Introduction
Trabeculectomy is considered the primary treatment
method for patients with glaucoma (1) but its efficacy is limited by the
frequency of postoperative subconjunctival fibrosis. Although
anti-fibrotic drugs including mitomycin C (MMC) and 5-fluorouracil
(5-FU) are commonly used (2), the
surgical failure rate remains high and may lead to
sight-threatening infections (3).
There is an urgent requirement to elucidate the molecular
mechanisms that drive the formation of fibrosis, and to explore
alternative intervention strategies.
Activated human tenon fibroblasts (HTFs),
characterized by myofibroblast transdifferentiation, are the
central effector cells during fibrosis (4). Myofibroblasts drive fibrosis
formation by increasing the production of extracellular matrix
proteins including type I, III, V and type VI collagens (5). In addition, myofibroblasts secrete
tissue inhibitors of metalloproteinases (TIMPs), decreasing the
activity of extracellular matrix-degrading enzymes (6). Multiple stimuli have been identified
to trigger fibroblasts activation, particularly growth factors and
cytokine family members (7), of
which the transforming growth factor-β (TGF-β) family are the most
potent and well-documented mediators (8).
The human TGF-β family comprises 3 isoforms,
designated TGF-β1, TGF-β2, and TGF-β3 (9). Despite previous data suggesting that
all 3 TGF-β isoforms are identifiable in HTFs (10), TGF-β1 is the predominant isoform
associated with subconjunctival scarring (11,12).
Typically, TGF-β1 functions by binding to its heterodimeric
receptor complex and causing phosphorylation of the downstream
signal transduction proteins mothers against decapentaplegic
homolog (Smad) 2 and Smad3, which in turn form complexes with
Smad4, translocate to the nucleus and regulate target gene
transcription (13). In addition
to the canonical Smad pathway, members of the mitogen-activated
protein kinase (MAPK) signaling pathways, including p38MAPK,
extracellular signal-regulated kinase (ERK)1/2 and c-Jun-N-terminal
kinase (JNK), are activated by TGF-β (14). Although the role of canonical Smad
signaling in fibrosis has been well established, whether
non-canonical MAPK signaling is involved in this process remains
unverified.
The present study aimed to investigate the role of
the MEK1/2 inhibitor U0126 on HTF activation, including
proliferation, migration, and α-smooth muscle actin (α-SMA)
expression, and collagen contraction induced by TGF-β1. In
addition, the present study also aimed to elucidate the underlying
mechanisms of action by examining the activation statuses of the
Smad2/3, ERK1/2 and p38MAPK signaling pathways.
Materials and methods
Cell culture
Samples of human Tenon's capsule were obtained from
patients from the Zhongshan Ophthalmic Center (Sun Yat-sen
University, Guangzhou, China), with the approval of the Ethical
Committee of the Zhongshan Ophthalmic Center and in accordance with
the Declaration of Helsinki. Informed consent was gained. Fresh
tissues were cut into 1–2 mm pieces under a stereo microscope
(×1.5), then placed in culture dishes with Dulbecco's modified
Eagle's medium (DMEM)/F12 supplemented with 10% fetal bovine serum
(both from Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA,
USA), 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C and 5%
CO2. After 8 days, primary cultures (~60% confluence)
were passaged with 0.25% trypsin-EDTA (Invitrogen; Thermo Fisher
Scientific, Inc.). Cells at passages 3–6 were used in subsequent
experiments. Cell identification was performed by immunostaining
with the fibroblast marker vimentin and cytokeratin (Abcam,
Cambridge, UK). Briefly, the cells were seeded on coverslip with
80–90%, and were fixed with 4% paraformaldehyde at room temperature
for 30 min. After washing with PBS, the cells were permeated with
0.1% TritonX-100 for 15 min at room temperature. After blocking
with 5% BSA for 30 min at room temperature, cells were incubated
with antibody against vimentin (cat. no. 5741; Cell Signaling
Technology, Inc., Danvers, MA, USA; 1:200) at 4°C overnight. Cells
were then incubated with the secondary antibody Alexa Fluor 488 of
goat anti-rabbit IgG antibody (cat. no. ab150077; Abcam; 1:500) at
room temperature for 1 h, The nuclei were stained with DAPI (5
mg/ml) at room temperature for 1 min. Immunofluorescence was
visualized using a fluorescence microscope (LSM710; Zeiss AG,
Oberkochen, Germany) at 512×512 pixel resolution. Images were
collected using a ×20 apochromatic objective.
Cell proliferation assay
HTFs proliferation was determined by Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan) assay. Briefly, a 96-well plate was seeded with HTFs
(1×104 cells/well) and incubated overnight at 37°C and
5% CO2. Following treatment with TGF-β1 (5 ng/ml) alone
or in the presence of 5 or 10 µM of U0126, 10 µl CCK-8 solution was
added to each well and incubated at 37°C for 4 h. A microplate
reader was used to measure the absorbance at 450 nm (Bio-Rad
Laboratories, Inc., Hercules, CA, USA).
Wound healing assay
HTFs (5×105) were seeded into 6-well
plates and incubated to 100% confluence. The cell monolayer was
scratched with a 200-µl pipette tip and washed with PBS, following
which fresh DMEM/F12 containing TGF-β1 (5 ng/ml) alone or combined
with 5 µM of U0126 was added and the plates incubated for 24 h. The
distance of cell migration was measured via phase-contrast
microscopy (magnification, ×20).
Fibroblast-mediated collagen
contraction assay
A collagen contraction assay was performed using a
Cell Biolabs Collagen Contraction Assay kit (Cell Biolabs, Inc.,
San Diego, CA, USA), according to the manufacturer's protocol.
Briefly, HTFs (5×105 cells/well) and 500 µl collagen
preparation were added to a 24-well plate for polymerization.
Following treatment with TGF-β1 (5 ng/ml) alone or combined with 5
µM of U0126 for 24 h, gels were detached from the wells and images
were captured under a light stereo microscope (magnification,
×1.5). Analyses were performed using ImageJ software 2.1.4.7
(National Institutes of Health, Bethesda, MD, USA).
