Introduction
The vestibular system is responsible for the
perception of spatial location, posture and movement, and its
dysfunction or selective impairment leads to vestibular deficits,
including motion impairment, postural deviation, disorientation,
nystagmus, vertigo and dizziness (1–3),
during which apoptotic mediators promote those pathological
alterations (4). Vestibular
compensation is considered to be behavioral recovery from lesions
to the peripheral vestibular system, and is widely used to
investigate the plasticity of central vestibular system and
recuperation of vestibular functions (5).
Brain-derived neurotrophic factor (BDNF) regulates
synaptic transmission and plasticity (6), contributes to neurogenesis (7), and is involved in spatial orientation
(8), motor and cognitive skills
(9); thus, it has been closely
associated with the development of the vestibular system. BDNF, an
activity-dependent release substance, functions through binding to
its affinity receptor tyrosine receptor kinase B (TrkB) in the
vestibular system (5). BDNF
deficiency causes severe loss and degeneration of vestibular
neurons, and contributes to vestibular dysfunction (10,11).
A previous study demonstrated that the upregulation of BDNF
accelerates the process of vestibular compensation (7). Therefore, BDNF appears to be
important in vestibular compensation.
In previous years, corticosteroids have been
identified to have beneficial effects on symptoms and vestibular
functions (12), however, evidence
of corticosteroid therapy for vestibular disorders remains
insufficient (13,14). Dexamethasone (DXMS), a well-known
glucocorticoid, exerts a high therapeutic index for activation of
vestibular nucleus neurons, and subsequently accelerates vestibular
compensation (15). However, to
the best of our knowledge, detailed elucidation of the roles of
DXMS and its mechanisms in vestibular compensation has been rare.
In the present study, an arsanilic acid (AA)-induced rat vestibular
dysfunction model was established to investigate whether DXMS is a
therapeutic option for this disease. Following DXMS (1 and 3
mg/kg/day) treatment, vestibular symptoms, behavioral scores and
brain damage were assessed, and the expression levels and
distributions of BDNF, tyrosine receptor kinase B (TrkB) and
K+/Cl− cotransporter isoform 2 (KCC2),
cleaved-caspase 3 and c-Jun NH 2-terminal kinase (JNK)1/2 were
detected in brain sections. In addition, RU486, an inhibitor of
glucocorticoid receptor, was introduced for intervention of the
effects of DXMS. The data obtained indicated that DXMS was
effective in alleviating AA-induced rat vestibular dysfunction, and
the activation of BDNF and inhibition of JNK1/2 singling pathways
were the underlying mechanism. Further investigations of DXMS in
human vestibular compensation are required.
Materials and methods
Rat behavioral observation
A total of 30 male Sprague-Dawley rats (6–8 weeks
old; weighing 200–250 g) were purchased from Jrdun Biotechnology
(Shanghai) Co., Ltd., Shanghai, China. The rats were kept at 21±2°C
and 50% humidity with 12-h light/dark cycles for a week prior to
treatment, with free access to food and water. Vestibular
dysfunction was established in rats by intratympanic injection of
AA (100 mg/ml; 0.05 ml; Aladdin Bio-Chem Technology Co., Ltd.,
Shanghai, China) (16). After 3
days, behavioral observation was performed for 1 h, and vestibular
scores were subsequently assessed through the following behavioral
assessments.
Air righting reflex
A sponge was spread on a flat surface and the
animals were placed in a supine position in the air 50 cm away
above the sponge, following which the animal was dropped
horizontally. Four-legged smooth landing on the ground was
considered as positive (normal animal), whereas, the body or other
regions landing first were considered vestibular dysfunction
animals. The positive landing rate (%) of each rat in each group
was recorded.
Contact righting reflex
The rats were placed in a syringe core tube in a
supine position, following which the recovery time for position
correction was recorded. Normal rats turned over their body
position in a few seconds. However, rats with vestibular
dysfunction exhibited difficultly in identifying the inverted
position, and thus failed to flip, or turn over their body position
with prolonged time.
Head tilt and swimming behavior
rating
The rats were placed in the center of a water
bottle. The swimming behavior of animals within 2 min was observed.
The grading standards for rat behaviors were as follows: 0 points,
no dysfunction (no head tilt, straight swimming, reaching the wall
or climbing); 1 point, marginal dysfunction (no obvious head
deviation, some circular swimming, not always reaching the wall); 2
points, moderate dysfunction (marginal head deviation, frequent
circular swimming, not always reaching the wall); and 3 points,
serious dysfunction (obvious head deviation, circular swimming, not
reaching the wall).
Experimental groups
The rats were randomly divided into the following
groups (n=6/group): i) Rats received vehicle (physiological saline)
and considered the sham group; ii) Rats injected with AA to
successfully induce vestibular dysfunction, considered the AA
group; ii) AA + DXMS (1 mg/kg/day) group rats were subjected to
AA-induced vestibular dysfunction, and then received DXMS
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at a dose of 1
mg/kg/day for 3 days through intraperitoneal injection; iv) AA +
DXMS (3 mg/kg/day) group rats were treated as for the AA + DXMS (1
mg/kg/day) group, but were administered with DXMS (3 mg/kg/day)
following AA-induced vestibular dysfunction; v) AA + DXMS + RU486
group rats were treated as the AA + DXMS (3 mg/kg/day) group, but
were additionally administered RU486 (Sigma-Aldrich; Merck KGaA) at
a dose of 100 mg/kg/day through subcutaneous injection into the
nape of the neck. On the fourth day, behavioral observations of the
rats in each group were performed, followed by the vestibular score
assay. Subsequently, the rats were sacrificed, and brain tissue was
collected for the subsequent experiments.
Hematoxylin-eosin (HE) staining
A total of six rats in each group were sacrificed to
undergo thoracotomy. A tube was inserted at ascending aorta,
perfused with 4% paraformaldehyde and the brain tissue was removed.
At 4 mm from the optic chiasm, coronal sections (4-mm) were
archived, routinely fixed with 10% formalin for 48 h at room
temperature, washed three times with PBS (3 min/wash), dehydrated,
paraffin-embedded and subsequently cut into sections. The 4-µm
sections underwent HE staining at room temperature for 30 min and
an Olympus CX41 light microscope (Olympus Corporation, Tokyo,
Japan; magnification, ×200) was used for visualization.
