Introduction
Acute lymphoblastic leukaemia (ALL) is the most
common malignancy in children; the 5-year disease-free survival
probability of children with ALL has been improved to approximately
80%. While ALL is less prevalent in adults, the long-term survival
rate is only 35–50% even with the adoption of allogeneic
haematopoietic stem cell transplantation (allo-HSCT) treatment
(1–3). Recently, chimeric antigen receptor
(CAR) T-cell therapy was induced to relapsed and refractory B-ALL
patients, resulting in remission rates of 67–90% (4). However, the incidence of relapse
after CAR T-cell treatment exceeds 40% (5). Thus, novel strategies and drugs for
ALL treatment need further exploration.
c-Myc, an oncogene that encodes a 64–67 kDa
transcription factor, has been known for a few decades. c-Myc plays
an important role in cell-fate decisions, including proliferation,
differentiation and apoptosis. The abnormal expression of the c-Myc
gene is critically involved in tumourigenesis (6,7).
c-Myc expression increases in primary T-ALL and B-ALL cells and
indicates a poor prognosis (8).
Interestingly, c-Myc inhibition by small hairpin RNA or BET
bromodomain have been shown to prevent leukaemia initiation in mice
by eliminating leukaemia-initiating cell (LIC) activity and
suppressing the growth of relapsed of paediatric T-ALL cells
(9). Thus, c-Myc inhibition
combination with other traditional chemotherapy agents could more
effectively eliminate ALL cells.
Glucocorticoids (GCs) can induce G1 cell cycle
arrest and the apoptosis of lymphoid cells; GCs are the preferred
drugs in the traditional regimens of ALL patient treatments, such
as VP (Vincristine, VCR + Prednison, P), VDP (Vincristine, VCR +
Daunorubicin, DNR + Prednison, P), and VDLP (Vincristine, VCR +
Daunorubicin, DNR + L-asparaginase, L-ASP + Prednison, P) (10–12).
The direct binding of GCs to cytosolic glucocorticoid receptors
(GRs) can induce the release of the latter from its protein complex
and subsequent dimerization and translocation to the nucleus. The
dimerization of GRs by binding to GC response elements regulates a
series of gene expression inducing apoptosis and cycle arrest
(13). Among these genes, c-Myc
inhibition plays a critical role in the apoptosis and cell cycle
arrest of ALL cells induced by GCs (14,15).
Thus, we speculated that the combination of GCs with c-Myc
inhibitors could synergistically kill ALL cells by down-regulating
further c-Myc expression.
In this study, we investigated the inhibition of
cell viability and the induction of cell cycle and apoptosis in ALL
cell lines treated with dexamethasone (DXM) alone or in combination
with c-Myc inhibitor 10058-F4. Our results for the first time
demonstrated that 10058-F4 increased the growth inhibition of ALL
cells induced with DXM by cell cycle arrest and the promotion of
apoptosis.
Materials and methods
Cell lines and reagents
NALM-6 (a B-ALL cell line) and CEM cells (a T-ALL
cell line) were purchased from the American Type Culture Collection
(Manassas, VA, USA). These cells were maintained in RPMI-1640
culture medium (GE Healthcare, Chicago, IL, USA) supplemented with
10% heat-inactivated foetal bovine serum (PAN-Biotech GmbH,
Aidenbach, Germany), 100 units/ml penicillin, and 100 units/ml
streptomycin (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA). All cell cultures were carried out at 37°C in a humidified
atmosphere with 5% CO2. According to manufacturer
instructions, DXM and 10058-F4 (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) were dissolved in dimethylsulphoxide (DMSO) at
10 and 20 mM, respectively. Reagents were stored at −20°C and
further diluted to indicated concentrations in culture medium
before use.
Cell viability assay
Cell viability was measured by MTS method (CellTiter
96®Aqueous One Solution, cat. no. 207284; Promega
Corporation, Madison, WI, USA). Cells were seeded in 96-well plates
(100 µl per well at 2×105/ml) and were treated with DXM
(0–0.8 mM) alone or in combination with 30 and 60 µM 10058-F4 for
24, 48 and 72 h and the concentration of 10058-F4 was used in our
previous study (16), which
produced a better sensitization effect in combination with valproic
acid. Besides, this concentration of 10058-F4 was also chosen in
another study (17). In each well,
20 µl MTS solution were added, and the cells were incubated at 37°C
for 3 h. The absorbance values of each well were measured by
spectrophotometry (iMark; Bio-Rad Laboratories, Inc., Hercules, CA,
USA) at 490 nm.
Cell cycle analysis
Cell cycle stage was detected by Cell Cycle Staining
kit [propidium iodide (PI); cat. no. CCS012; MultiSciences Biotech
Co., Ltd., Hangzhou, China]. Cells were treated with DXM (0–0.8 mM)
with or without 60 µM 10058-F4 for 24 h, collected in a tube using
pipet tips, washed twice in 4°C PBS solution and resuspended in 1
ml DNA staining solution. Subsequently, 10 µl permeabilization
solution were added, and these samples were incubated in the dark
at room temperature for 30 min. Cell cycle distribution was
assessed by FACScan (BD FACSCanto™ II; BD Biosciences, Franklin
Lakes, NJ, USA) analysis. The data were analysed by ModFit LT
programme (Verity Software House Inc., Topsham, ME, USA).