Western blot analysis
Extracts were prepared by lysing cells with
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) on ice for 10 min. Protein
concentrations were quantified with a Micro bicinchoninic acid
Protein Assay kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Protein lysates (30 µg) were separated
via SDS-PAGE with 10% of separation gel and 3% stacking gel and
then transferred to a polyvinylidene fluoride membrane (Thermo
Fisher Scientific, Inc.). Proteins of interest were detected using
primary antibodies against human α-SMA (cat. no. ab5694; 1:1,000)
and GAPDH (cat. no. ab9485; 1:1,000; both Abcam), TGF-β1 (cat. no.
sc-130348; 1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX,
USA) Smad2 (cat. no. 5339; 1:1,000), Smad3 (cat. no. 9523;
1:1,000), phosphorylated (p)-Smad2/3 (cat. no. 8828; 1:1,000),
p38MAPK (cat. no. 8690; 1:1,000), p-p38MAPK (cat. no. 4511;
1:1,000), ERK1/2 (cat. no. 4695; 1:1,000), p-ERK1/2 (cat. no. 4370;
1:1,000; all Cell Signaling Technology, Inc.) and zinc finger
protein SNAI1 (cat. no. 3879; Cell Signaling Technology, Inc.;
1:1000), followed by incubation with mouse peroxidase-conjugated
secondary antibodies (cat. no. sc-2357; Santa Cruz Biotechnology,
Inc.; 1:2,000) for 1 h at room temperature. Specific proteins were
visualized by enhanced chemiluminescence (cat. no. WBKLS0500; Merck
KGaA, Darmstadt, Germany).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells using
RNeasy spin columns (Qiagen Ltd., Crawley, UK) according to the
manufacturer's protocol. cDNA was synthesized by reverse
transcriptase from total RNA with PrimeScript RT Master Mix (Takara
Bio Inc., Otsu, Japan). The RT reaction was conducted at 25°C, 5
min, followed by 55°C, 10 min, and the reaction was terminated by
heating at 85°C for 5 min. qPCR was subsequently performed using
the SYBR® Premix Ex Taq™ kit (RR820A; Takara
Biotechnology Co., Ltd). The thermocycling conditions for qPCR is
as follows: Initial denaturation at 95°C for 30 sec; 40 cycles of
denaturing the cDNA template at 95°C for 10 sec and
annealing/extension at 60°C for 30 sec. The gene expression levels
were normalized to GAPDH according to the 2−∆∆Cq
relative quantification method (15). The primers used in the experiment
were as follows: GAPDH forward, 5′-GGAGCGAGATCCCTCCAAAAT-3′ and
reverse, 5′-GGCTGTTGTCATACTTCTCATGG-3′; α-SMA forward,
5′-AAAAGACAGCTACGTGGGTG-3′ and reverse,
5′-GCCATGTTCTATCGGGTACTTC-3′; Snail forward,
5′-TCGGAAGCCTAACTACAGCGA-3′ and reverse,
5′-AGATGAGCATTGGCAGCGAG-3′; TGF-β1 forward,
5′-GGCCAGATCCTGTCCAAGC-3′ and reverse,
5′-GTGGGTTTCCACCATTAGCAC-3′.
Immunofluorescence staining
Cells were seeded onto coverslips once they reached
40–60% confluence. Upon treatment with the indicated agents, cells
were fixed in 4% paraformaldehyde for 15 min at room temperature
and then permeated with 0.1% Triton X-100 for 10 min. Following
blocking with 3% bovine serum albumin (Sigma-Aldrich; Merck KGaA)
for 1 h at room temperature, the cells were incubated with a
primary antibody against α-SMA (cat. no. ab5694; Abcam; 1:200) or
cytokeratin (cat. no. 5741; 1:200; Cell Signaling Technology, Inc.)
at 4°C overnight, and then exposed to the Alexa Fluor®
488-conjugated green fluorescence secondary antibodies (cat. no.
ab150077; 1:500) or Alexa Fluor® 555-conjugated of red
fluorescence secondary antibodies (cat. no. ab150158; 1:500; both
Abcam) for 1 h at room temperature. The nuclei were stained with
DAPI (5 mg/ml) for 1 min at room temperature and images were
captured using a Leica fluorescence microscope (magnification,
×20).
Statistical analysis
All experiments were performed at least three times
and the data are presented as the means ± standard deviation.
Comparisons among multiple groups were performed by one-way
analysis of variance with Tukey's post-hoc test using GraphPad
Prism version 7.0 (GraphPad Software, Inc., La Jolla, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Morphology and molecular
characterization of HTFs
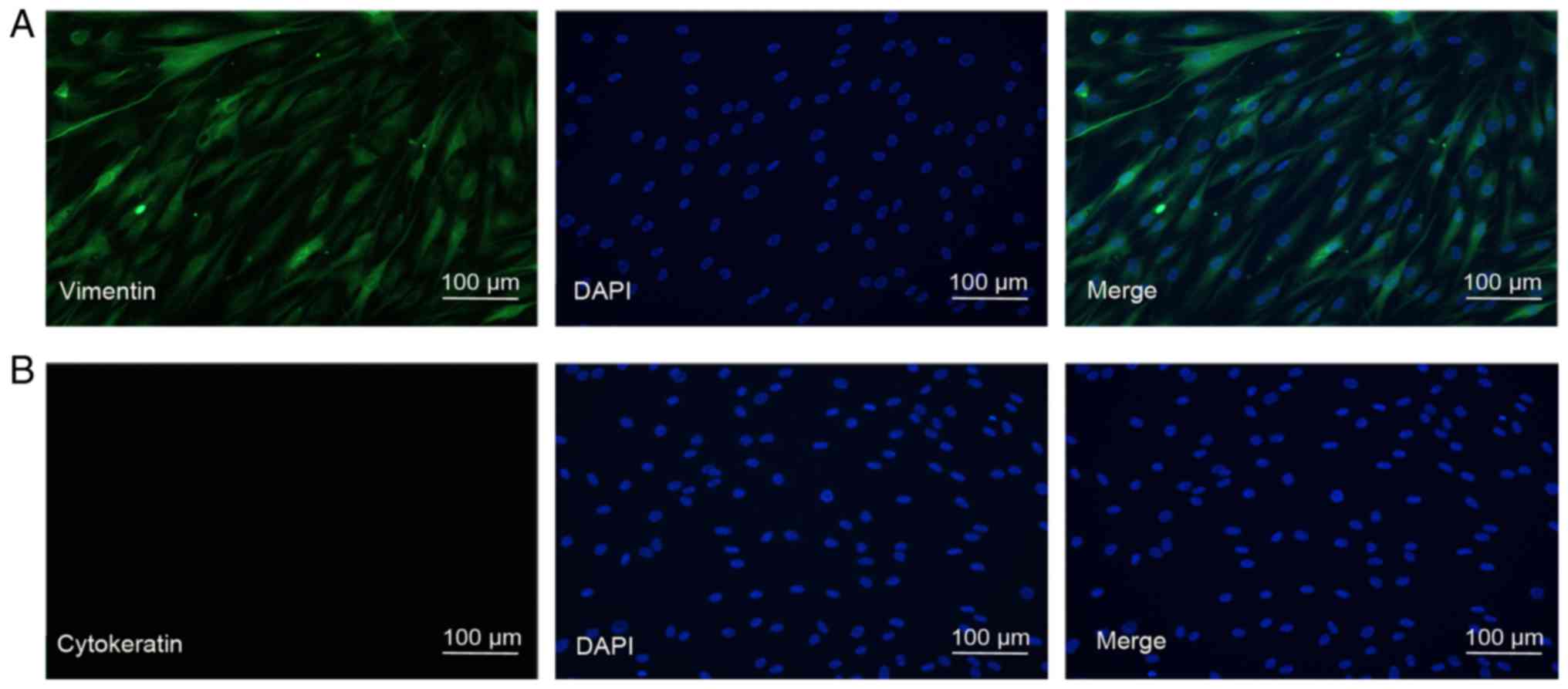
First, the present study aimed to identify the
primary cultured HTFs. The cells were grown in a monolayer and
exhibited a typical spindly, generally flat and elongated shape
(Fig. 1). To identify HTFs at a
molecular level, immunostaining using vimentin and cytokeratin,
cell surface markers for fibroblasts and the epithelium,
respectively, was performed. As indicated in Fig. 1, the HTFs exhibited extensive
staining for vimentin and not for cytokeratin, suggesting that the
isolated HTFs were of high purity.