Terminal deoxyribonucleotidyl
transferase-mediated biotinylated dUTP nick-end labeling (TUNEL)
analysis
Following deparaffinization in xylene and
rehydration in 100, 95, 85 and 75% gradient alcohol and double
distilled water for 3 min each time, brain tissue sections (4–7 µm)
placed on glass slides were incubated with 50 µl TUNEL solution
(Roche Diagnostics, Basel, Switzerland) at 37°C for 1 h. Subsequent
to washing three times in PBS (3 min/wash), the slides were stained
using a DAB substrate kit (cat. no. FL-6001; Shanghai Long Island
Biotech Co., Ltd.) for 5 min at 37°C followed by counterstaining
with hematoxylin (cat. no. 714094; BASO Diagnostic, Inc.) to stain
nuclei at 37°C for 10 min, and finally mounted with neutral balsam
(Beijing Solarbio Science & Technology Co., Ltd., Beijing,
China) at 37°C for another 24 h.
Apoptosis-positive cells were characterized by
brown-yellow granules in the nucleus. A light microscope Nikon
Eclipse Ni-U (Nikon Corporation. Tokyo, Japan; magnification, ×200)
equipped with a Nikon Ds-Ri2 microscopy digital camera was used for
visualization. The area of apoptosis-positive cells was calculated
using Image Pro-Plus 6.0 software (Media Cybernetics, Inc.,
Rockville, MD, USA) in 10 randomly selected microscopic fields per
specimen.
Immunohistochemistry (IHC) assay
Following blocking with 1% bovine serum albumin
[Sangon Biotech (Shanghai) Co., Ltd., Shanghai, China] at room
temperature for 30 min, the slides were incubated with primary
antibodies: Mouse anti-BDNF (1:10,000; cat. no. ab203573; Abcam,
Cambridge, MA, USA), rabbit anti-TrkB (1:100; cat. no. ab18987;
Abcam) and mouse anti-KCC2 (1:200; cat. no. NBP2-59337; Novus
Biologicals, LLC, Littleton, CO, USA) for 1 h at room temperature,
followed by horseradish peroxidase (HRP)-conjugated mouse
anti-rabbit antibody (cat. no. D-3004; Shanghai Long Island Biotech
Co., Ltd., Shanghai, China) for 20 min at 25°C. The slides were
stained using a DAB-containing substrate kit (cat. no. FL-6001;
Shanghai Long Island Biotech Co., Ltd.) to develop a brown color
followed by hematoxylin staining (cat. no. 714094; BASO Diagnostic,
Inc., Wuhan, China) for 3 min at room temperature. A fluorescence
microscope (Leica Microsystems, Inc., Buffalo Grove, IL, USA;
magnification, ×200) was used to calculate regions of BDNF-, TrKB-
or KCC2-positive cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The mRNA and protein expression levels of BDNF,
TrkB, KCC2, cleaved-caspase 3, JNK1/2 and phosphorylated (p)-JNK1/2
were assessed by RT-qPCR and western blot analyses, respectively,
according to a previous study (5).
Total RNA from brain tissues was extracted using TRIzol®
Reagent (cat. no. 1596-026; Invitrogen; Thermo Fisher Scientific,
Inc.) and reverse transcribed using a cDNA synthesis kit (cat. no.
K1622; Fermentas; Thermo Fisher Scientific, Inc.). mRNA expression
levels of BDNF, TrkB, KCC2 and cleaved-caspase 3 were determined
using a SYBR Green PCR kit (Thermo Fisher Scientific, Inc.) on an
ABI Prism 7300 SDS Software (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with amplification condition programed as: 95°C
for 10 min; 40 cycles for 95°C for 15 sec; 60°C for 45 sec; 95°C
for 15 sec; 60°C for 1 min; 95°C for 15 sec; and 60°C for 15 sec.
mRNA expression levels of GAPDH served as an internal control.
Expression was calculated using the 2−∆∆Cq method
(17). The primer pairs targeting
BDNF (GenBank NM_001270630.1), TrkB (GenBank NM_001163168.2), KCC2
(GenBank NM_ NM_134363.1), cleaved-caspase 3 (GenBank NM_012922.2)
and an internal standard GAPDH were as follows: BDNF, forward
5′-GACAAGGCAACTTGGCCTAC-3′ and reverse 5′-TCCAGCAGCTCTTCGATCAC-3′
(position: 452-590; product size: 139 bp); TrkB, forward
5′-ACTGGACCACGCCAACTGAC-3′ and reverse 5′-TCACCACCACGGCATAGACC-3′
(position: 1872-1988; product size: 117 bp); KCC2, forward
5′-ATCGAGATCCTGCTGGCTTAC-3′ and reverse 5′-CTTGACGCCCACAAAGACTAC-3′
(position: 713-865; product size: 153 bp); cleaved-caspase 3,
forward 5′-GGCATCTCCTGTGATTGG-3′ and reverse
5′-CTCAGCACTCTGGGAAAG-3′ (position: 1780-1964; product size: 185
bp); GAPDH, forward 5′-GGAGTCTACTGGCGTCTTCAC-3′ and reverse
5′-ATGAGCCCTTCCACGATGC-3′ (position: 357-593; product size: 237
bp).
Western blot analysis
A total of 30 µg total protein underwent western
blot analysis. Following full lysis of brain tissue (20 mg) in
radioimmunoprecipitation assay buffer (Jrdun Biotechnology
(Shanghai) Co., Ltd.), samples were centrifuged (12,000 × g; 10
min; 4°C). Total protein content in the supernatant was determined
using a Bicinchoninic Acid Protein Assay kit (Beyotime Institute of
Biotechnology, Haimen, China). In total, 30 µg total protein in the
supernatant was separated using 15% SDS-PAGE, and
electrophoretically pure of BDNF, TrkB, KCC2, cleaved-caspase 3,
JNK1/2 and p-JNK1/2 were transferred onto nitrocellulose (NC)
membranes (cat. no. HATF0001; EMD Millipore, Billerica, MA, USA).
Subsequent to blocking with 5% nonfat milk at room temperature for
1 h, NC membranes were incubated with antibody against BDNF (1:300
dilution; cat. no. ab203573; Abcam), antibody against TrKB (1:500;
cat. no. ab18987; Abcam), antibody against KCC2 (1:200; cat. no.
NBP2-59337; Novus Biologicals, LLC), antibody against caspase 3
(1:500; cat no. ab44976; Abcam), antibody against JNK1/2 (1:1,000;
cat. no. 9252; Cell Signaling Technology, Inc., Danvers, MA, USA),
antibody against p-JNK1/2 (1:1,000; cat. no. 9255; Cell Signaling
Technology, Inc.) and antibody against GAPDH (1:2,000; cat. no.
