Introduction
Acute lung injury (ALI) is the early stage of acute
respiratory distress syndrome (ARDS), which is a severe
inflammatory injury to the lung (1). ALI/ARDS is characterized by the
accumulation of protein-rich edema fluid in the alveolar
compartments of the lung (2). The
mechanism of lung injury varies with the cause; for example,
increased vascular permeability, overproduction of cytokines,
leukocyte recruitment and surfactant dysfunction, leading to
interstitial and alveolar pulmonary edema, alveolar collapse and
hypoxemia. Damage to epithelial and endothelial cells serves an
important role in the development of ALI/ARDS (3). The clinical manifestation is
characterized by regeneration and healing via resection or repair,
frequently leading to persistent intra-alveolar and interstitial
fibrosis (4). Survivors of ARDS
are often exhibit chronic pulmonary fibrosis, reduced pulmonary
function and diminished health-related quality of life (5). Indeed, chronic inflammation and
tissue fibrosis are principal causes of morbidity and mortality in
the chronic stage of ARDS, which is responsible for ~30% of
hospital mortalities following lung resection (6).
Pulmonary fibrosis is a devastating lung problem
characterized by diffuse interstitial inflammation and exaggerated
collagen accumulation, which in turn leads to the destruction of
alveolar structures and remodeling (7). Extracellular matrix (ECM)
accumulation includes the accumulation of collagen-IV (Col-IV) and
fibronectin (FN). The pathogenesis of pulmonary fibrosis in acute
lung injury, including ARDS, is poorly understood at present;
therefore, there is an urgent need to expand the present
understanding of the pathogenesis of ALI.
Interleukin (IL)-6 is a pleiotropic cytokine
implicated in the pathogenesis of numerous infectious diseases and
a number of immune-mediated disorders (8,9).
IL-6 binds to an IL-6 receptor, and associates with a dimer of the
ubiquitously-expressed gp130 receptor subunit, which initiates
intracellular signaling (9).
Certain studies have indicated that IL-6 is involved in pulmonary
fibrosis in vivo (10–12).
Prohibitin (PHB) is a highly-conserved protein that
has multiple functions in the cell (10–12).
The PHB1 gene is located on chromosome 17q21 and encodes a 30 kDa
protein that associates with prohibitin 2 (PHB2) forming 16–20-mer
ring-like structures with chaperone or scaffolding activities
within the mitochondria (13).
These structures share >50% identical amino acid residues and
are two members of the PHB family (14). PHB1, along with the highly
homologous PHB2, is ubiquitously expressed in an array of
eukaryotic organisms (15). PHB1
is involved in multiple cellular functions and the subcellular
localization of prohibitin may determine its function (16). PHB in membranes regulates the
cellular signaling of membrane transport, nuclear PHB controls
transcription activation and the cell cycle, and the mitochondrial
PHB complex stabilizes the mitochondrial genome and modulates
mitochondrial dynamics, mitochondrial morphology, mitochondrial
biogenesis and the mitochondrial intrinsic apoptotic pathway
(16). Alterations in PHB1 levels
have been associated with pathologies, including inflammation,
obesity, autoimmunity or cancer (17–19).
PHB1, which may be a potential therapeutic target for the treatment
of a variety of diseases, has been reported to prevent
inflammation-associated oxidative stress and injury due to its
antioxidant properties (20,21).
Previously, certain studies identified that PHB was
associated with pulmonary disease. Soulitzis et al (22) reported that, non-chronic
obstructive pulmonary disease (COPD) smokers exhibited lower PHB1
mRNA expression levels when compared with non-smokers, while PHB1
expression was even further decreased in patients with COPD. By
contrast, PHB2 levels were similar among the three study groups.
Agrawal et al (23)
demonstrated that in a mouse model of allergic airway inflammation,
vitamin D deficiency decreased the expression of vitamin D receptor
(VDR) and PHB. Supplementation with vitamin D may increase the
expression of VDR and PHB, which may be responsible for reducing
allergic airway inflammation. Meanwhile, certain studies identified
that PHB was associated with inflammation and fibrosis. PHB
expression has been proven to be negatively correlated with
hepatic, intestinal and renal interstitial fibrosis (24–26).