Apoptosis detection by flow
cytometry
Apoptotic cells were detected on a FACScan flow
cytometer (BD FACSCanto™ II; BD Biosciences) using Annexin
V-fluorescein isothiocyanate (FITC) and PI (BD Pharmingen, San
Diego, CA, USA) staining. Cells treated with DXM (0–0.8 mM), alone
or in combination with 60 µM 10058-F4 for 24 h, were collected,
washed twice in 4°C PBS and resuspended in 50 µl Annexin V binding
buffer. Next, 5 µl Annexin V-FITC and 5 µl PI were added, and these
samples were incubated in the dark at room temperature for 15 min.
Finally, 450 µl Annexin V binding buffer were added, and cell death
was measured by flow cytometry.
Western blot analysis
Cells were treated with DXM (0–0.8 mM), alone or in
combination with 60 µM 10058-F4 for 24 h, and lysed using sodium
dodecyl sulphate (SDS) buffer containing proteinase inhibitors
(PMSFs). The total protein concentrations of the cells were
determined by BCA Protein Assay kit (Beyotime Institute of
Biotechnology, Haimen, China). The samples were separated on 12%
SDS polyacrylamide gel electrophoresis (SDS-PAGE) gels and
transferred onto polyvinylidene fluoride (PVDF) membranes (Bio-Rad
Laboratories, Inc.). The PVDF membranes were blocked with 5%
non-fat milk for 2 h at room temperature and incubated with primary
antibodies, i.e., rabbit anti-human polyclonal c-Myc, rabbit
anti-human monoclonal cyclin-dependent kinase (CDK)-4, rabbit
anti-human monoclonal CDK6, rabbit anti-human monoclonal cleaved
PARP, rabbit anti-human monoclonal cleaved caspase-3 and rabbit
anti-human monoclonal β-actin (dilution, 1:1,000; Cell Signaling
Technology, Inc., Danvers, MA, USA), overnight at 4°C. After
washing three times for 10 min each time with TBST solution, these
PVDF membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (dilution, 1:5,000; Cell
Signaling Technology, Inc.) for 2 h at room temperature. The