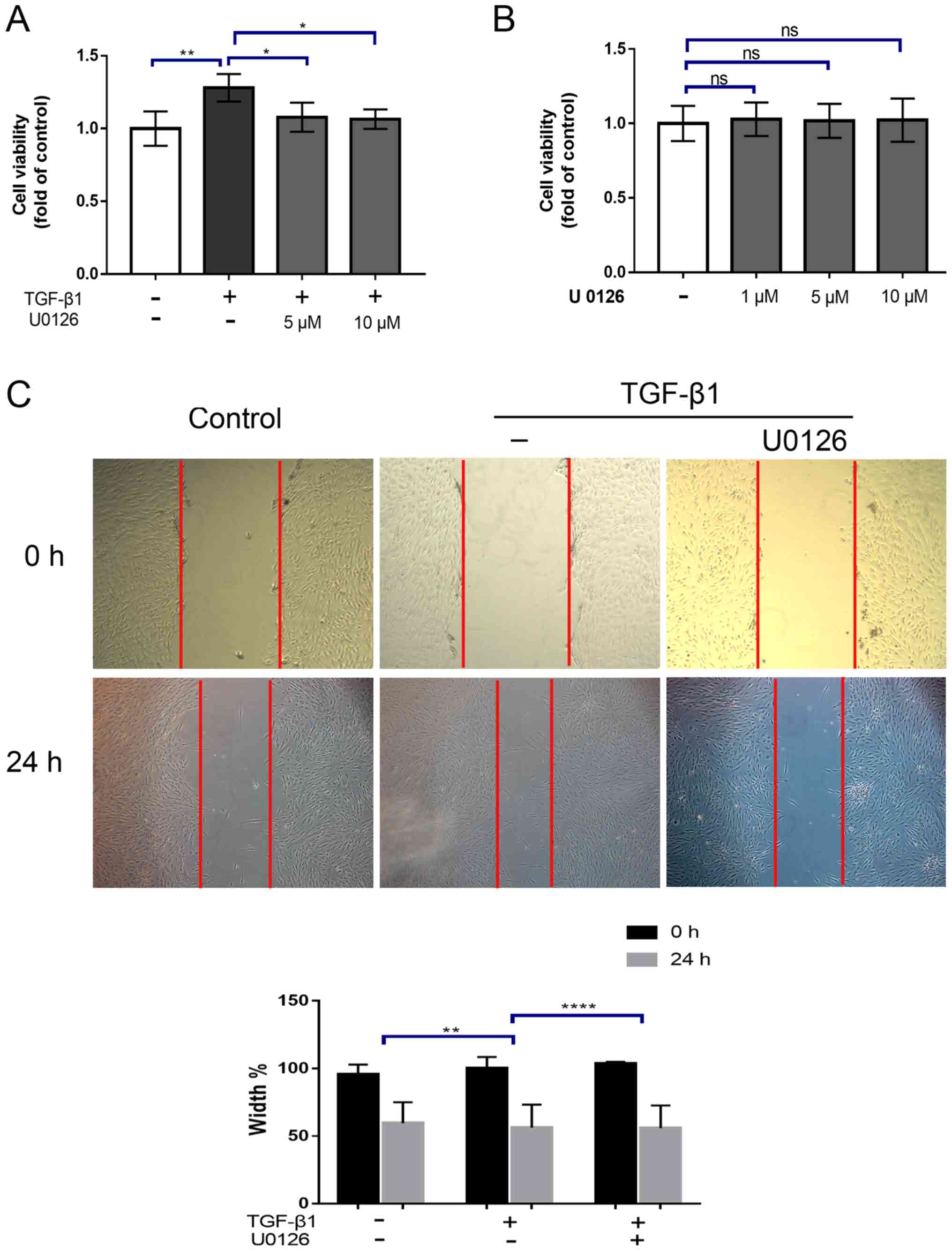
U0126 attenuates TGF-β1-induced HTF
proliferation and migration
Activated fibroblasts generally exhibit a high
degree of proliferation and migration. To ascertain whether the
inhibition of MEK signaling may suppress the HTF proliferation
induced by TGF-β1, HTFs were incubated with TGF-β1 (5 ng/ml) alone
or in the presence of 5 or 10 µM of the MEK1/2 inhibitor U0126.
Cell proliferation was determined using a CCK-8 assay. It was
identified that treatment with TGF-β1 for 24 h significantly
increased the proliferation of HTFs (P<0.01; Fig. 2A), which was abrogated by
pre-treatment with U0126. (P<0.05; Fig. 2A). However, no significant
difference in the effects between 5 and 10 µM treatments was
observed. Notably, treatment with U0126 alone had no significant
effect on cell viability (Fig.
2B). Next, the effect of U0126 on HTF migration was evaluated
using a scratch wound assays. As indicated in Fig. 2, cell migration was significantly
increased upon treatment with TGF-β1 for 24 h (relative wound
width=44.2±4.5% TGF-β1 vs. 57.6±2.1% vehicle control; P<0.001;
Fig. 2C). This effect was also
substantially attenuated by pre-treatment with 5 µM U0126 (relative
wound width =73.6±6.1% TGF-1β+U0126 vs. 41.1±4.8% TGF-1β;
P<0.01; Fig. 2C). Collectively,
these results indicated that U0126 may markedly inhibit HTF
proliferation and migration simulated by TGF-β1.