5174; Cell Signaling Technology, Inc.) at 4°C overnight followed by
incubation with HRP-conjugated secondary antibody (1:1,000;
Beyotime Institute of Biotechnology) at 37°C for 1 h.
Immunoreactive bands were analysed using an enhanced
chemiluminescent system (GE Healthcare, Chicago, IL, USA) with
ImageJ software (Version 1.6; National Institutes of Health,
Bethesda, MD, USA). Protein expression levels of BDNF, TrKB, KCC2,
caspase 3 and JNK1/2 were normalized using GAPDH, whereas, p-JNK1/2
was normalized using JNK1/2.
Statistical analysis
GraphPad Prism 7 software (GraphPad Software, Inc.,
La Jolla, CA, USA) was used for graphing. Values are presented as
the mean ± standard deviation (n=3). Significant differences
between groups were calculated using one-way analysis of variance
with Tukey's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference. For IHC analysis, the areas
of BDNF-, TrkB- and KCC2-positive cells were detected in five
high-power fields (HPF; magnification, ×200). The results based on
the percentage of stained cells were graded as weakly positive
(<25%), medium positive (25–49%) and strongly positive
(>50%). For TUNEL analysis, the percentage of apoptotic cells
were measured in 10 HPF (magnification, ×200), which were graded as
scores of 0 (<10%), 1 (10–25%), 2 (26–50%) and 3 (>50%). The
degree of staining was divided into scores of 0 (none), 1 (light
yellow), 2 (brown yellow) and 3 (brown). The results based on the
addition of those two criteria were considered as positive (≥4
score) and negative (<4 score).
Results
Successful establishment of vestibular
dysfunction
At 3 days following the administration of AA (98%;
100 mg/ml), the rats in the AA group manifested classic symptoms of
vestibular injury, including dizziness, nausea, vomiting,
nystagmus, head-shaking nystagmus, head tilt and postural
instability.
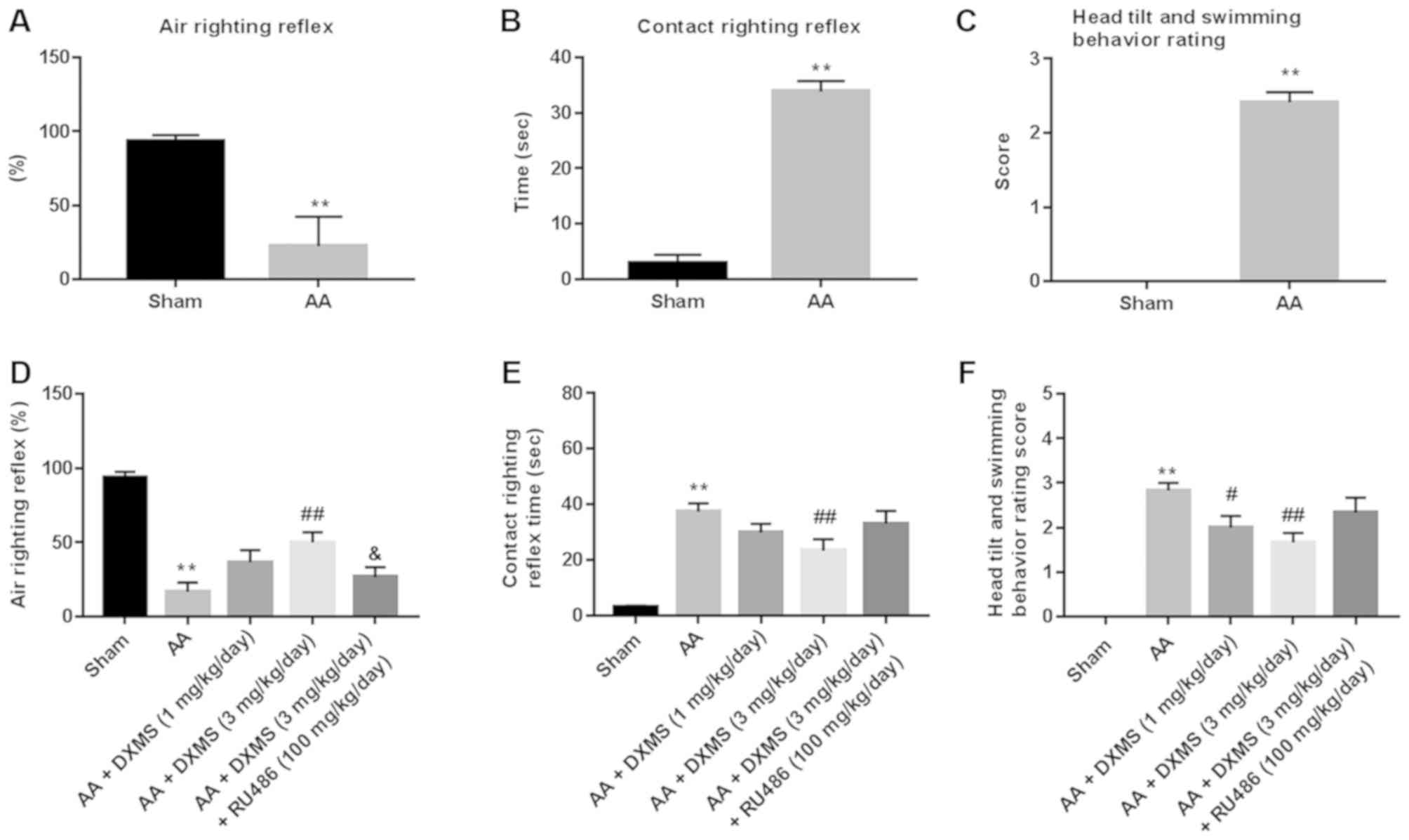
In the air righting reflex test (Fig. 1A), the positive rate of the
AA-treated group was ~25±2%, which was significantly lower compared
with the control group (87±1%). In the contact righting reflex test
(Fig. 1B), the rats in the sham
group returned from a supine position to a normal posture within 5
sec; however, the rats in the AA group took ~30-50 sec, indicating
a significant difference in the righting reflex time between the
two groups (P<0.01). In the head tilt and swimming behavior
rating test (Fig. 1C), the score
of the AA group (score of 3) was significantly higher compared with
the sham group (score of 0). Overall, the symptom observation and
behavioral investigations suggested the successful establishment of
AA-induced rat vestibular dysfunction.