membranes were washed three times again with TBST solution, and
protein bands were visualized with an enhanced chemiluminescence
detecting kit. Densitometry quantification of immunoblot analyses
was performed using Image lab software (v. 5.2.1; Bio-Rad
Laboratories, Inc.).
Statistical analysis
Significant differences between the means ± standard
deviation of experimental and control groups were compared by
Student's t-test. Two-way analysis of variance and Tukey's post hoc
test were performed for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference. All
statistical analyses were performed using SPSS Statistics 18.0
software (SPSS, Inc., Chicago, IL, USA).
Results
10058-F4 increases the growth
inhibition of NALM6 and CEM cells induced by DXM
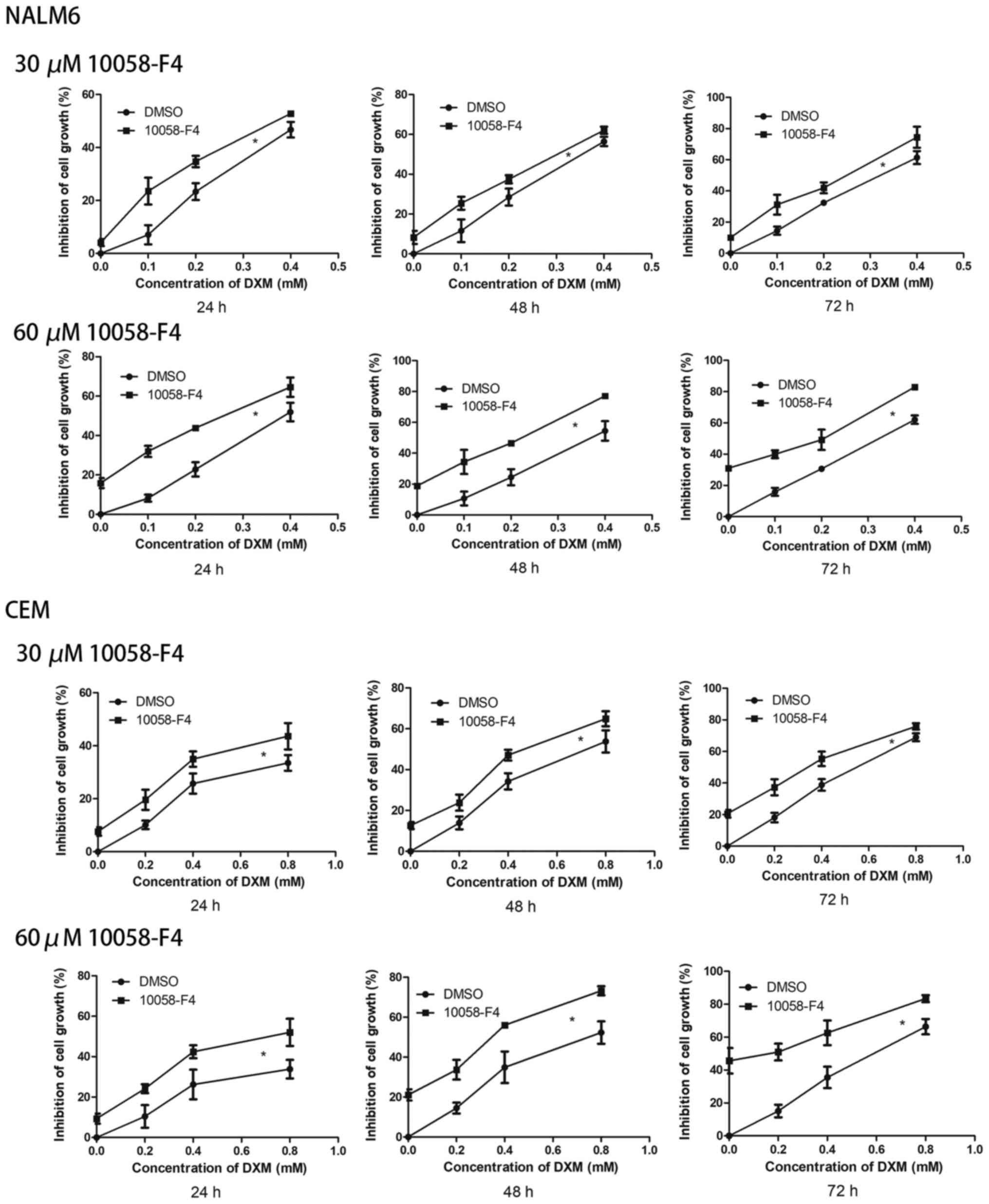
Cell growth was analysed by MTS assay. The growth
inhibition rates of the NALM6 cells treated with DXM (0, 0.1, 0.2
and 0.4 mM) combined with 10058-F4 were higher than those treated
with corresponding concentrations of DXM (P<0.05 for all). In
the CEM cells, the growth inhibition rates of DXM (0, 0.2, 0.4 and
0.8 mM) plus 10058-F4 were also higher than those of the
corresponding concentrations of DXM alone (P<0.05 for all;
Fig. 1).
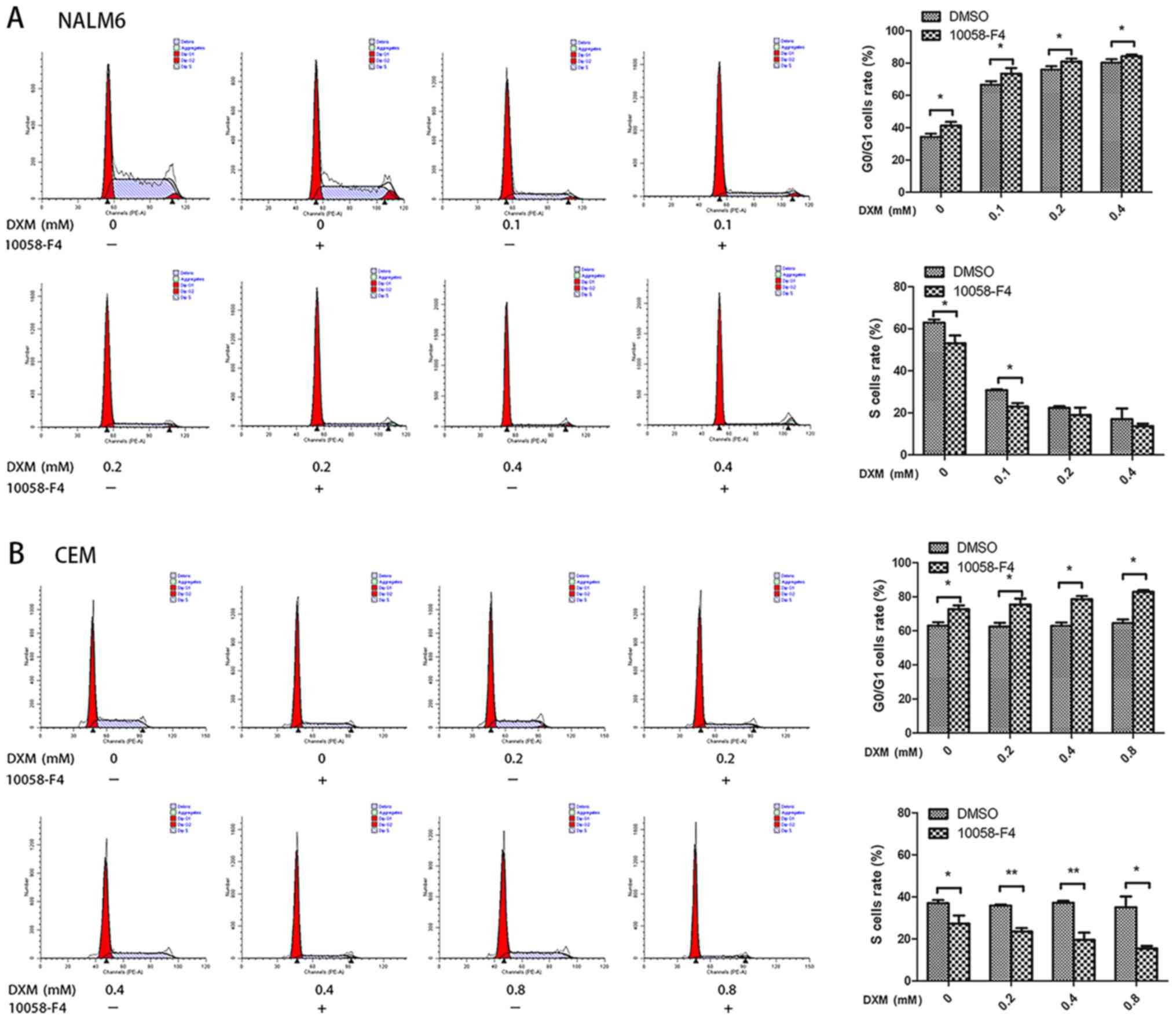
10058-F4 increases the cell-cycle
arrest of NALM6 and CEM cells induced by DXM
To explore whether the combination of 10058-F4 with
DXM induced further cell cycle arrest, we compared the cell cycle
distribution of NALM6 and CEM cells treated with DXM alone or in
combination with 10058-F4. As shown in Fig. 2A, the rates of
G0/G1 cells in the groups treated with DXM
(0, 0.1, 0.2 and 0.4 mM) in combination with 60 µM 10058-F4 for 24
h were higher than those in the corresponding groups treated with
DXM alone (P<0.05 for all). The same results were observed in
the CEM cells (P<0.05 for all; Fig.