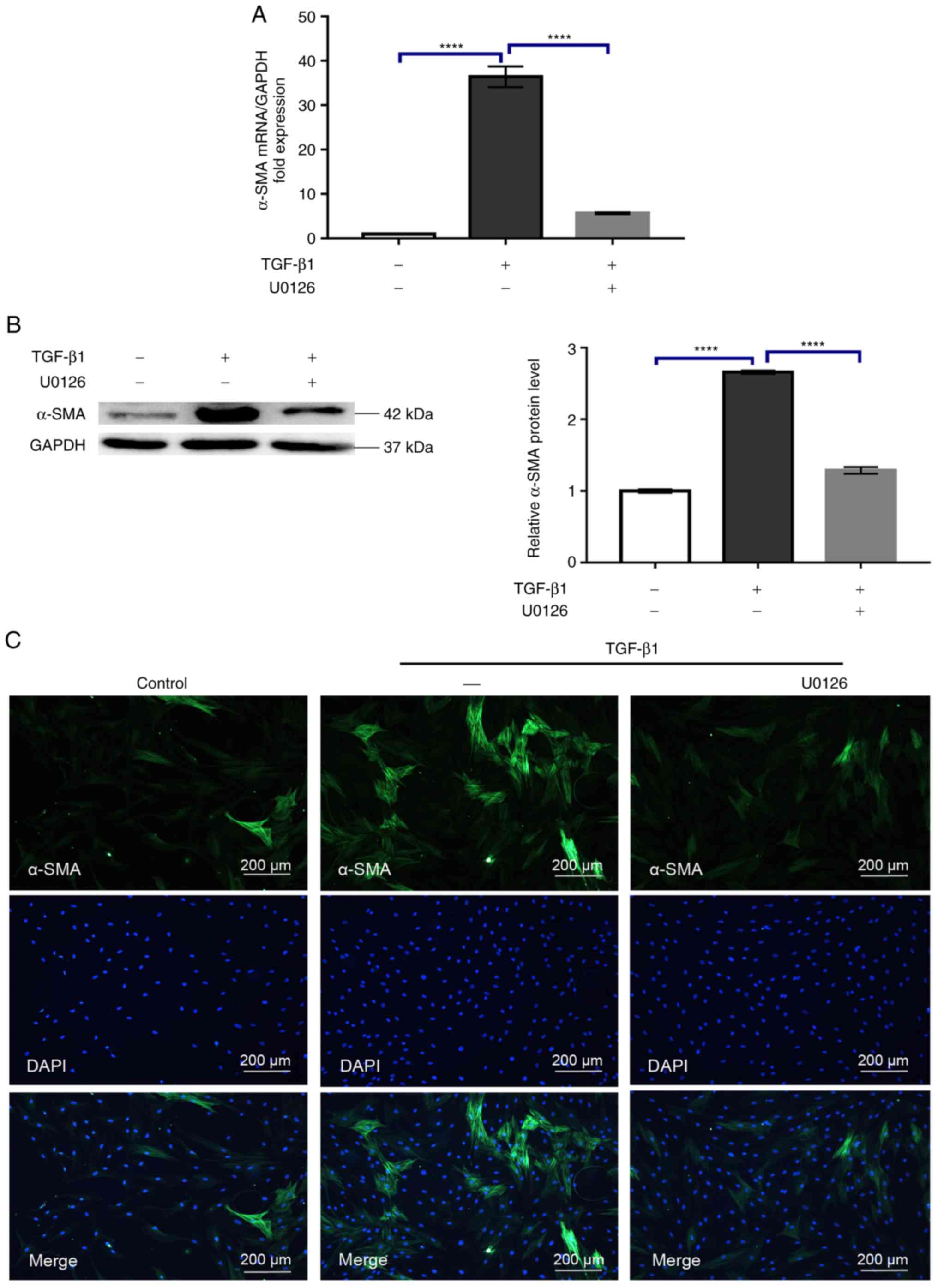
U0126 prevents the conversion of HTFs
into myofibroblasts by TGF-β1
The transdifferentiation of fibroblasts into
myofibroblasts is considered a pivotal step during fibrosis. At the
molecular level, this process is characterized by the upregulation
of α-SMA expression (16). To
determine the effect of U0126 on the TGF-β1-induced conversion of
fibroblasts into myofibroblasts, the changes in α-SMA mRNA and
protein expression were evaluated via RT-qPCR and western blot
analysis, respectively. As indicated in Fig. 3A and B, while TGF-β1 exposure
increased α-SMA mRNA and protein expression, as expected, this
effect was compromised by 5 µM U0126 (P<0.0001). Additional
immunofluorescence was performed to monitor the expression of α-SMA
following treatment with 5 µM U0126, anda similar result was
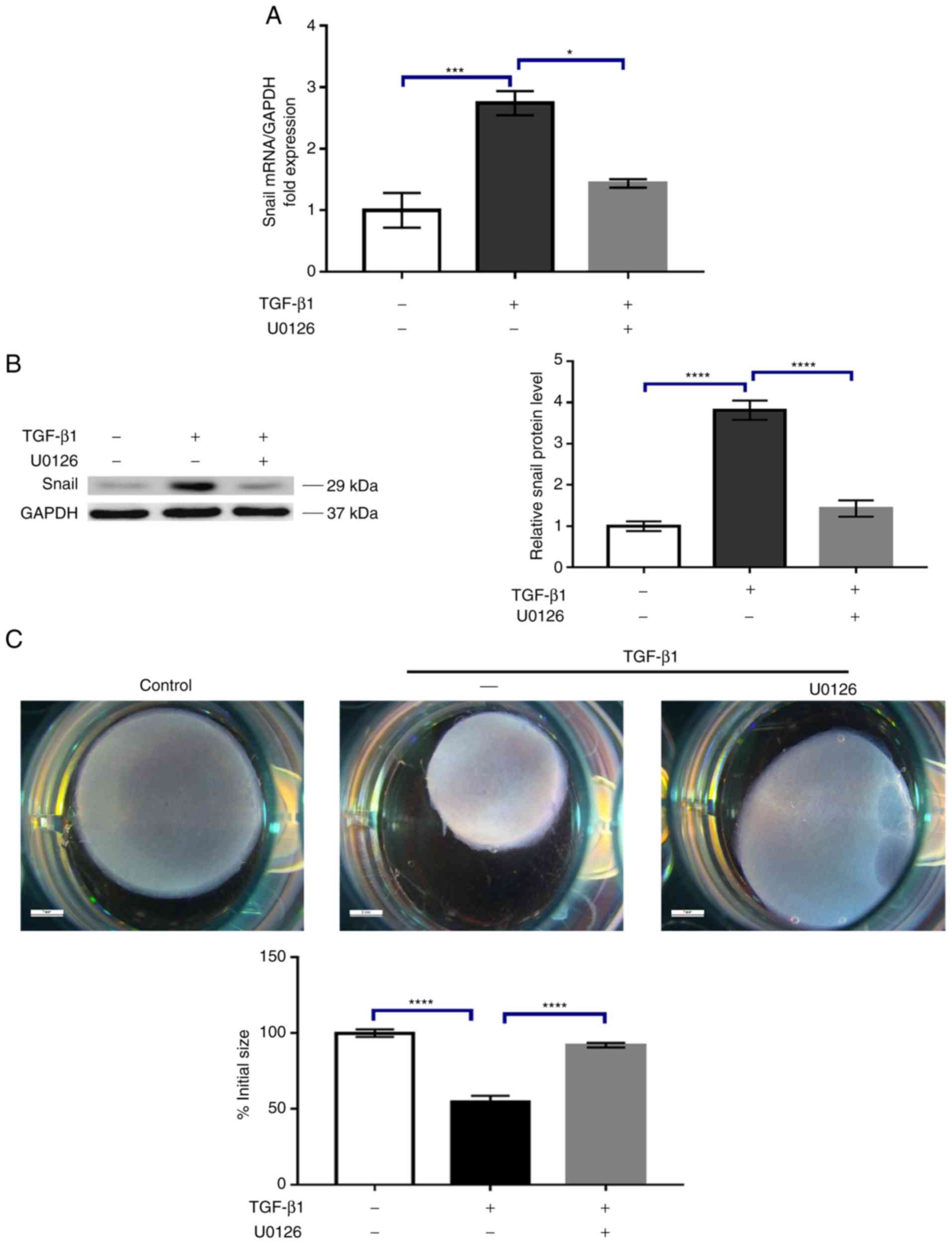
obtained (Fig. 3C). In addition to
α-SMA, previous studies have indicated that Snail, a transcription
factor that serves a critical role in epithelial-mesenchymal
transition (EMT), is also involved in the switching of fibroblasts
into myofibroblasts. Therefore, the effect of U0126 on the
expression of Snail induced by TGF-β1 was examined. The results
demonstrated that, while untreated HTF cells exhibited low levels
of Snail expression, incubation with 5 ng/ml TGF-β1 resulted in a
marked increase in Snail expression (Fig. 4A and B). Similarly, 5 µM U0126
largely attenuated TGF-β1-induced Snail mRNA and protein expression
(Fig. 4A and B). At the functional
level, activated HTFs contribute to contraction of the
extracellular matrix, a hallmark of scar formation (17). Therefore, whether U0126 may inhibit