Lipopolysaccharide (LPS) is widely accepted in the
establishment of ALI models and has the ability to induce the
release of numerous inflammatory mediators, including tumor
necrosis factor (TNF)-α, IL-1β, IL-6, NO and superoxide anions
(27). Intratracheal
administration of LPS increases cytokine levels in bronchial
alveolar lavage fluids, whereas LPS challenge in lung endothelial
or bronchial epithelial cells enhances barrier permeability and
IL-6 release (28).
At present, to the best of our knowledge, there have
been no studies confirming the roles served by PHB in ALI. Based on
the LPS-induced cell model of ALI, the present study explored the
expression of PHB and ECM in the process of ALI. The results of the
present study revealed that PHB and ECM increased in an LPS-induced
acute injury cell model.
Materials and methods
Cell culture and treatments
The murine alveolar epithelial cell line MLE-12 was
purchased from the American Type Culture Collections (ATCC;
Manassas, VA, USA) and cultured in Dulbecco's modified Eagle's
medium/F12 supplemented with 10% fetal bovine serum (both from
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA), 0.1 mg/ml
streptomycin and 100 U/ml penicillin (Gibco; Thermo Fisher
Scientific, Inc.) and maintained in a 5% CO2 and 95% air
atmosphere at 37°C. The culture medium was replaced with fresh
medium every 2 days. Cells were plated at a density of
4×105 cells/well in 6-well plates overnight. Once the
cells reached 80% confluence, they were divided into two groups and
treated as follows: The control group was treated with sterile PBS
and the treatment group was stimulated with 500 ng/ml LPS
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 12 h
(determined by our pre-experiment) (Zang et al, unpublished
study). Total proteins and mRNA were extracted from cells following
the 12 h incubation.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
To detect the mRNA expression levels of IL-6, PHB1
and PHB2, total RNA was extracted from each group using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions, and the quality and concentration
were assessed. RNA (1 µg) was reverse-transcribed using the
ReverTra Ace qPCR RT kit (cat. no. FSQ-101; Toyobo Co., Ltd.,
Osaka, Japan), according to the manufacturer's instructions. qPCR
was performed with SYBR Green Real-time PCR Master Mix (Takara Bio,
Inc., Otsu, Japan) on an Applied Biosystems 7500 Real-time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.). The
qPCR thermal cycling was performed as follows: Initial incubation
for 10 min at 95°C, followed by denaturing for 40 cycles at 95°C
for 15 sec and annealing for 60 sec at 60°C, and elongation at 60°C
for 15 sec. All reactions were performed in triplicate, with three
samples from different groups. The quantification of target mRNAs
was normalized to β-actin, an internal control gene. The average
quantification cycle (Cq; the cycles of template amplification to
the threshold) was worked out as the value of each sample. The data
for fold change was analyzed using the 2−ΔΔCq method
(29). The primer sequences were
as follows: β-actin forward, 5′-ATGGAGGGGAATACAGCCC-3′ and reverse,
5′-TTCTTTGCAGCTCCTTCGTT-3′; PHB1 forward,
5′-GGGAAGGAGTTCACAGAGGCAGTA-3′ and reverse,
5′-CACCCTCAGCAGAGATGATGGC-3′; PHB2 forward,
5′-CAAGAACCCCACCACCAGAGAA-3′ and reverse,
5′-TCCAAGAGGGCAGATACAGAAAAG-3′; IL-6 forward,
5′-ACCAGAGGAAATTTTCAATAGGC-3′ and reverse,
5′-TGATGCACTTGCAGAAAACA-3′.
Western blot analysis
Protein was extracted using 1X SDS sample buffer
(Sigma-Aldrich; Merck KGaA) with a cocktail of protease and
phosphatase inhibitors. Following the measurement of protein
concentration using a bicinchoninic acid kit (Beijing Dingguo
Changsheng Biotechnology Co., Ltd., Beijing, China) with a
microplate spectrophotometer (Thermo Fisher Scientific, Inc.), the
protein samples were denatured by heating at 95°C for 10 min, and
30 µg/lane protein was separated by 9–15% SDS-PAGE (ranging
according to the molecular weight of the target protein) and then
transferred onto nitrocellulose membranes (EMD Millipore,
Billerica, MA, USA). Membranes were blocked with 5% non-fat milk in
TBS with 0.1% Tween-20 (TBST; Sigma-Aldrich; Merck KGaA) at 4°C for
60 min. Following rinsing with TBST, the membranes were probed with
primary antibodies against PHB1 (cat. no. ab75771; 1:1,000), PHB2
(cat. no. ab154992; 1:1,000) (both from Abcam, Cambridge, UK), IL-6
(cat. no. 12,912; 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA), Col-IV (cat. no. ab6586; 1:5,000) and FN (cat.
no. ab45688; 1:5,000) (both from Abcam) at 4°C overnight, followed
by incubation with IRDye 800CW goat anti-rabbit secondary antibody
(cat. no. 042-07-15-06; 1:10,000; LI-COR Biosciences, Lincoln, NE,
USA) at 4°C for 30 min protected from light. β-actin (cat. no.