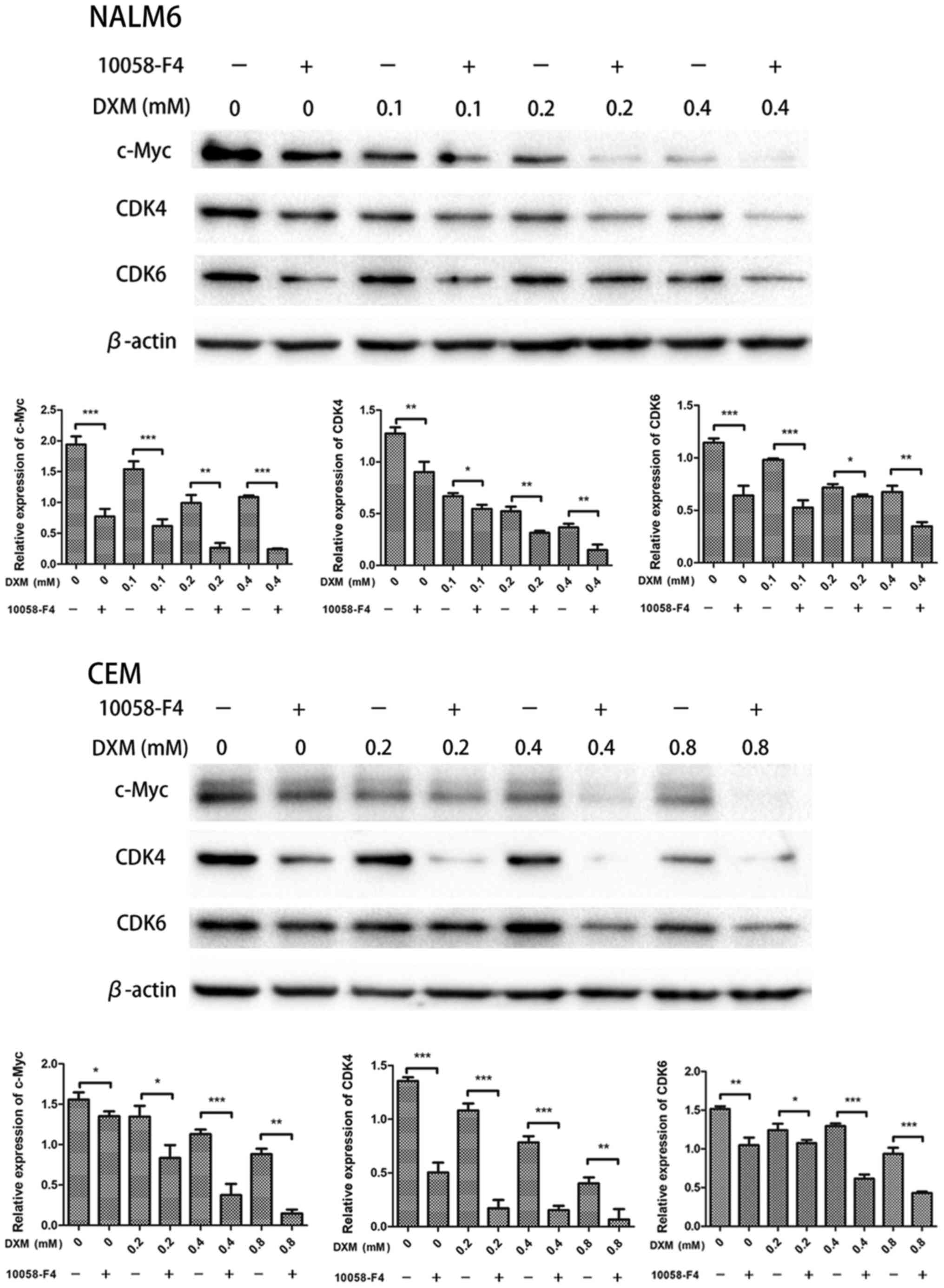
2B). Additionally, compared with those in the groups treated
with DXM alone, the protein expressions of c-Myc, CDK4 and CDK6 in
the groups treated with 10058-F4 and DXM decreased in the two cell
lines (P<0.05 for all; Fig. 3).
These findings indicated that 10058-F4 dramatically increased the
cell-cycle arrest induced by DXM.