the collagen matrix contraction response induced by TGF-β1 was
determined. As expected, TGF-β1 treatment lead to enhanced collagen
matrix contraction compared with the vehicle control (P<0.0001;
Fig. 4C). Treatment with 5 µM
U0126 significantly decreased the collagen matrix contraction
induced by TGF-β1 (P<0.0001; Fig.
4C). Altogether, these results support the hypothesis that
U0126 may potently prevent HTF transdifferentiation into
myofibroblasts.
U0126 suppressed the TGF-β1-stimulated
phosphorylation of Smad2/3, P38MAPK, and ERK1/2 in HTFs
TGF-β1 has been demonstrated to mediate the
canonical Smad2/3 and the non-canonical MAPK pathways in various
cell types. To reveal the mechanism underlying U0126-inhibition of
HTF activation, the activation status of these signaling pathways
in HTFs was evaluated. Smad2/3 and P38MAPK phosphorylation levels
were detected by western blot analysis, and the results suggested
that treatment with 5 ng/ml TGF-β1 induced activation of the
Smad2/3 signaling pathway, as reflected by the increased levels of
TGF-β1 mRNA and protein expression and Smad2/3 phosphorylation,
which was in agreement with previous studies (18,19)
(Fig. 5A). Furthermore, the
phosphorylation of non-canonical p38MAPK and ERK1/2 proteins was
also stimulated by TGF-β1 (Fig.
5B). Exposure to 5 µM U0126 markedly impaired TGF-β1-induced
TGF-β1 mRNA and protein expression, and the phosphorylation of
Smad2/3, p38MAPK and ERK1/2 (Fig. 5A
and B). Therefore, the data suggested that U0126 prevents HTF
activation through suppression of the Smad2/3, p38MAPK and ERK1/2
signaling pathways.