4970; 1:1,000; Cell Signaling Technology, Inc.) was used as an
internal loading control. Immunoblot detection was performed with
the Odyssey Imaging System (LI-COR Biosciences) and densitometric
analysis was performed using Adobe Photoshop (version 13.0; Adobe
Systems Inc., San Jose, CA, USA).
Statistical analysis
Data are expressed as mean ± standard deviation of
>3 replicates. All statistical analyses were performed using
SPSS version 19.0 statistical software (IBM Corp., Armonk, NY,
USA). Comparisons between two groups were performed using the
Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
mRNA and IL-6 protein expression are
upregulated following stimulation with LPS
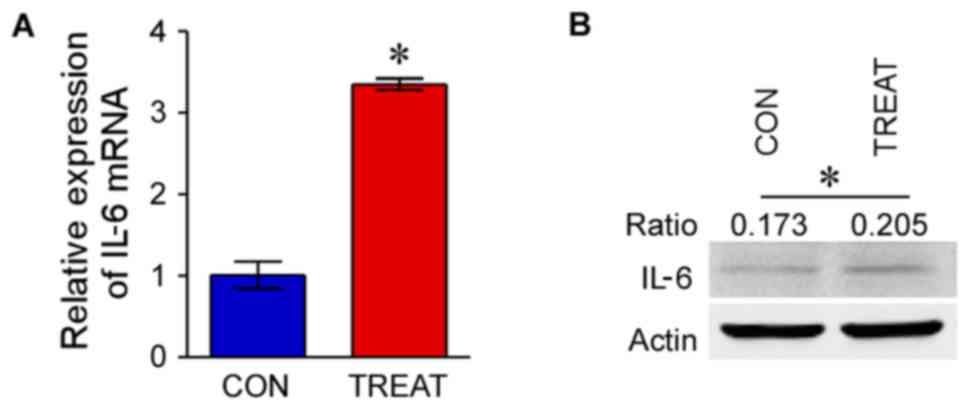
The mRNA expression of IL-6 in MLE-12 following
treatment with LPS (treatment group) was significantly upregulated
compared with the control group (P<0.05; Fig. 1A). Western blot analysis indicated
that the protein expression of IL-6 in the treatment group was
markedly increased compared with the control group (P<0.05;
Fig. 1B).
ECM components Col-IV and FN
accumulate in murine alveolar epithelial cells following acute
injury
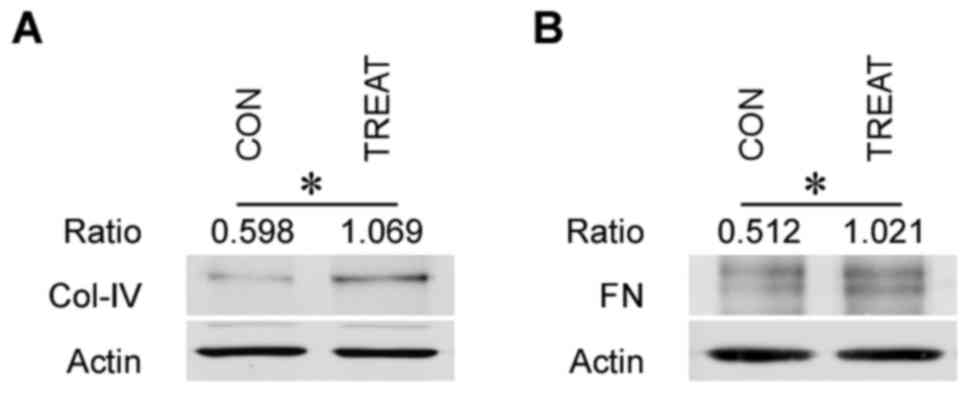
The protein expression levels of ECM components
(Col-IV and FN) were detected via western blotting. ECM components,
including Col-IV and FN, in the treatment group were markedly
increased when compared with the control group (P<0.05; Fig. 2). This indicated that ECM
components were accumulated in alveolar epithelial cells following
acute inflammatory injury.