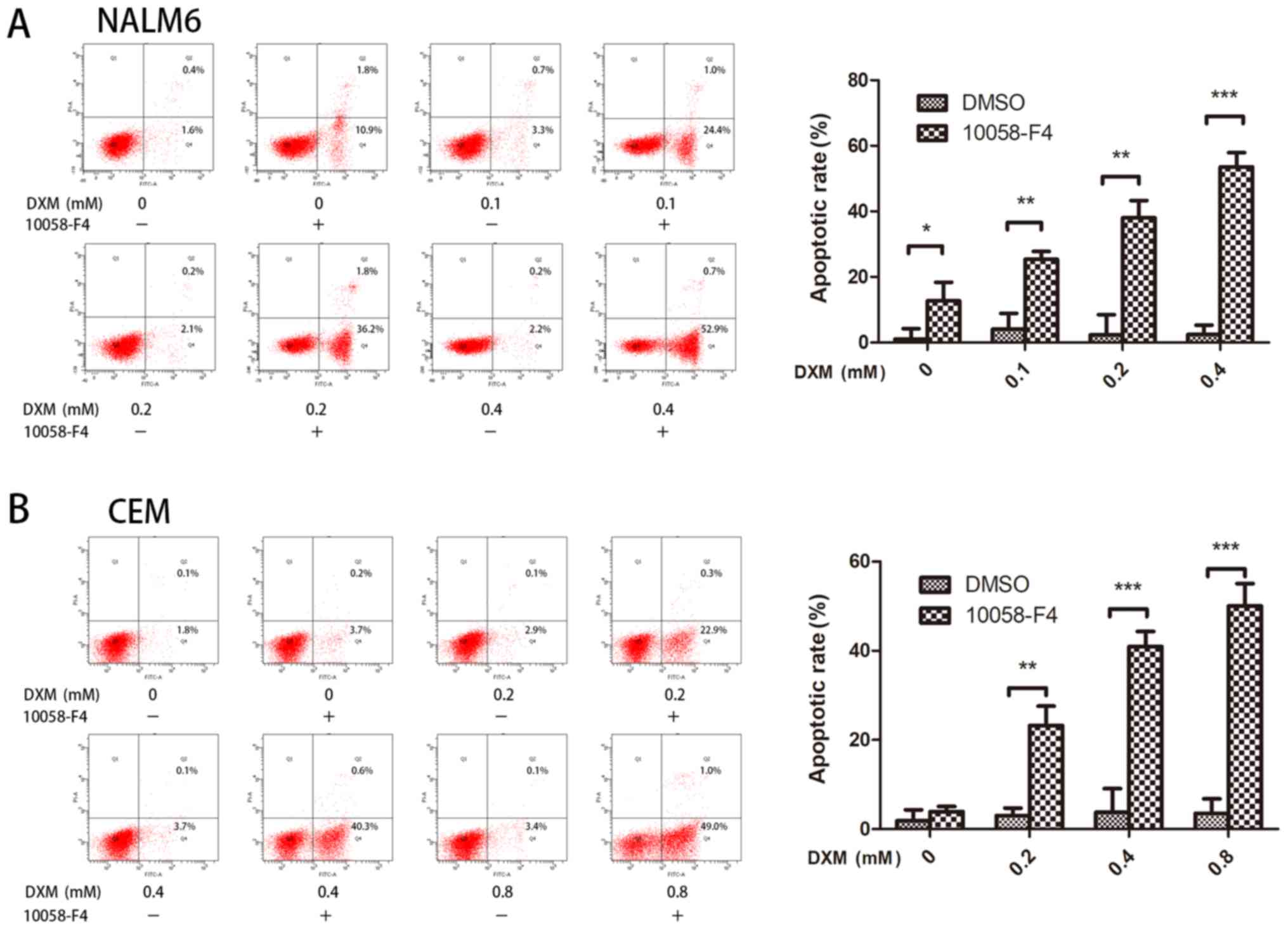
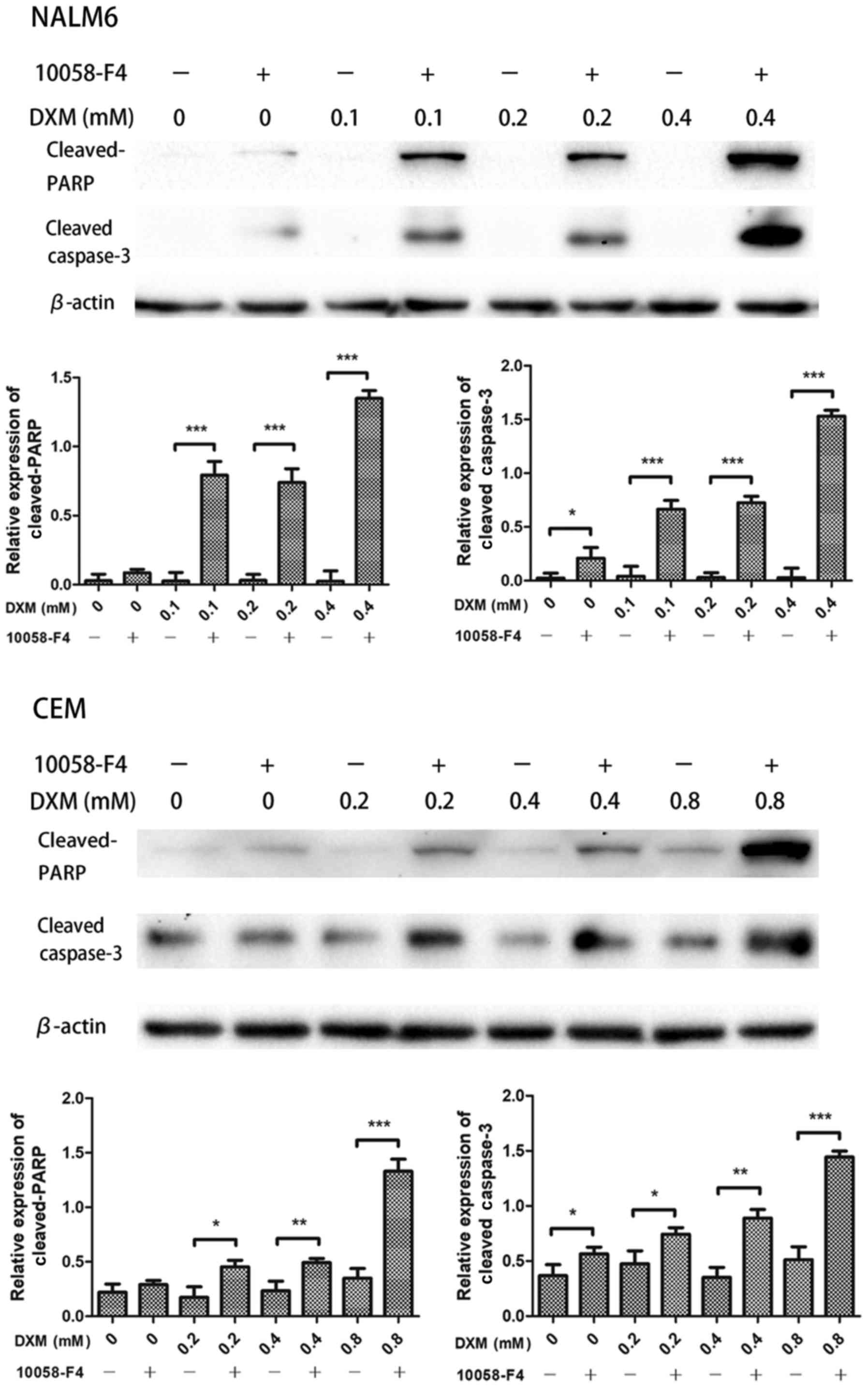
Combined 10058-F4 and DXM treatment
greatly induces apoptosis of NALM6 and CEM cells
Previous studies have demonstrated 10058-F4 as an
effective agent in inducing cell death in myeloma and AML cells
(17,18). Based on these reports, we
determined whether 10058-F4 increased the apoptosis of ALL cells
induced by DXM. In the NALM6 and CEM cells, the apoptotic rates of
groups treated with DXM and 10058-F4 significantly increased when
compared with those of groups treated with DXM alone. As shown in
Fig. 4A, compared with treatments
with corresponding concentrations of DXM (0, 0.1, 0.2 and 0.4 mM)
alone, the cell death rates (Annexin V+/PI+ and Annexin V+/PI-) of
NALM6 cells treated with 10058-F4 combined with DXM significantly
increased (P<0.05 for all). Similarly, the cell death rates of
the CEM cells treated with DXM (0, 0.2, 0.4 and 0.8 mM) in
combination with 60 µM 10058-F4 for 24 h were significantly higher
than those treated with corresponding concentrations of DXM alone
(P<0.05 for all; Fig. 4B). The
Western blot results also showed that 10058-F4 further promoted the
cleavages of caspase-3 and cleaved-PARP in NALM6 and CEM cells
induced by DXM (P<0.05 for all; Fig. 5), suggesting that 10058-F4
significantly increased the apoptosis of ALL cells induced by
DXM.
Discussion
c-Myc, a helix-loop-helix-leucine zipper (HLH-ZIP)
oncoprotein, dimerizes with another HLH-ZIP protein, Max, and
subsequently binds DNA. This c-Myc-Max interaction regulates target