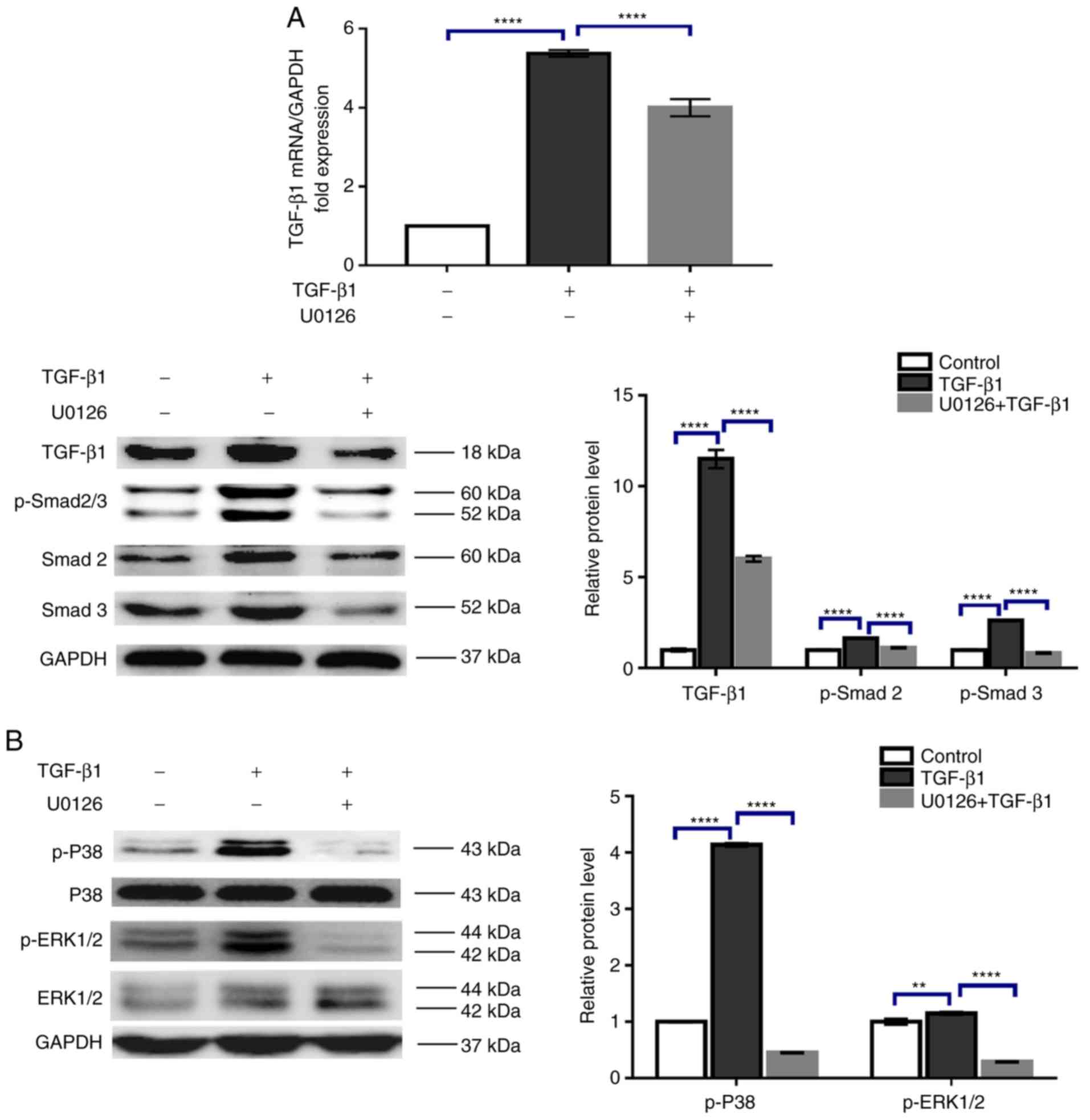 | Figure 5.U0126 counteracts the Smad2/3 and
P38MAPK phosphorylation induced by TGF-β1. HTFs were exposed to
TGF-β1 (5 ng/ml) in the presence or absence of U0126 (5 µM) for 24
h. The mRNA and protein expression levels of (A) TGF-β1 and
p-Smad2/3, and (B) p-P38MAPK and p-ERK1/2 were analyzed by reverse
transcription quantitative polymerase chain reaction and western
blot analysis, and normalized to total Smad2/3, ERK1/2 or GAPDH,
respectively. Data are presented as the means ± standard deviation
of 3 independent repeats. **P<0.01 and ****P<0.0001 vs. the
TGF-β1 or control group. HTFs, human tenon fibroblasts; TGF-β1,
transforming growth factor β1; Smad 2/3, mothers against
decapentaplegic homolog 2/3; P38MAPK, p38 mitogen-activated protein
kinase; ERK1/2, extracellular signal-regulated kinase 1/2; p-,
phosphorylated. |
Discussion
In the present study, it was observed that U0126, a
specific MEK1/2 inhibitor, has a potent antifibrotic effect among
cultured HTFs stimulated by TGF-β1. Functionally, U0126 prevented
TGF-β1-induced HTF proliferation, migration and 3D collagen
contraction responses. U0126 may inhibit HTF conversion into
myofibroblasts by downregulating α-SMA and Snail expression. The
phosphorylation of Smad2/3, ERK1/2 and P38MAPK was significantly
attenuated by U0126 treatment, suggesting that U0126 inhibits HTF
activation via suppressing the classical and non-classical
signaling pathways of TGF-β1.
The activation of fibroblasts is a typical
characteristic of fibrotic injury, not only in the ocular region
but in the whole body (20).
Therefore, targeting the inhibition of fibroblast activation has
been regarded as a critical intervention strategy for fibrotic
diseases. In ocular tissue, although certain anti-mitotic agents
including MMC and 5-FU have been employed to contract
trabeculectomy-associated subconjunctival fibroblasts (21,22),
they often result in serious postoperative complications, including
bleb leakage, hypotony maculopathy and endophthalmitis (23,24).
The data indicating that U0126 potently prevents HTF activation
indicates an alternative targeting approach for this disease. It
should be noted that a concentration-dependent inhibitory effect of
U0126 on HTF activation, at least for cell proliferation, was not
observed. Previous studies in other cell types suggested that U0126
prevented TGF-β-induced cell proliferation in a dose-dependent
manner, but not α-SMA and Collagen 1 expression (25,26).
Therefore, it was hypothesized that the concentration-dependent
effect of U0126 may depend on the specific cellular events
evaluated.
As with any drug, the potential side effects of
U0126 should be considered when assessing its effect on the
Ras/rapidly accelerated fibrosarcoma (Raf)/MEK/ERK signaling
pathway. In particular, the possible effect of U0126 on retinal
ganglion cell (RGC) survival, which is closely associated with
glaucoma pathogenesis (27).
However, given that the role of Raf/MEK/ERK signaling pathway
inhibition in RGC survival remains controversial, with several
studies indicating a pro-apoptosis effect and others a protective
action (28–30), whether the use of U0126 may have an
adverse or beneficial effect on RGC survival requires additional
investigation.
The transdifferentiation of fibroblasts into
myofibroblasts is a hallmark of fibroblast activation. At the
molecular level, myofibroblasts are characterized by increased
α-SMA expression (31), the actin
isoform typically expressed in smooth muscle cells. Furthermore,
the Snail transcription factor usually involved in EMT has been
demonstrated to participate in myofibroblast transdifferentiation
(32). In the present study, it
was identified that TGF-β1 augmented the expression of α-SMA and
Snail at both the mRNA and protein levels in HTFs. These effects
were primarily inhibited by pre-treatment with U0126. Furthermore,
the inhibitory effect of U0126 at the functional level was verified
by performing 3D collagen lattice contraction experiments, which
mimic the situation in vivo during wound healing, and
obtained a consistent result. The results of the present study are
in agreement with several other studies indicating that U0126
inhibits fibroblast transdifferentiation in other tissues (33,34),
suggesting that U0126 may serve as an anti-fibrotic agent in
general.
