protein aggregates to
promyelocytic leukemia bodies and may aid in their proteasomal
degradation in the nucleus, indicating the potential nuclear roles
of this protein. However, the function of p62 transported into the
nucleus during endotoxemia is unknown.
At present, the majority of studies have focused on
the association between cytoplasmic proteins and AKI. Nuclear
autophagy has rarely been reported. Whether the increased
expression of p62 in the nucleus of renal medullar cells during
LPS-induced AKI is associated with nuclear autophagy remains
unknown. In addition to mediating selective autophagy as an
autophagy adaptor protein, p62 may act as an important signaling
hub to control cell survival and apoptosis (38,39).
For example, p62 may promote apoptosis by mediating the aggregation
of caspase-8 (39). Whether the
enhanced p62 expression observed in the present study may promote
the apoptosis of medullar cells by activating an apoptotic pathway
remains unknown. Therefore, further studies are required to better
understand the role of p62 in LPS-induced AKI.
In conclusion, the mRNA and protein levels of p62
decreased in the kidney during early inflammation and subsequently
increased at 24 h following treatment with LPS.
Immunohistochemistry indicated that p62 protein was predominantly
expressed in the cytoplasm of renal tubules in control mice,
although it was redistributed to the medullas following LPS
injection. In vitro experiments demonstrated that p62
overexpression was able to aggravate LPS-induced injury in renal
tubular epithelial cells. The results of the present study
supported a potential role of p62 in the kidney during
LPS-stimulated endotoxemia via alterations in the expression level
and location of p62. Further studies are required to elucidate the
function of p62 in the cytoplasm and nucleus of renal tubular
epithelial cells during endotoxemia.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81571880, 81373147,
81671895, 30901555 and 30972870), the Natural Science Foundation of
Hunan Province (grant no. 2016JJ2157) and the Changzhi Medical
College Innovation Team Project (grant no. CX201501).
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
TL, JZ, YL and XX conceived and designed the
experiments. TL, JZ, SM and YX executed the experiments and
analyzed the data. TL wrote manuscript. All authors interpreted the
data, critically revised the manuscript for important intellectual
contents and approved the final version.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee of the Animal Experimental Institute of Central South
University, Changsha, China, and carried out in accordance with the
approved protocol.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mehta RL, Bouchard J, Soroko SB, Ikizler
TA, Paganini EP, Chertow GM and Himmelfarb J: Program to Improve
Care in Acute Renal Disease (PICARD) Study Group: Sepsis as a cause
and consequence of acute kidney injury: Program to improve care in
acute renal disease. Intensive Care Med. 37:241–248. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lopes JA, Jorge S, Resina C, Santos C,
Pereira A, Neves J, Antunes F and Prata MM: Acute kidney injury in
patients with sepsis: A contemporary analysis. Int J Infect Dis.
13:176–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cruz MG, Dantas JG, Levi TM, Mde Rocha S,
de Souza SP, Boa-Sorte N, de Moura CG and Cruz CM: Septic versus
non-septic acute kidney injury in critically ill patients:
Characteristics and clinical outcomes. Rev Bras Ter Intensiva.
26:384–391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jiang M, Liu K, Luo J and Dong Z:
Autophagy is a renoprotective mechanism during in vitro hypoxia and
in vivo ischemia-reperfusion injury. Am J Pathol. 176:1181–1192.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kimura T, Takabatake Y, Takahashi A,
Kaimori JY, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T,
Soga T, et al: Autophagy protects the proximal tubule from
degeneration and acute ischemic injury. J Am Soc Nephrol.
22:902–913. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leventhal JS, Ni J, Osmond M, Lee K,
Gusella GL, Salem F and Ross MJ: Autophagy limits endotoxemic acute
kidney injury and alters renal tubular epithelial cell cytokine
expression. PLoS One. 11:e01500012016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Komatsu M and Ichimura Y: Physiological
significance of selective degradation of P62 by autophagy. FEBS
Lett. 584:1374–1378. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shaid S, Brandts CH, Serve H and Dikic I:
Ubiquitination and selective autophagy. Cell Death Differ.
20:21–30. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Moscat J and Diaz-Meco MT: P62 at the
crossroads of autophagy, apoptosis, and cancer. Cell.
137:1001–1004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lippai M and Lőw P: The role of the
selective adaptor P62 and ubiquitin-like proteins in autophagy.