gene expression (19). In T-ALL,
55% of the patients harbour Notch1 mutations that activate Notch
signalling and promote c-Myc expression (20). Additionally, 10 to 20% of T-ALL
patients have FWB mutations, an E3 ubiquitin ligase responsible for
the degradation of c-Myc; this also increases c-Myc expression
(21). Hence, c-Myc deregulation
is a common phenomenon in T-ALL. Furthermore, an increase in c-Myc
expression has been observed in B-ALL cells (8). A c-Myc inhibitor (e.g., 10058-F4) can
inhibit c-Myc-Max association, decrease c-Myc expression and
prevent transactivation by c-Myc-Max heterodimers (22). Previous studies have implied that
c-Myc inhibition eliminated both B-ALL and T-ALL cells (16,23).
Our results demonstrated that 10058-F4 alone could suppress the
growth of B-ALL cell line NALM6 cells and T-ALL cell line CEM
cells.
Because GCs decrease c-Myc expression, we speculated
that c-Myc inhibitors enhanced the growth suppression of ALL cells
induced by GCs. In our study, 10058-F4 increased significantly
inhibited growth, G0/G1 arrest and especially
the apoptosis of NALM6 and CEM cells induced by DXM. Furthermore,
c-Myc downregulation induced by DXM was reinforced by 10058-F4 and
was accompanied by deceased CDK4/CDK6 expression. c-Myc has been
demonstrated to regulate CDK6 activity; furthermore, CDK4 has been
considered a target of c-Myc (24,25).
Hence, our results indicated that the c-Myc-CDK4/CDK6 axis could
play an important role in G0/G1 arrest
induced by combined DXM and 10058-F4 treatment and its exact
mechanism need to be further explored.