TGF-β1 acts by binding to two serine/threonine
receptors, namely TGF-β type I and type II receptors, in target
cells, which results in the activation of multiple signaling
cascade pathways, including the canonical Smad2/3 pathway and
non-canonical MAPK signaling pathways including p38MAPK, ERK1/2 and
JNK (35,36). In general, activated Smad2/3 form
complexes with Smad4, subsequently translocating to the nucleus and
regulating the expression of genes associated with fibroblast
proliferation, migration and extracellular matrix deposition
(37). In addition to the
canonical pathway, there is increasing evidence to support that the
non-canonical pathway also has a vital role in mediating
TGF-β1-induced cellular functions in the fibroblasts of numerous
tissue types (38,39). As for HTFs, it has been suggested
that the inhibition of p38MAPK is sufficient to prevent the
conversion of HTFs into myofibroblasts (14). In the present study, it was
demonstrated that treatment with U0126 significantly impeded
TGF-β1-induced Smad2/3, p38MAPK and ERK1/2 activation. The data
suggested that the canonical Smad2/3 and non-canonical p38MAPK and
ERK1/2 signaling pathways are critical for maintaining the
activated phenotype of HTFs, and that the inhibition of one of
these pathways is sufficient to restore the phenotype activated by
TGF-β1. We speculate that this phenomenon may be explained by
potential crosstalk between Smad2/3, p38MAPK and ERK1/2 pathways in
HTFs. For example, data obtained with other cell types indicate
that p38MAPK may phosphorylate and increase the transcription of
Smad2/3 (40,41). Furthermore, ERK1/2 has been
revealed to regulate Smad2/3 activity in chondrocytes and hepatoma
cells (42,43). Therefore, ERK1/2 may function as an
upstream signal kinase of Smad2/3 in HTFs.
In summary, the present study provided evidence that
the inhibition of the MEK signaling pathway has an effect on
TGF-β1-induced HTF activation. This result provides an experimental
basis for the clinical application of MEK inhibitors in the
treatment of trabeculectomy-associated fibrotic disease.
Acknowledgements
Not applicable.
Funding
This work is funded by Guangdong Natural Science
Foundation (grant no. 2015A030313171), National Natural Science
Foundation of China (grant no. 81200685), Guangdong Province
Science & Technology Plan (grant no. 2014B020228002) and
Program for Outstanding Young Teacher of Sun Yat-sen University
(grant no. 17ykpy76). The funders had no role in study design, data
collection, and analysis, decision to publish, or preparation of
the manuscript.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
JW and XL conceived and performed the experiments.
JW performed the statistical analysis. XL and MY wrote the
manuscript with input from WG, BQ, YL and RL. All authors approved
the final version of the manuscript for publication.
Ethics approval and consent to
participate
The present study was approved by the Ethical
Committee of the Zhongshan Ophthalmic Center and performed in
accordance with the Declaration of Helsinki. Informed consent was
obtained.
Patient consent for publication
Written informed consent was obtained.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Fan Gaskin JC, Nguyen DQ, Soon Ang G,
O'Connor J and Crowston JG: Wound healing modulation in glaucoma
filtration surgery-conventional practices and new perspectives: The
role of antifibrotic agents (Part I). J Curr Glaucoma Pract.
8:37–45. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pimentel E and Schmidt J: Is mytomicyn
better than 5-fluorouracil as antimetabolite in trabeculectomy for
glaucoma? Medwave. 18:e71372018.(In English, Spanish). View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kitazawa Y, Kawase K, Matsushita H and
Minobe M: Trabeculectomy with mitomycin. A comparative study with
fluorouracil. Arch Ophthalmol. 109:1693–1698. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi H, Wang H, Fu S, Xu K, Zhang X, Xiao Y
and Ye W: Losartan attenuates scar formation in filtering bleb
after trabeculectomy. Invest Ophthalmol Vis Sci. 58:1478–1486.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chang L, Crowston JG, Cordeiro MF, Akbar
AN and Khaw PT: The role of the immune system in conjunctival wound
healing after glaucoma surgery. Surv Ophthalmol. 45:49–68. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hinz B, Phan SH, Thannickal VJ, Prunotto
M, Desmoulière A, Varga J, De Wever O, Mareel M and Gabbiani G:
Recent developments in myofibroblast biology: Paradigms for
connective tissue remodeling. Am J Pathol. 180:1340–1355. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hartupee J and Mann DL: Role of
inflammatory cells in fibroblast activation. J Mol Cell Cardiol.
93:143–148. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carthy JM: TGFβ signaling and the control
of myofibroblast differentiation: Implications for chronic
inflammatory disorders. J Cell Physiol. 233:98–106. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nickel J, Ten Dijke P and Mueller TD:
TGF-β family co-receptor function and signaling. Acta Biochim
Biophys Sin (Shanghai). 50:12–36. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin X, Yu M, Wu K, Yuan H and Zhong H:
Effects of pirfenidone on proliferation, migration, and collagen
contraction of human Tenon's fibroblasts in vitro. Invest
Ophthalmol Vis Sci. 50:3763–3770. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li DQ, Lee SB and Tseng SC: Differential
expression and regulation of TGF-beta1, TGF-beta2, TGF-beta3,
TGF-betaRI, TGF-betaRII and TGF-betaRIII in cultured human corneal,
limbal, and conjunctival fibroblasts. Curr Eye Res. 19:154–161.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fu S, Sun L, Zhang X, Shi H, Xu K, Xiao Y
and Ye W: 5-Aza-2′-deoxycytidine induces human Tenon's capsule
fibroblasts differentiation and fibrosis by up-regulating TGF-β
type I receptor. Exp Eye Res. 165:47–58. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stahnke T, Kowtharapu BS, Stachs O,
Schmitz KP, Wurm J, Wree A, Guthoff RF and Hovakimyan M:
Suppression of TGF-β pathway by pirfenidone decreases extracellular
matrix deposition in ocular fibroblasts in vitro. PLoS One.
12:e01725922017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Meyer-Ter-Vehn T, Gebhardt S, Sebald W,
Buttmann M, Grehn F, Schlunck G and Knaus P: p38 inhibitors prevent
TGF-beta-induced myofibroblast transdifferentiation in human tenon
fibroblasts. Invest Ophthalmol Vis Sci. 47:1500–1509. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shu DY and Lovicu FJ: Myofibroblast
transdifferentiation: The dark force in ocular wound healing and
fibrosis. Prog Retin Eye Res. 60:44–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hsu CK, Lin HH, Harn HI, Hughes MW, Tang
MJ and Yang CC: Mechanical forces in skin disorders. J Dermatol
Sci. 90:232–240. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Y, Kimura K, Orita T, Teranishi S,
Suzuki K and Sonoda KH: Inhibition by all-trans-retinoic acid of
transforming growth factor-β-induced collagen gel contraction
mediated by human tenon fibroblasts. Invest Ophthalmol Vis Sci.