DXMS is a therapeutic drug for
AA-induced rat vestibular dysfunction
Syndromes accompanied with vestibular dysfunction in
the A A + DXMS (1 mg/kg/day) group were marginally alleviated, with
significant improvements in terms of gradual disappearance of head
tilt, postural instability and nystagmus in the AA + DXMS (3
mg/kg/day) group. DXMS moderately increased scores in the air
righting reflex test, reduced the times in the contact righting
reflex test and decreased scores in the head tilt and swimming
behavior rating test, however, with additional RU486 treatment, the
DXMS-induced disappearance on symptoms of vestibular dysfunction
and improvement of vestibular scores were significantly reversed
(Fig. 1D-F).
DXMS reduces damage to brain tissues
in AA-treated rats
In the histopathological analysis (Fig. 1G), the AA group exhibited
disorganized neuron arrangement, widened extracellular spaces,
retracted nuclei, and disappearance of nucleoli when compared with
the corresponding sham group, demonstrating severe damage in the
brain tissue. In the DXMS-treated groups (1 and 3 mg/kg/day), the
extracellular spaces and nuclear condensation were significantly
reduced in a dose-dependent manner, and the cytoplasm and nucleus
were clearer when compared with those in the AA group,
demonstrating that DXMS reduced damage to the brain tissues in the
AA-treated rats. In the TUNEL analysis (Fig. 1H), the apoptotic area (%) of the
brain tissue in the AA group was larger (83.5±1%) compared with the
sham group (33.5±1.7%). However, DXMS significantly decreased the
apoptotic area in a dose-dependent manner with a minimum value
(39.6±0.8%) observed at 3 mg/kg/day. The DXMS-induced
histopathological and apoptotic changes were significantly reversed
with additional RU486 treatment, suggesting that RU486 was an
inhibitor of the therapeutic effect of DXMS on AA-induced rat
vestibular dysfunction.
DXMS activates the BDNF signaling
pathway in AA-treated rats
Activating the BDNF signaling pathway contributed to
vestibular compensation. To examine whether the BDNF signaling
pathway was involved in the protective effect of DXMS on AA-induced
vestibular dysfunction, BDNF signaling pathway-associated BDNF,
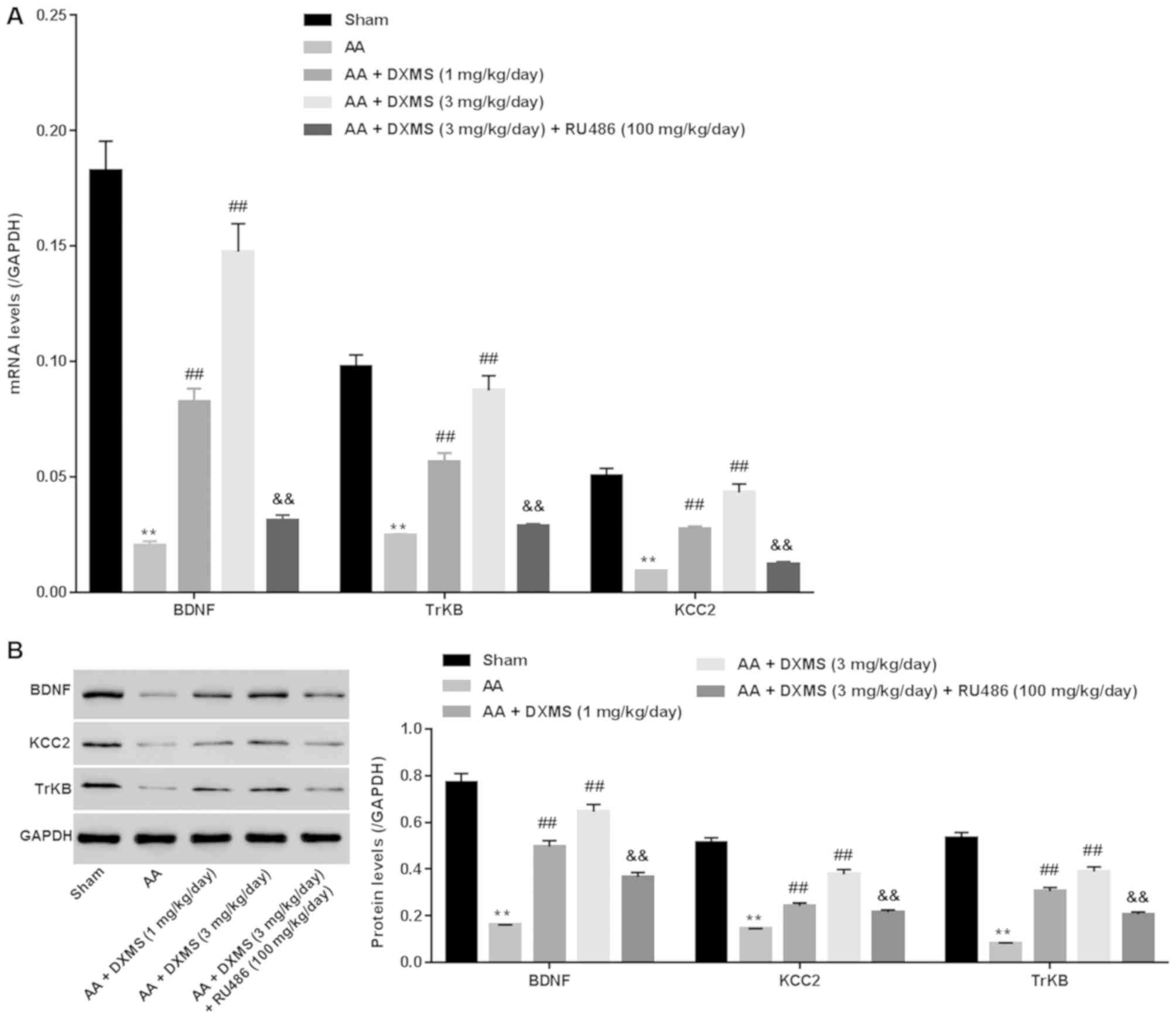
TrkB and KCC2 were assessed. As presented in Fig. 2A and B, significantly decreased
mRNA and protein levels of BDNF, TrkB and KCC2 were obtained in the
AA group when compared with the sham group (P<0.01), confirming
that inhibition of the BDNF signaling pathway was involved in
AA-induced vestibular dysfunction. By contrast, DXMS significantly
increased the mRNA and protein expression levels of BDNF, TrkB and
KCC2 in a dose-dependent manner, the alterations of which were
markedly reversed following additional RU486 treatment. This
suggested that activating the BDNF signaling pathway was the
mechanism by which DXMS exerted its protective effect on AA-induced
vestibular dysfunction.