GCs are the basic drugs in the treatment of ALL;
novel combinations have been focused on these drugs. LEE011, a
CDK4/CDK6 inhibitor, is synergistic with the GC DXM in
vitro. Their combination has been shown to prolong the survival
of T-ALL mice model cells (26).
Liu et al (27) reported
that low-dose anisomycin promoted apoptosis and cell cycle arrest
induced by GCs in GC-resistant T-ALL CEM-C1 cells via the
activation of GRs and p38-MAPK/JNK. The constitutive activation of
NOTCH1 signalling plays a vital role in the pathogenesis of T-ALL.
Thus, PF-03084014, a γ-secretase inhibitor, was found to have a
synergistic antileukaemic effect on T-ALL cells when combined with
GCs in vitro and in vivo (28). However, to the best of our
knowledge, we are the first to show that c-Myc inhibitors increased
the sensitivity of B-ALL cell line NALM6 cells and T-ALL cell line
CEM cells to the anti-neoplastic effects of GCs.
The role of the therapeutic targeting of c-Myc on
antineoplastic activity in vivo has been debated due to
reported rapid metabolism, inadequate target site penetration and
possible liver and kidney toxicity (29,30);
however, small molecule c-Myc inhibitors conjugated to
integrin-targeted nanoparticles have been shown to overcome these
defects (31). Notably, a recent
study showed that the dual targeting of p53 and c-Myc selectively
eradicated leukaemic stem cells in chronic myeloid leukaemia (CML)
in the treatment of refractory haematological malignancies
(32). The present study
demonstrated that the c-Myc inhibitor 10058-F4 promoted
G0/G1 arrest and cell death induced by DXM in
the ALL cell lines NALM6 and CEM. These findings indicated that the
combination of GCs with c-Myc inhibitors may be a novel potent
therapeutic strategy for ALL. However, further clinical
investigations on their combination are necessary.
Acknowledgements
Not applicable.
Funding
The present study was supported by The Zhejiang
Provincial Natural Science Foundation of China (grant nos.
LY17H160005 and LY14H080001), The National Natural Science
Foundation of China (grant no. 81401321), The Project from
Traditional Chinese Medicine Administration of Zhejiang Province
(grant no. 2015ZZ018) and The Natural Science Foundation of Ningbo
(grant no. 2014A610217).
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
QM and GO designed the experiments and revised the
manuscript. ML performed the experiments and wrote the manuscript.
YW, WW, SY, HZ, BH, YC, CS and YZ performed the experiments. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Goldstone AH, Richards SM, Lazarus HM,
Tallman MS, Buck G, Fielding AK, Burnett AK, Chopra R, Wiernik PH,
Foroni L, et al: In adults with standard-risk acute lymphoblastic
leukemia, the greatest benefit is achieved from a matched sibling
allogeneic transplantation in first complete remission and an
autologous transplantation is less effective than conventional
consolidation/maintenance chemotherapy in all patients: Final
results of the International ALL Trial (MRC UKALL XII/ECOG E2993).
Blood. 111:1827–1833. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pui CH, Sandlund JT, Pei D, Campana D,
Rivera GK, Ribeiro RC, Rubnitz JE, Razzouk BI, Howard SC, Hudson
MM, et al: Improved outcome for children with acute lymphoblastic
leukemia: Results of total therapy study XIIIB at St Jude
children's research hospital. Blood. 104:2690–2696. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bassan R: Evolving strategies for the
management of high-risk adult acute lymphoblastic leukemia.
Haematologica. 90:12992005.PubMed/NCBI
|
|
4
|
Frey NV and Porter DL: CAR T-cells merge
into the fast lane of cancer care. Am J Hematol. 91:146–150. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park JH, Geyer MB and Brentjens RJ:
CD19-targeted CAR T-cell therapeutics for hematologic malignancies:
Interpreting clinical outcomes to date. Blood. 127:3312–3320. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schreiber-Agus N and DePinho RA:
Repression by the Mad(Mxi1)-Sin3 complex. Bioessays. 20:808–818.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou F, Medh RD and Thompson EB:
Glucocorticoid mediated transcriptional repression of c-myc in
apoptotic human leukemic CEM cells. J Steroid Biochem Mol Biol.
73:195–202. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ge Z, Guo X, Li J, Hartman M, Kawasawa YI,
Dovat S and Song C: Clinical significance of high c-MYC and low
MYCBP2 expression and their association with Ikaros dysfunction in
adult acute lymphoblastic leukemia. Oncotarget. 6:42300–42311.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Roderick JE, Tesell J, Shultz LD, Brehm
MA, Greiner DL, Harris MH, Silverman LB, Sallan SE, Gutierrez A,
Look AT, et al: c-Myc inhibition prevents leukemia initiation in
mice and impairs the growth of relapsed and induction failure
pediatric T-ALL cells. Blood. 123:1040–1050. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gaynon PS and Carrel AL:
Glucocorticosteroid therapy in childhood acute lymphoblastic
leukemia. Adv Exp Med Biol. 457:593–605. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Distelhorst CW: Recent insights into the
mechanism of glucocorticosteroid-induced apoptosis. Cell Death
Differ. 9:6–19. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tissing WJ, Meijerink JP, den Boer ML and
Pieters R: Molecular determinants of glucocorticoid sensitivity and
resistance in acute lymphoblastic leukemia. Leukemia. 17:17–25.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saffar AS, Ashdown H and Gounni AS: The
molecular mechanisms of glucocorticoids-mediated neutrophil
survival. Curr Drug Targets. 12:556–562. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thulasi R, Harbour DV and Thompson EB:
Suppression of c-myc is a critical step in glucocorticoid-induced
human leukemic cell lysis. J Biol Chem. 268:18306–18312.