mRNA expression levels of PHB1 and
PHB2, and western blot analysis of PHB1 and PHB2
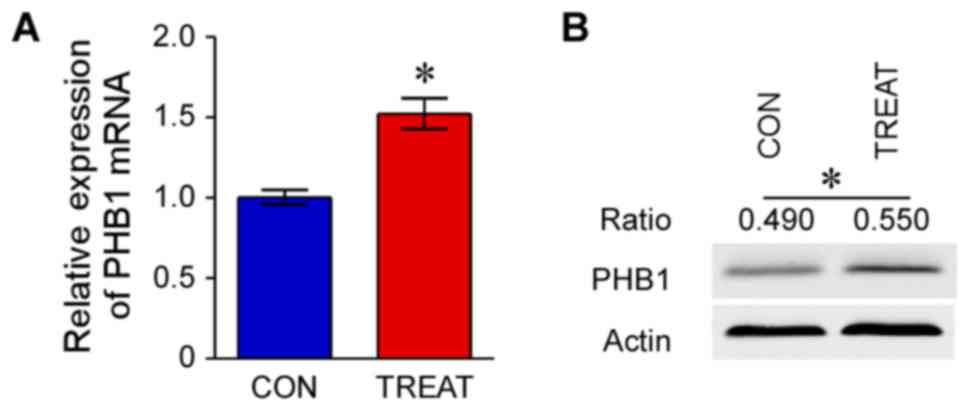
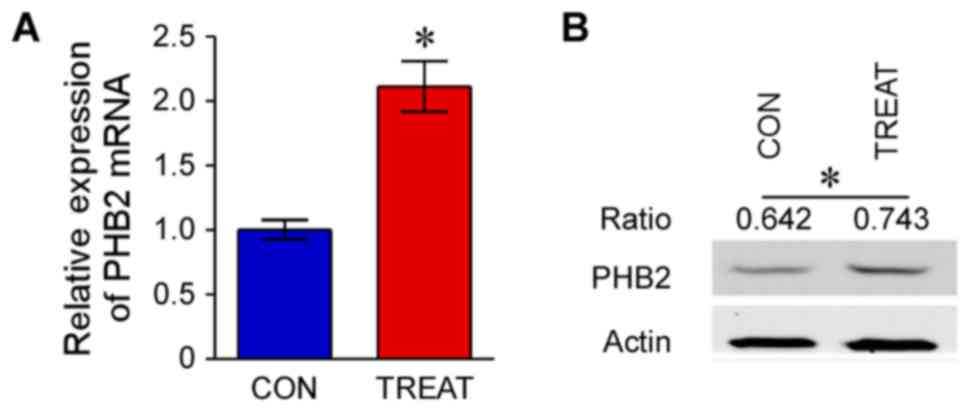
The mRNA expression levels of PHB1 and PHB2 in the
treatment group were significantly upregulated compared with the
control group (P<0.05; Figs. 3
and 4). Furthermore, western blot
analysis indicated that protein expression levels of PHB1 and PHB2
in the treatment group were increased compared with the control
group (P<0.05; Figs. 3 and
4).
Discussion
Pulmonary fibrosis is a chronic, progressive and
irreversible process. Severe inflammatory injury in the lungs
frequently leads to persistent fibrosis (4). Patients with ARDS frequently exhibit
chronic pulmonary fibrosis. The presence of fibrosis is
significantly correlated with the duration of ARDS. Additionally,
fibrosis may be an important reason for the poor prognosis of
patients with ARDS, since it leads to decreases in lung compliance
and oxygenation (30,31). Pulmonary fibrosis is characterized
by the activation of myofibroblasts, which may originate from
epithelial cells via the onset of epithelial to mesenchymal
transition (EMT). The persistence of the myofibroblasts beyond a
period of normal repair has been associated with excessive ECM
accumulation, finally resulting in pulmonary fibrosis (32,33).
A previous study demonstrated that LPS induces EMT and pulmonary
fibrosis in vivo (34).