Biomed Res Int. 2014:8327042014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
DeVorkin L and Gorski SM: Monitoring
autophagic flux using Ref(2)P, the Drosophila P62 ortholog. Cold
Spring Harb Protoc. 2014:959–966. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin X, Li S, Zhao Y, Ma X, Zhang K, He X
and Wang Z: Interaction domains of P62: A bridge between P62 and
selective autophagy. DNA Cell Biol. 32:220–227. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rogov V, Dötsch V, Johansen T and Kirkin
V: Interactions between autophagy receptors and ubiquitin-like
proteins form the molecular basis for selective autophagy. Mol
Cell. 53:167–178. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Manley S, Williams JA and Ding WX: Role of
p62/SQSTM1 in liver physiology and pathogenesis. Exp Biol Med
(Maywood). 238:525–538. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Luo RZ, Yuan ZY, Li M, Xi SY, Fu J and He
J: Accumulation of P62 is associated with poor prognosis in
patients with triple-negative breast cancer. Onco Targets Ther.
6:883–888. 2013.PubMed/NCBI
|
|
18
|
Laurin N, Brown JP, Morissette J and
Raymond V: Recurrent mutation of the gene encoding sequestosome 1
(SQSTM1/p62) in paget disease of bone. Am J Hum Genet.
70:1582–1588. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang L, Li P, Fu S, Calay ES and
Hotamisligil GS: Defective hepatic autophagy in obesity promotes ER
stress and causes insulin resistance. Cell Metab. 11:467–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Codogno P and Meijer AJ: Autophagy: A
potential link between obesity and insulin resistance. Cell Metab.
11:449–451. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Y, Jia Z, Sun Y, Zhou L, Downton M,
Chen R, Zhang A and Yang T: Postnatal regulation of
15-hydroxyprostaglandin dehydrogenase in the rat kidney. Am J
Physiol Renal Physiol. 307:F388–F395. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li T, Liu Y, Zhao J, Miao S, Xu Y, Liu K,
Liu M, Wang G and Xiao X: Aggravation of acute kidney injury by
mPGES-2 down regulation is associated with autophagy inhibition and
enhanced apoptosis. Sci Rep. 7:102472017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kadowaki M and Karim MR: Cytosolic LC3
ratio as a quantitative index of macroautophagy. Methods Enzymol.
452:199–213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang P and Mizushima N: LC3- and
P62-based biochemical methods for the analysis of autophagy
progression in mammalian cells. Methods. 75:13–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choe JY, Jung HY, Park KY and Kim SK:
Enhanced P62 expression through impaired proteasomal degradation is
involved in caspase-1 activation in monosodium urate
crystal-induced interleukin-1b expression. Rheumatology (Oxford).
53:1043–1053. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kovacs JR, Li C, Yang Q, Li G, Garcia IG,
Ju S, Roodman DG, Windle JJ, Zhang X and Lu B: Autophagy promotes
T-cell survival through degradation of proteins of the cell death
machinery. Cell Death Differ. 19:144–152. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Komatsu M, Kageyama S and Ichimura Y:
P62/SQSTM1/A170: Physiology and pathology. Pharmacol Res.
66:457–462. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wu Y, Zhang Y, Wang L, Diao Z and Liu W:
The role of autophagy in kidney inflammatory injury via the NF-κB
route induced by LPS. Int J Med Sci. 12:655–667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mei S, Livingston M, Hao J, Li L, Mei C
and Dong Z: Autophagy is activated to protect against endotoxic
acute kidney injury. Sci Rep. 6:221712016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sansanwal P and Sarwal MM: P62/SQSTM1
prominently accumulates in renal proximal tubules in nephropathic
cystinosis. Pediatr Nephrol. 27:2137–2144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han J, Hou W, Goldstein LA, Lu C, Stolz
DB, Yin XM and Rabinowich H: Involvement of protective autophagy in
TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem.
283:19665–19677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Huang S and Sinicrope FA:
Celecoxib-induced apoptosis is enhanced by ABT-737 and by
inhibition of autophagy in human colorectal cancer cells.
Autophagy. 6:256–269. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Huang S, Okamoto K, Yu C and Sinicrope FA:
P62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8
aggregation/activation on the autophagosome. J Biol Chem.
288:33654–33666. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kose S and Imamoto N: Nucleocytoplasmic
transport under stress conditions and its role in HSP70 chaperone
systems. Biochim Biophys Acta. 1840:2953–2960. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pankiv S, Lamark T, Bruun JA, Øvervatn A,
Bjørkøy G and Johansen T: Nucleocytoplasmic shuttling of p62/SQSTM1
and its role in recruitment of nuclear polyubiquitinated proteins
to promyelocytic leukemia bodies. J Biol Chem. 285:5941–5953. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sanz L, Diaz-Meco MT, Nakano H and Moscat
J: The atypical PKC-interacting protein P62 channels NF-kappaB
activation by the IL-1-TRAF6 pathway. EMBO J. 19:1576–1586. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC,
Lill JR and Ashkenazi A: Cullin3-based polyubiquitination and
P62-dependent aggregation of caspase-8 mediate extrinsic apoptosis
signaling. Cell. 137:721–735. 2009. View Article : Google Scholar : PubMed/NCBI
|