55:4199–4205. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Meyer-ter-Vehn T, Sieprath S, Katzenberger
B, Gebhardt S, Grehn F and Schlunck G: Contractility as a
prerequisite for TGF-beta-induced myofibroblast
transdifferentiation in human tenon fibroblasts. Invest Ophthalmol
Vis Sci. 47:4895–4904. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wallace HA and Bhimji SS: Wound, Healing,
Phases, in StatPearls. StatPearls Publishing StatPearls Publishing
LLC; Treasure Island (FL): 2017
|
|
21
|
Reiter C, Wimmer S, Schultheiss A, Klink
T, Grehn F and Geerling G: Corneal epitheliopathy following
trabeculectomy with postoperative adjunctive 5-fluorouracil. Klin
Monbl Augenheilkd. 227:887–891. 2010.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fan Gaskin JC, Nguyen DQ, Soon Ang G,
O'Connor J and Crowston JG: Wound healing modulation in glaucoma
filtration surgery-conventional practices and new perspectives:
Antivascular endothelial growth factor and novel agents (Part II).
J Curr Glaucoma Pract. 8:46–53. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bindlish R, Condon GP, Schlosser JD,
D'Antonio J, Lauer KB and Lehrer R: Efficacy and safety of
mitomycin-C in primary trabeculectomy: Five-year follow-up.
Ophthalmology. 109:1336–1342. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Khaw PT, Sherwood MB, Doyle JW, Smith MF,
Grierson I, McGorray S and Schultz GS: Intraoperative and post
operative treatment with 5-fluorouracil and mitomycin-c: Long term
effects in vivo on subconjunctival and scleral fibroblasts. Int
Ophthalmol. 16:381–385. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang HH, Chang MC, Wu IH, Huang GF, Huang
WL, Wang YL, Lee SY, Yeh CY, Guo MK, Chan CP, et al: Role of
ALK5/Smad2/3 and MEK1/ERK signaling in transforming growth factor
beta 1-modulated growth, collagen turnover and differentiation of
stem cells from apical papilla of human tooth. J Endod.
41:1272–1280. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen X, Ye S, Xiao W, Wang W, Luo L and
Liu Y: ERK1/2 pathway mediates epithelial-mesenchymal transition by
cross-interacting with TGFβ/Smad and Jagged/Notch signaling
pathways in lens epithelial cells. Int J Mol Med. 33:1664–1670.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nickells RW: The cell and molecular
biology of glaucoma: mechanisms of retinal ganglion cell death.
Invest Ophthalmol Vis Sci. 53:2476–2481. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gupta VK, You Y, Li JC, Klistorner A and
Graham SL: Protective effects of 7,8-dihydroxyflavone on retinal
ganglion and RGC-5 cells against excitotoxic and oxidative stress.
J Mol Neurosci. 49:96–104. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei J, Jiang H, Gao H and Wang G: Raf-1
kinase inhibitory protein (RKIP) promotes retinal ganglion cell
survival and axonal regeneration following optic nerve crush. J Mol
Neurosci. 57:243–248. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Luo JM, Cen LP, Zhang XM, Chiang SW, Huang
Y, Lin D, Fan YM, van Rooijen N, Lam DS, Pang CP and Cui Q:
PI3K/akt, JAK/STAT and MEK/ERK pathway inhibition protects retinal
ganglion cells via different mechanisms after optic nerve injury.
Eur J Neurosci. 26:28–42. 2007. View Article : Google Scholar
|
|
31
|
Hinz B, Mastrangelo D, Iselin CE,
Chaponnier C and Gabbiani G: Mechanical tension controls
granulation tissue contractile activity and myofibroblast
differentiation. Am J Pathol. 159:1009–1020. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Biswas H and Longmore GD: Action of SNAIL1
in cardiac myofibroblasts is important for cardiac fibrosis
following hypoxic injury. PLoS One. 11:e01626362016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reichard JF and Petersen DR: Involvement
of phosphatidylinositol 3-kinase and extracellular-regulated kinase
in hepatic stellate cell antioxidant response and myofibroblastic
transdifferentiation. Arch Biochem Biophys. 446:111–118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Makino T, Jinnin M, Muchemwa FC, Fukushima
S, Kogushi-Nishi H, Moriya C, Igata T, Fujisawa A, Johno T and Ihn
H: Basic fibroblast growth factor stimulates the proliferation of
human dermal fibroblasts via the ERK1/2 and JNK pathways. Br J
Dermatol. 162:717–723. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guo X and Wang XF: Signaling cross-talk
between TGF-beta/BMP and other pathways. Cell Res. 19:71–88. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Pardali E, Sanchez-Duffhues G,
Gomez-Puerto MC and Ten Dijke P: TGF-β-induced
endothelial-mesenchymal transition in fibrotic diseases. Int J Mol
Sci. 18(pii): E21572017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zeglinski MR, Roche P, Hnatowich M, Jassal
DS, Wigle JT, Czubryt MP and Dixon IM: TGFβ1 regulates Scleraxis
expression in primary cardiac myofibroblasts by a Smad-independent
mechanism. Am J Physiol Heart Circ Physiol. 310:H239–H249. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Che X, Wang Q, Xie Y, Xu W, Shao X, Mou S
and Ni Z: Astragaloside IV suppresses transforming growth factor-β1
induced fibrosis of cultured mouse renal fibroblasts via inhibition
of the MAPK and NF-κB signaling pathways. Biochem Biophys Res
Commun. 464:1260–1266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kamaraju AK and Roberts AB: Role of
Rho/ROCK and p38 MAP kinase pathways in transforming growth
factor-beta-mediated Smad-dependent growth inhibition of human
breast carcinoma cells in vivo. J Biol Chem. 280:1024–1036. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Iwayama H, Sakamoto T, Nawa A and Ueda N:
Crosstalk between Smad and mitogen-activated protein kinases for
the regulation of apoptosis in cyclosporine A- induced renal
tubular injury. Nephron Extra. 1:178–189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhu Y, Gu J, Zhu T, Jin C, Hu X and Wang
X: Crosstalk between Smad2/3 and specific isoforms of ERK in
TGF-β1-induced TIMP-3 expression in rat chondrocytes. J Cell Mol
Med. 21:1781–1790. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Boye A, Kan H, Wu C, Jiang Y, Yang X, He S
and Yang Y: MAPK inhibitors differently modulate TGF-β/Smad
signaling in HepG2 cells. Tumour Biol. 36:3643–3651. 2015.
View Article : Google Scholar : PubMed/NCBI
|