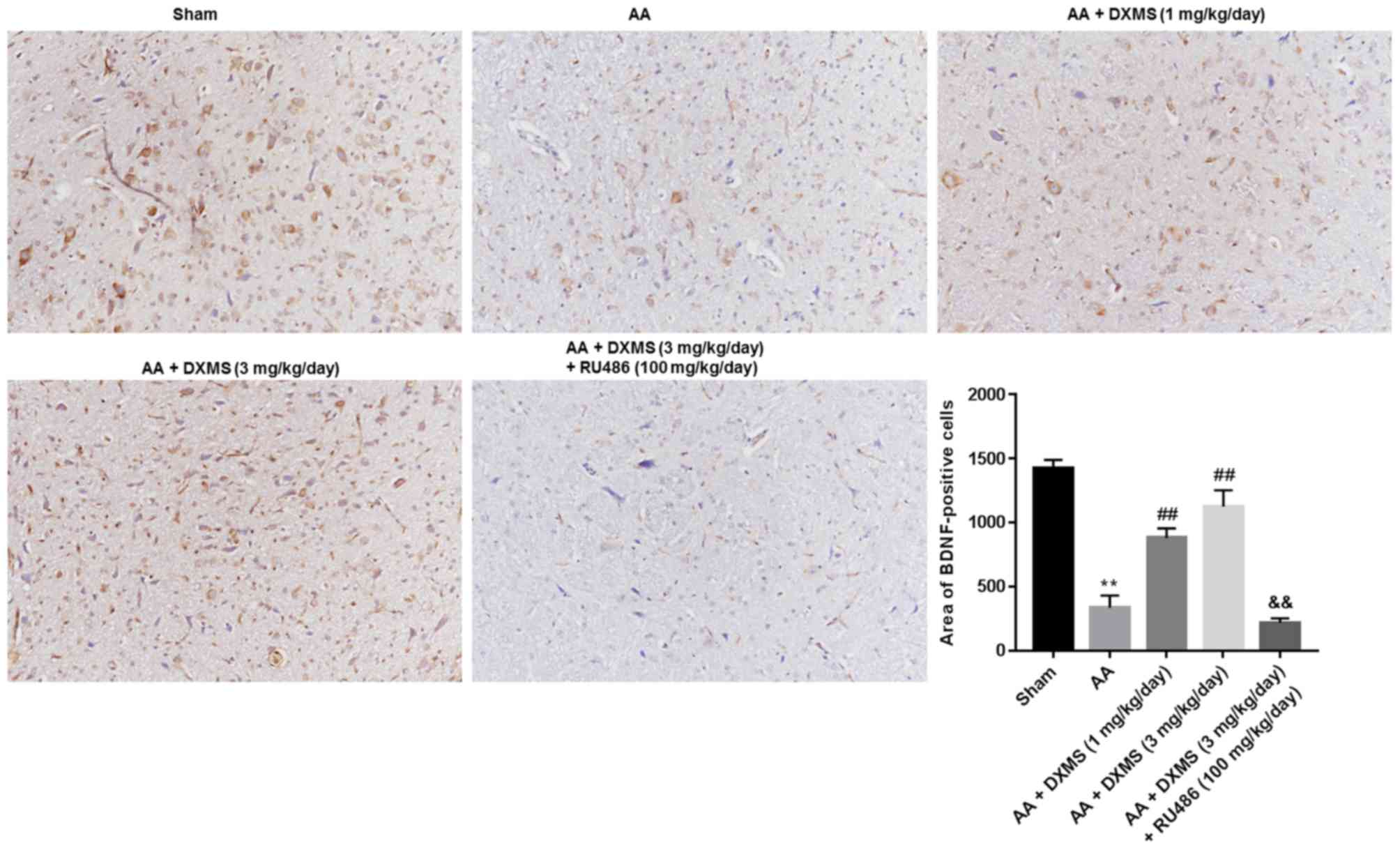
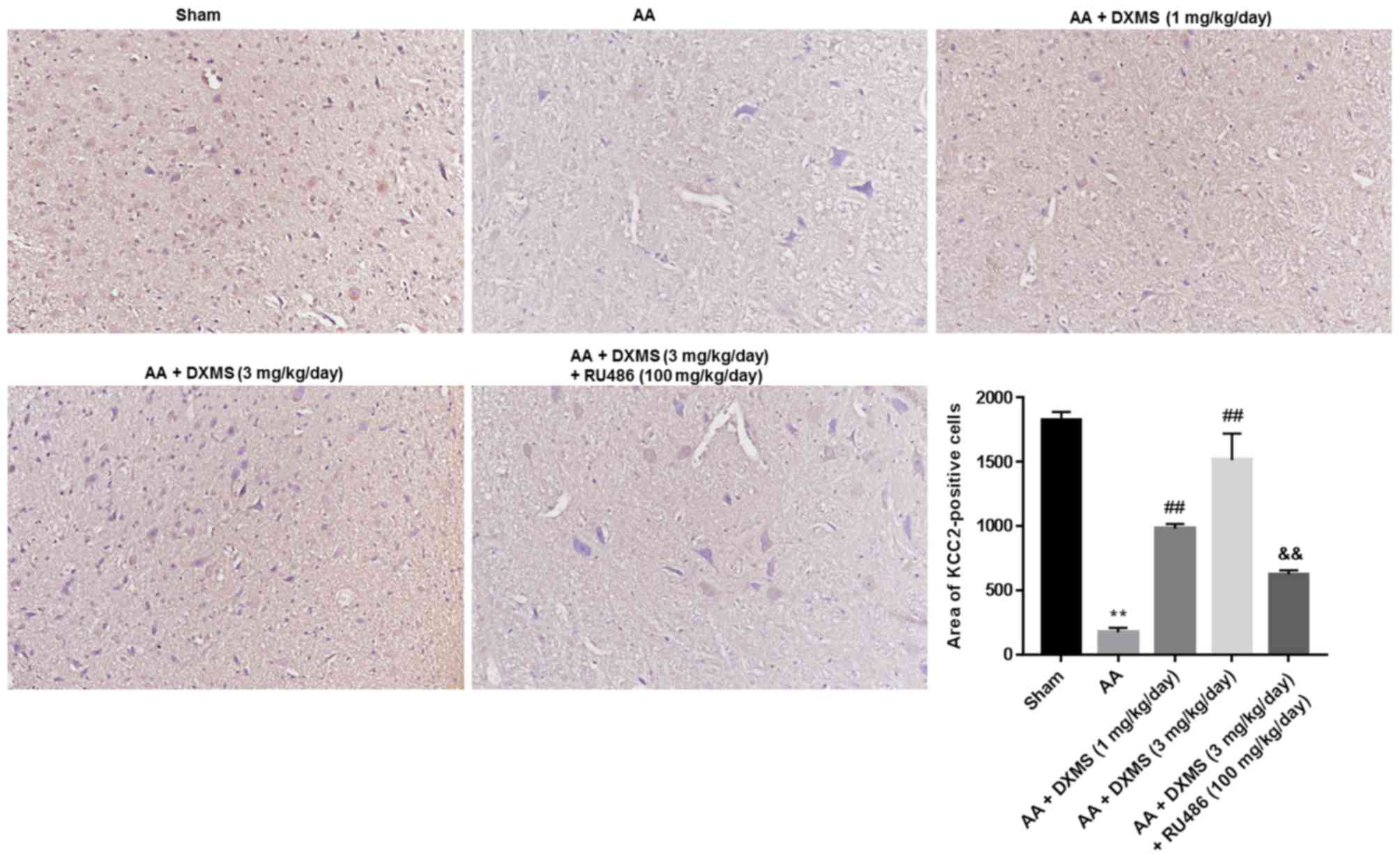
The expression levels and distribution of BDNF, TrkB
and KCC2 in the brain sections were separately determined by an IHC
assay. As demonstrated in Fig. 3,
similar to the mRNA and protein expression levels of BDNF, the
areas of BDNF-positive cells were significantly decreased in the AA