LPS, a major component of Gram-negative bacteria, is
one of the principal pro-inflammatory reaction factors in infection
diseases, leading to inflammatory overreactions in vitro and
in vivo. It has been reported to be one of the primary
factors that induces the inflammatory response, and the LPS-induced
murine model of lung injury has been widely used to investigate the
mechanisms of ALI (28,35). In the model of ALI, LPS
significantly increased the production of inflammatory cytokines
including TNF-α, IL-1β and IL-6, which has been demonstrated to be
involved in the development of ALI (36). In the present study, a similar ALI
cell model was created via stimulation with 500 ng/ml LPS,
according to the protocol of Zhu et al (37). IL-6 was considered to be a marker
of the LPS-induced inflammatory response (38). According to the data from the
present study, the mRNA and protein expression levels of IL-6 were
notably increased via LPS stimulation, compared with the control
group. This finding was consistent with previous studies and
indicated that the model of LPS-induced acute injury was
successfully established.
A previous study (39) demonstrated that IL-6 response
element (IL-6RE) is the essential transcription regulatory site for
maximal basal and IL-6-induced PHB promoter activity. Signal
transducer and activator of transcription 3 mediates basal and
IL-6-induced PHB transcription and binds to IL-6RE in the PHB
promoter. It may be hypothesized that IL-6 increases PHB protein
and mRNA expression by activating the PHB promoter in vitro
and in vivo (39). In the
present study, the mRNA and protein expression levels of PHB1 and
PHB2 in alveolar epithelial cells stimulated with LPS were
increased compared with those in the control group. The present
study revealed that increased IL-6 was associated with the
upregulation of PHB in murine alveolar epithelial cells.
Certain studies have identified that PHB is
associated with inflammation and fibrosis in multiple diseases. Ko
et al (24) suggested that
a liver-specific deletion of PHB1 results in marked liver fibrosis.
Yuan et al (40) reported
that treatment with IL-10 is associated with increased PHB, and may
decrease inflammation and fibrosis in an animal model of Crohn's
disease; PHB may be a potential target for intestinal fibrosis
associated with inflammatory bowel disease (IBD). Zhou et al
(38) demonstrated that lower
expression of PHB was associated with increased renal interstitial
fibrosis and ECM accumulation. Previous studies (20,21)
suggested that PHB serves an important role in combating oxidative
stress, by interacting with the NADH dehydrogenase subunits of
mitochondrial respiratory complex I. Following the above studies,
it was hypothesized that, in the LPS-induced ALI cell model, PHB
may alleviate the process of pulmonary fibrosis due to its role in
antioxidant stress.
In the present ALI model of alveolar epithelial
cells, ECM and PHB were upregulated simultaneously. This appears to
contradict the effect of PHB identified in previous reports on
pulmonary disease and fibrosis, where PHB1 was proven to be a
protective factor in patients with COPD (22) and the deletion of PHB1 was
demonstrated to exacerbate liver fibrosis (24). The present study suggested that
this discrepancy may be attributed to the protective effect of PHB,
which may not reverse the process of LPS-induced fibrosis.
In a previous study of IBD, decreased expression of
prohibitin was associated with intestinal fibrosis progression, and
treatment with IL-10 was associated with increased prohibitin,
thereby ameliorating intestinal fibrosis (40). There has been no study, to the best
of our knowledge, which has investigated the potential association
of PHB with ECM accumulation in murine alveolar epithelial cells
following treatment with LPS. Following previous studies, the
present study hypothesized that, in an LPS-induced alveolar
epithelial cell injury model, upregulation of PHB expression may
effectively alleviate pulmonary fibrosis and become a novel
therapeutic target.
Although the mechanism underlying PHB and ECM
accumulation in pulmonary fibrosis remains to be elucidated, PHB
and ECM components were upregulated in the ALI cell model in the
present study. The possible signaling pathway merits investigation
in further research into the process of pulmonary fibrosis, which
includes pulmonary inflammation, apoptosis and fibrosis.
In conclusion, the present study identified that the
PHB expression level increased and ECM components accumulated in
murine alveolar epithelial cells with LPS-induced acute injury, and
further investigations may be performed to verify the detailed
mechanism.
Acknowledgements
The present study was supported by the Natural
Science Foundation of China under grant no. 81400719.
References
|
1
|
Luh SP and Chiang CH: Acute lung
injury/acute respiratory distress syndrome (ALI/ARDS): The
mechanism, present strategies and future perspectives of therapies.