1993.PubMed/NCBI
|
|
15
|
Helmberg A, Auphan N, Caelles C and Karin
M: Glucocorticoid-induced apoptosis of human leukemic cells is
caused by the repressive function of the glucocorticoid receptor.
EMBO J. 14:452–460. 1995.PubMed/NCBI
|
|
16
|
Mu Q, Ma Q, Lu S, Zhang T, Yu M, Huang X,
Chen J and Jin J: 10058-F4, a c-Myc inhibitor, markedly increases
valproic acid-induced cell death in Jurkat and CCRF-CEM
T-lymphoblastic leukemia cells. Oncol Lett. 8:1355–1359. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang MJ, Cheng YC, Liu CR, Lin S and Liu
HE: A small-molecule c-Myc inhibitor, 10058-F4, induces cell-cycle
arrest, apoptosis, and myeloid differentiation of human acute
myeloid leukemia. Exp Hematol. 34:1480–1489. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Holien T, Våtsveen TK, Hella H, Rampa C,
Brede G, Groseth LA, Rekvig M, Borset M, Standal T, Waage A and
Sundan A: Bone morphogenetic proteins induce apoptosis in multiple
myeloma cells by Smad-dependent repression of MYC. Leukemia.
26:1073–1080. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luscher B and Larsson LG: The basic
region/helix-loop-helix/leucine zipper domain of Myc
proto-oncoproteins: Function and regulation. Oncogene.
18:2955–2966. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weng AP, Ferrando AA, Lee W, Morris JP IV,
Silverman LB, Sanchez-Irizarry C, Blacklow SC, Look AT and Aster
JC: Activating mutations of NOTCH1 in human T cell acute
lymphoblastic leukemia. Science. 306:269–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
O'Neil J, Grim J, Strack P, Rao S,
Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters
R, et al: FBW7 mutations in leukemic cells mediate NOTCH pathway
activation and resistance to gamma-secretase inhibitors. J Exp Med.
204:1813–1824. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin X, Giap C, Lazo JS and Prochownik EV:
Low molecular weight inhibitors of Myc-Max interaction and
function. Oncogene. 22:6151–6159. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ott CJ, Kopp N, Bird L, Paranal RM, Qi J,
Bowman T, Rodig SJ, Kung AL, Bradner JE and Weinstock DM: BET
bromodomain inhibition targets both c-Myc and IL7R in high-risk
acute lymphoblastic leukemia. Blood. 120:2843–2852. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mateyak MK, Obaya AJ and Sedivy JM: c-Myc
regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle
progression at multiple independent points. Mol Cell Biol.
19:4672–4683. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hermeking H, Rago C, Schuhmacher M, Li Q,
Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F,
et al: Identification of CDK4 as a target of c-MYC. Proc Natl Acad
Sci USA. 97:2229–2234. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pikman Y, Alexe G, Roti G, Conway AS,
Furman A, Lee ES, Place AE, Kim S, Saran C, Modiste R, et al:
Synergistic drug combinations with a CDK4/6 inhibitor in T-cell
acute lymphoblastic leukemia. Clin Cancer Res. 23:1012–1024. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu Y, Ge J, Li Q, Guo X, Gu L, Ma ZG, Li
XH and Zhu YP: Low-dose anisomycin sensitizes
glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1
cells to dexamethasone-induced apoptosis through activation of
glucocorticoid receptor and p38-MAPK/JNK. Leuk Lymphoma.
55:2179–2188. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samon JB, Castillo-Martin M, Hadler M,
Ambesi-Impiobato A, Paietta E, Racevskis J, Wiernik PH, Rowe JM,
Jakubczak J, Randolph S, et al: Preclinical analysis of the
γ-secretase inhibitor PF-03084014 in combination with
glucocorticoids in T-cell acute lymphoblastic leukemia. Mol Cancer
Ther. 11:1565–1575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Prochownik EV and Vogt PK: Therapeutic
targeting of Myc. Genes Cancer. 1:650–659. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guo J, Parise RA, Joseph E, Egorin MJ,
Lazo JS, Prochownik EV and Eiseman JL: Efficacy, pharmacokinetics,
tisssue distribution, and metabolism of the Myc-Max disruptor,
10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in
mice. Cancer Chemother Pharmacol. 63:615–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Soodgupta D, Pan D, Cui G, Senpan A, Yang
X, Lu L, Weilbaecher KN, Prochownik EV, Lanza GM and Tomasson MH:
Small molecule MYC inhibitor conjugated to integrin-targeted
nanoparticles extends survival in a mouse model of disseminated
multiple myeloma. Mol Cancer Ther. 14:1286–1294. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abraham SA, Hopcroft LE, Carrick E, Drotar
ME, Dunn K, Williamson AJ, Korfi K, Baquero P, Park LE, Scott MT,
et al: Dual targeting of p53 and c-MYC selectively eliminates
leukaemic stem cells. Nature. 534:341–346. 2016. View Article : Google Scholar : PubMed/NCBI
|