group when compared with the sham group. DXMS enhanced the areas of
BDNF-positive cells in a dose-dependent manner, and reached a peak
value at 3 mg/kg/day, which was significantly decreased with
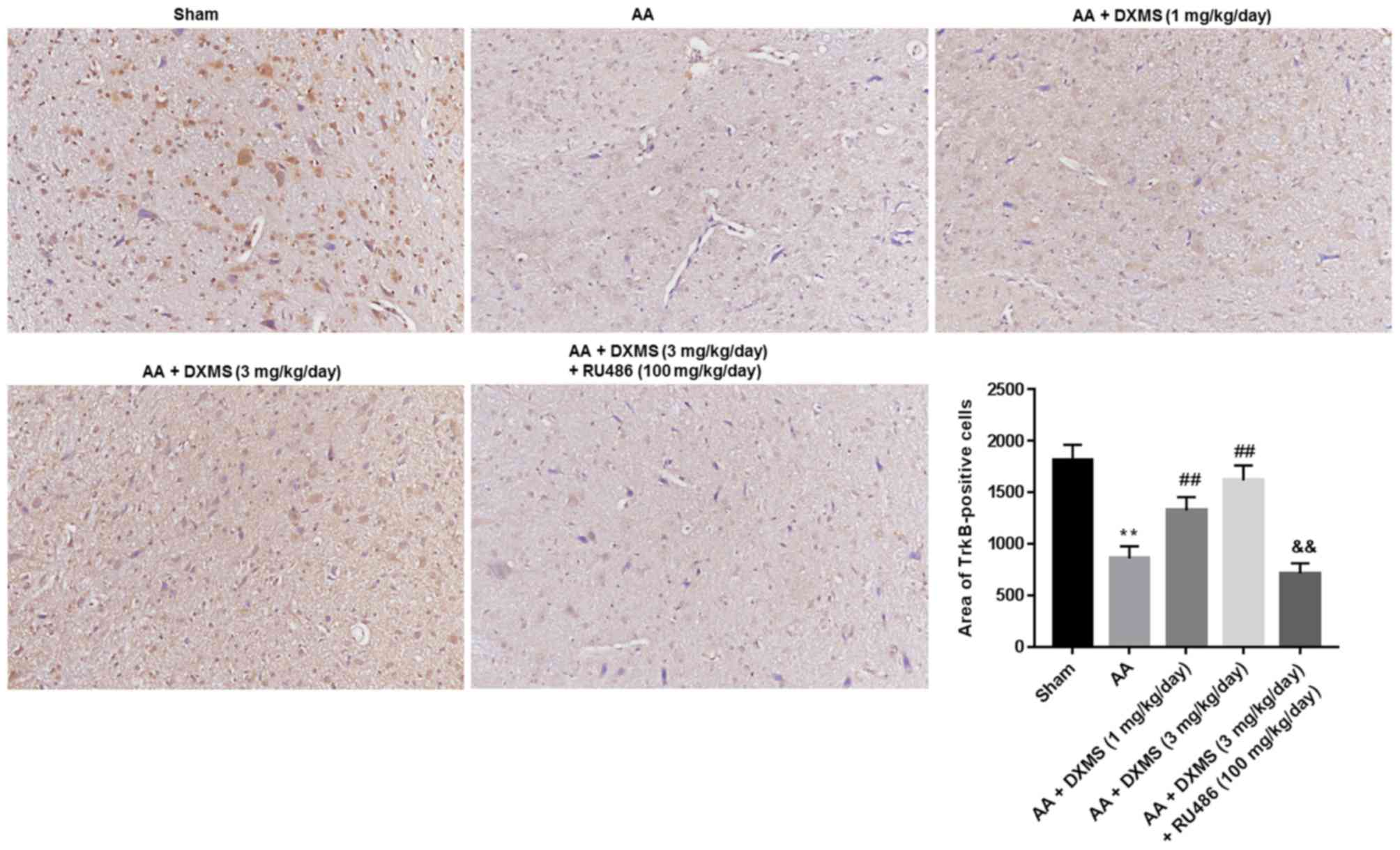
additional RU486 treatment (P<0.01). As presented in Figs. 4 and 5, similar trends of change were observed
in the areas of TrkB or KCC2-positive cells.