J Zhejiang Univ Sci B. 8:60–69. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tomashefski JF Jr: Pulmonary pathology of
acute respiratory distress syndrome. Clin Chest Med. 21:435–466.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Orme JJ, Romney JS, Hopkins RO, Pope D,
Chan KJ, Thomsen G, Crapo RO and Weaver LK: Pulmonary function and
health-related quality of life in survivors of acute respiratory
distress syndrome. Am J Respir Crit Care Med. 167:690–694. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chida M, Ono S, Hoshikawa Y and Kondo T:
Subclinical idiopathic pulmonary fibrosis is also a risk factor of
postoperative acute respiratory distress syndrome following
thoracic surgery. Eur J Cardiothorac Surg. 34:878–881. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukuda Y, Ishizaki M, Masuda Y, Kimura G,
Kawanami O and Masugi Y: The role of intraalveolar fibrosis in the
process of pulmonary structural remodeling in patients with diffuse
alveolar damage. Am J Pathol. 126:171–182. 1987.PubMed/NCBI
|
|
8
|
Lin P: Targeting interleukin-6 for
noninfectious uveitis. Clin Ophthalmol. 9:1697–1702. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rose-John S: The soluble interleukin-6
receptor and related proteins. Best Pract Res Clin Endocrinol
Metab. 29:787–797. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
De Lauretis A, Sestini P, Pantelidis P,
Hoyles R, Hansell DM, Goh NS, Zappala CJ, Visca D, Maher TM, Denton
CP, et al: Serum interleukin 6 is predictive of early functional
decline and mortality in interstitial lung disease associated with
systemic sclerosis. J Rheumatol. 40:435–446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
O'Donoghue RJ, Knight DA, Richards CD,
Prêle CM, Lau HL, Jarnicki AG, Jones J, Bozinovski S, Vlahos R,
Thiem S, et al: Genetic partitioning of interleukin-6 signalling in
mice dissociates Stat3 from Smad3-mediated lung fibrosis. EMBO Mol
Med. 4:939–951. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshizaki A, Yanaba K, Ogawa A, Asano Y,
Kadono T and Sato S: Immunization with DNA topoisomerase I and
Freund's complete adjuvant induces skin and lung fibrosis and
autoimmunity via interleukin-6 signaling. Arthritis Rheum.
63:3575–3585. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Snedden WA and Fromm H: Characterization
of the plant homologue of prohibitin, a gene associated with
antiproliferative activity in mammalian cells. Plant Mol Biol.
33:753–756. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Merkwirth C and Langer T: Prohibitin
function within mitochondria: Essential roles for cell
proliferation and cristae morphogenesis. Biochim Biophys Acta.
1793:27–32. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koushyar S, Jiang WG and Dart DA:
Unveiling the potential of prohibitin in cancer. Cancer Lett.
369:316–322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peng YT, Chen P, Ouyang RY and Song L:
Multifaceted role of prohibitin in cell survival and apoptosis.
Apoptosis. 20:1135–1149. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sharma A and Qadri A: Vi polysaccharide of
Salmonella typhi targets the prohibitin family of molecules
in intestinal epithelial cells and suppresses early inflammatory
responses. Proc Natl Acad Sci USA. 101:17492–17497. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Theiss AL, Idell RD, Srinivasan S,
Klapproth JM, Jones DP, Merlin D and Sitaraman SV: Prohibitin
protects against oxidative stress in intestinal epithelial cells.
FASEB J. 21:197–206. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gamble SC, Chotai D, Odontiadis M, Dart
DA, Brooke GN, Powell SM, Reebye V, Varela-Carver A, Kawano Y,
Waxman J and Bevan CL: Prohibitin, a protein downregulated by
androgens, represses androgen receptor activity. Oncogene.
26:1757–1768. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Theiss AL and Sitaraman SV: The role and
therapeutic potential of prohibitin in disease. Biochim Biophys
Acta. 1813:1137–1143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Theiss AL, Vijay-Kumar M, Obertone TS,
Jones DP, Hansen JM, Gewirtz AT, Merlin D and Sitaraman SV:
Prohibitin is a novel regulator of antioxidant response that
attenuates colonic inflammation in mice. Gastroenterology.