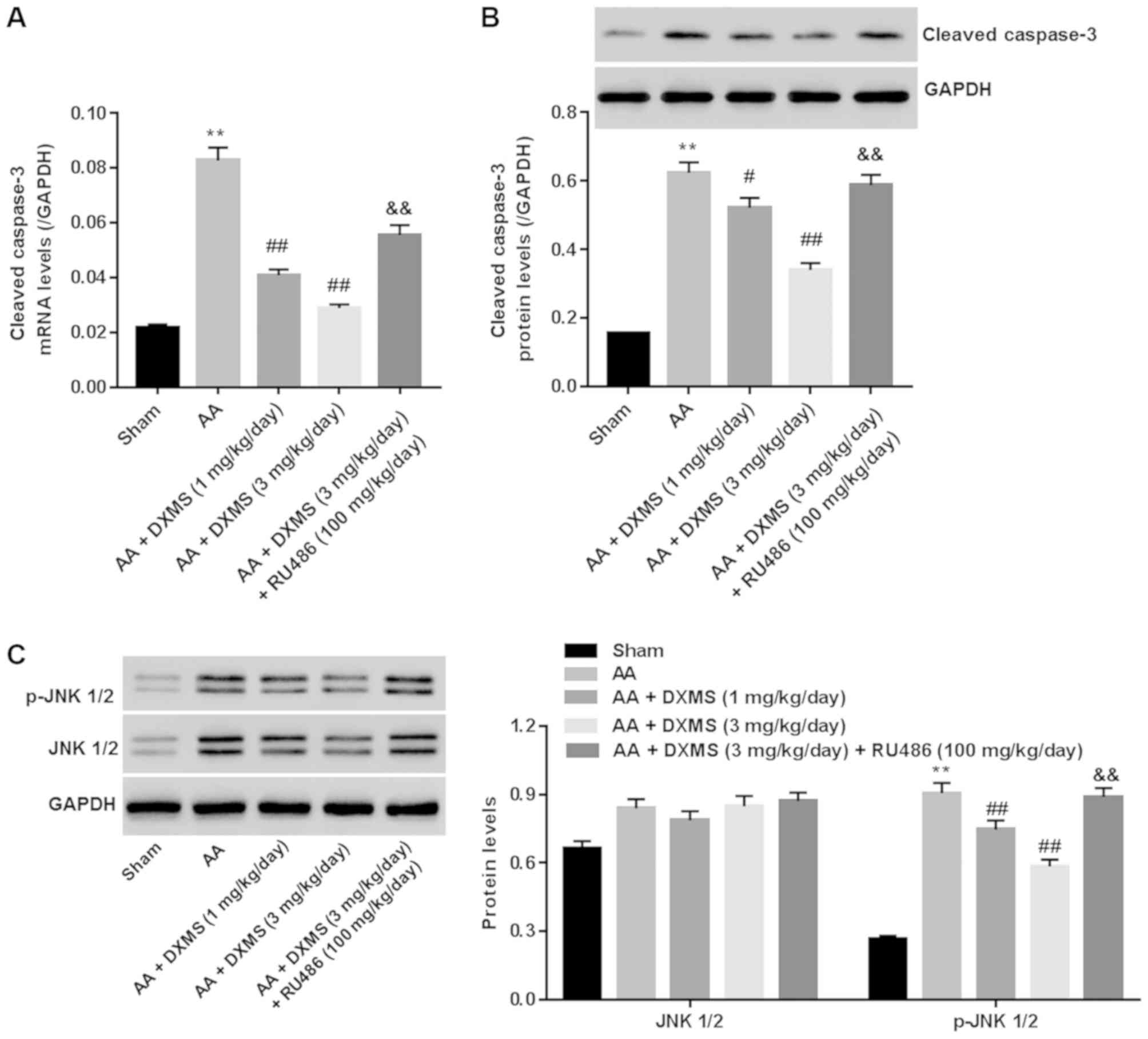
DXMS decreases cleaved-caspase 3 and
inhibits the JNK pathway in AA-treated rats
The JNK signaling pathway is essential for
triggering apoptosis in brain tissues following a variety of
injuries (18). Cleaved-caspase 3
is known as a key pro-apoptotic mediator. To assess the
anti-apoptotic mechanism of DXMS in AA-treated rats,
cleaved-caspase 3 and the JNK signaling pathway were detected. It
was identified that, in the AA group, a marked increase in the mRNA
level of cleaved-caspase 3 (Fig.
6A) and the protein expression levels of cleaved-caspase 3 and
p-JNK1/2 (Fig. 6B and C) were
observed when compared with those in the sham group, and these
alterations were significantly reversed with DXMS treatment in a
dose-dependent manner. In the AA + DXMS (3 mg/kg/day) + RU486
group, the expression levels of cleaved-caspase 3 and p-JNK1/2 were
markedly enhanced when compared with those of the AA + DXMS (3
mg/kg/day) group (P<0.01). There was approximately no
distinguished difference in the protein expression levels of JNK1/2
in the groups. These data suggested that reducing the expression of
cleaved-caspase 3 and preventing activation of the JNK1/2 pathway
were the anti-apoptotic mechanisms underlying the effect of DXMS in
AA-induced vestibular dysfunction.
Discussion
In the present study, rats were treated with AA at a
dose of 100 mg/ml to establish a rat vestibular lesioning model,
which was confirmed by observing syndromes that were associated
with vestibular dysfunction, including low-positive rates in the
air righting reflex test, prolonged times in the contact righting
reflex test, and high scores in the head tilt and swimming behavior
test. The histopathological evaluation and TUNEL analysis
demonstrated severe brain damage, and increased areas of apoptotic
cells in brain tissues, further substantiating vestibular deficits
in the AA-treated rats.
At present, DXMS, a known glucocorticoid, has been
reported to have a neuroprotective effect on
hypoxia/ischemia-stimulated, or traumatic brain injury (19–21).
In 1995, Yamanaka et al (15) reported that DXMS directly activates
the medial vestibular nucleus, thus accelerating vestibular
compensation. However, whether and how DXMS exerts its protective
effect on AA-induced vestibular lesions remains to be fully
elucidated. Intraperitoneal injection of dexamethasone into rats at
a dose of 1 mg/kg/day has been used to treat traumatic brain
injury, therefore, in the present study, doses of 1 and 3 mg/kg/day
were selected for the investigation (22). It was identified that, with DXMS
treatment, AA-induced rat vestibular dysfunction was significantly
alleviated in a dose-dependent manner, demonstrated by the
disappearance in classic signs of rat vestibular injury, improved
behavior scores, minor damage and decreased apoptotic cells in
brain tissues. In addition, RU486 serves as a glucocorticoid
receptor antagonist and is usually subcutaneously injected into
rats at a dose of 10 mg/kg/day; this which was additionally adopted
in the present experiment proposal for DXMS therapeutic
intervention (23,24). The resulting data suggested that
RU486 significantly worsened vestibular symptoms and behavior
scores, and reversed DXMS-induced histopathologic changes in brain
tissues, demonstrating that RU484 inhibited vestibular compensation
following DXMS treatment. Overall, the data indicated that DXMS was
an effective drug for recovery of AA-induced rat vestibular
dysfunction.