137(199–208): 208.e1–6. 2009.
|
|
22
|
Soulitzis N, Neofytou E, Psarrou M,
Anagnostis A, Tavernarakis N, Siafakas N and Tzortzaki EG:
Downregulation of lung mitochondrial prohibitin in COPD. Respir
Med. 106:954–961. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Agrawal T, Gupta GK and Agrawal DK:
Vitamin D deficiency decreases the expression of VDR and prohibitin
in the lungs of mice with allergic airway inflammation. Exp Mol
Pathol. 93:74–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ko KS, Tomasi ML, Iglesias-Ara A, French
BA, French SW, Ramani K, Lozano JJ, Oh P, He L, Stiles BL, et al:
Liver-specific deletion of prohibitin 1 results in spontaneous
liver injury, fibrosis and hepatocellular carcinoma in mice.
Hepatology. 52:2096–2108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou TB, Qin YH, Lei FY, Huang WF and
Drummen GP: Prohibitin attenuates oxidative stress and
extracellular matrix accumulation in renal interstitial fibrosis
disease. PLoS One. 8:e771872013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sanchez-Quiles V, Segura V, Bigaud E, He
B, O'Malley BW, Santamaría E, Prieto J and Corrales FJ:
Prohibitin-1 deficiency promotes inflammation and increases
sensitivity to liver injury. J Proteomics. 75:5783–5792. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hu X, Tian Y, Qu S, Cao Y, Li S, Zhang W,
Zhang Z, Zhang N and Fu Y: Protective effect of TM6 on LPS-induced
acute lung injury in mice. Sci Rep. 7:5722017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao J, He D, Su Y, Berdyshev E, Chun J,
Natarajan V and Zhao Y: Lysophosphatidic acid receptor 1 modulates
lipopolysaccharide-induced inflammation in alveolar epithelial
cells and murine lungs. Am J Physiol Lung Cell Mol Physiol.
301:L547–L556. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C (T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rocco PR, Dos SC and Pelosi P: Lung
parenchyma remodeling in acute respiratory distress syndrome.
Minerva Anestesiol. 75:730–740. 2009.PubMed/NCBI
|
|
31
|
Burnham EL, Janssen WJ, Riches DW, Moss M
and Downey GP: The fibroproliferative response in acute respiratory
distress syndrome: Mechanisms and clinical significance. Eur Respir
J. 43:276–285. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cottin V: Interstitial lung disease. Eur
Respir Rev. 22:26–32. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chapman HA: Epithelial-mesenchymal
interactions in pulmonary fibrosis. Annu Rev Physiol. 73:413–435.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dong WW, Zhang YQ, Zhu XY, Mao YF, Sun XJ,
Liu YJ and Jiang L: Protective effects of hydrogen-rich saline
against lipopolysaccharide-induced alveolar
epithelial-to-mesenchymal transition and pulmonary fibrosis. Med
Sci Monit. 23:2357–2364. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
He D, Su Y, Usatyuk PV, Spannhake EW,
Kogut P, Solway J, Natarajan V and Zhao Y: Lysophosphatidic acid
enhances pulmonary epithelial barrier integrity and protects
endotoxin-induced epithelial barrier disruption and lung injury. J
Biol Chem. 284:24123–24132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhao D, Ding R, Mao Y, Wang L, Zhang Z and
Ma X: Heparin rescues sepsis-associated acute lung injury and
lethality through the suppression of inflammatory responses.
Inflammation. 35:1825–1832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhu T, Wang DX, Zhang W, Liao XQ, Guan X,
Bo H, Sun JY, Huang NW, He J, Zhang YK, et al: Andrographolide
protects against LPS-induced acute lung injury by inactivation of
NF-kappaB. PLoS One. 8:e564072013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou TB, Qin YH, Li ZY, Xu HL, Zhao YJ and
Lei FY: All-trans retinoic acid treatment is associated with
prohibitin expression in renal interstitial fibrosis rats. Int J
Mol Sci. 13:2769–2782. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Theiss AL, Obertone TS, Merlin D and
Sitaraman SV: Interleukin-6 transcriptionally regulates prohibitin
expression in intestinal epithelial cells. J Biol Chem.
282:12804–12812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yuan C, Chen WX, Zhu JS, Chen NW, Lu YM,
Ou YX and Chen HQ: IL-10 treatment is associated with prohibitin
expression in the Crohn's disease intestinal fibrosis mouse model.
Mediators Inflamm. 2013:6171452013. View Article : Google Scholar : PubMed/NCBI
|