BDNF-triggered activation of TrkB reduces the
expression of KCC2, a specific marker of neuronal excitability
(25). At present, the
BDNF/TrkB/KCC2 pathway is known to be required for the development
of the vestibular system, and additionally for behavioral recovery
following vestibular dysfunction in adult mammals. Therefore, the
BDNF signaling pathway is widely recognized as a therapeutic target
for vestibular compensation (7).
The present study further investigated whether BDNF signaling
pathway was the mechanism that DXMS exerted its protective effect
on AA-induced rat vestibular dysfunction. It was identified that AA
significantly decreased the mRNA and protein expression levels of
BDNF, TrkB and KCC2, and the positive cells rates of BDNF, TrkB and
KCC2 (as determined by IHC staining) in brain tissues, confirming
that inhibition of the BDNF singling pathway was involved in
AA-induced vestibular dysfunction. However, DXMS markedly
up-regulated BDNF, TrkB and KCC2 in AA-induced rat vestibular
dysfunction, the effect of which was suppressed with additional
RU484 treatment. This demonstrated that DXMS confirmed AA-induced
rat vestibular dysfunction through the BDNF signaling pathway.
Caspases are principal executioners of various
apoptotic pathways (26). Evidence
indicates that the JNK pathway is involved in cell loss in the
central nervous system (27). In
2009, Shuto et al (28)
reported the anti-apoptotic effect of DXMS by inhibiting caspase-3
and retarding the JNK pathway in trimethyltin-induced neuronal
damage. The present study investigated whether cleaved-caspase 3
and JNK1/2 signals were involved in the anti-apoptotic mechanism of
DXMS in brain tissues of AA-stimulated rats. Firstly, it was
confirmed that AA triggered the activation of cleaved-caspase 3 and
the JNK1/2 pathway in brain tissues of rats with vestibular
dysfunction. Secondly, it was found that DXMS significantly
inhibited the mRNA expression of caspase-3, and suppressed the
protein levels of caspase-3 and p-JNK1/2 in the AA-treated rats,
and these alterations were approximately entirely reversed by
treatment with RU484. These data indicated that suppressing the
transcription and translation of cleaved-caspase 3 mRNA, and
inhibiting the JNK1/2 signaling pathway were involved in the
anti-apoptotic effect of DXMS in AA-treated rats.
In conclusion, DXMS demonstrated a therapeutic
effect against AA-induced dysfunction vestibular. The underlying
mechanisms involved activating BDNF and inhibiting JNK1/2 signaling
pathways. In vitro experiments are required to confirm this
conclusion, and experiment materials are being prepared in our
laboratory. Unfortunately, the results are unavailable at this
point.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Guidance
Projects for Science and Technology Development Plan of Suzhou
(grant no. SYSD2013086; China).
Availability of data and materials
The datasets used and/or analysed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
YL conducted the experiment and made data entry. DZ
conducted the statistical analysis and interpreted the data. Data
acquisition, analysis and interpretation were performed by YC and
ZC. ZF designed the study.
Ethics approval and consent to
participate
All experimental procedures involving rats followed
the local committee on the ethical use of animals of Shandong
Provincial Hospital (Jinan, China). The study was approved by the
Institutional Review Board of Shandong Provincial Hospital
Affiliated to Shandong University.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Cullen K and Sadeghi S: Vestibular system.
Scholarpedia. 3:2008. View Article : Google Scholar
|
|
2
|
Borel L, Lopez C, Péruch P and Lacour M:
Vestibular syndrome: A change in internal spatial representation.
Neurophysiol Clin. 38:375–389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hotson JR and Baloh RW: Acute vestibular
syndrome. N Engl J Med. 339:680–685. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wu X, Cai J, Li X, Li H, Li J, Bai X, Liu
W, Han Y, Xu L, Zhang D, et al: Allicin protects against
cisplatin-induced vestibular dysfunction by inhibiting the
apoptotic pathway. Eur J Pharmacol. 805:108–117. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zhou L, Zhou W, Zhang S, Liu B, Liang P,
Zhou Y, Zhou T, Zhang K, Leng Y and Kong W: BDNF signaling in the
rat cerebello-vestibular pathway during vestibular compensation:
BDNF signaling in vestibular compensation. FEBS J. 282:3579–3591.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tyler WJ and Pozzo-Miller L: Miniature
synaptic transmission and BDNF modulate dendritic spine growth and
form in rat CA1 neurones. J Physiol. 553:497–509. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dutheil S, Watabe I, Sadlaoud K, Tonetto A
and Tighilet B: BDNF signaling promotes vestibular compensation by
increasing neurogenesis and remodeling the expression of
potassium-chloride cotransporter KCC2 and GABAA receptor in the
vestibular nuclei. J Neurosci. 36:6199–6212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Botelho F, Lai C, Shum D and Chan Y:
BDNF-mediated GABAergic transmission in the rat vestibular nucleus
regulates the developmental recognition of spatial orientation. The
2012 Annual Meeting of the Society for Neuroscience: 735.11/A55.
2012.
|
|
9
|
Willson ML, Mcelnea C, Mariani J, Lohof AM
and Sherrard RM: BDNF increases homotypic olivocerebellar
reinnervation and associated fine motor and cognitive skill. Brain.
131:1099–1112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gómez-Casati ME, Murtie JC, Rio C,
Stankovic K, Liberman MC and Corfas G: Nonneuronal cells regulate
synapse formation in the vestibular sensory epithelium via
erbB-dependent BDNF expression. Proc Natl Acad Sci USA.
107:17005–17010. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
He YY, Zhang XY, Yung WH, Zhu JN and Wang
JJ: Role of BDNF in central motor structures and motor diseases.
Mol Neurobiol. 48:783–793. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trune DR and Canlon B: Corticosteroid
therapy for hearing and balance disorders. Anat Rec (Hoboken).
295:1928–1943. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Strupp M, Zwergal A, Feil K, Bremova T and
Brandt T: Pharmacotherapy of vestibular and cerebellar disorders
and downbeat nystagmus: Translational and back-translational
research. Ann N Y Acad Sci 1343. 27–36. 2015. View Article : Google Scholar
|
|
14
|
Wegner I, van Benthem PP, Aarts MC,
Bruintjes TD, Grolman W and van der Heijden GJ: Insufficient
evidence for the effect of corticosteroid treatment on recovery of
vestibular neuritis. Otolaryngol Head Neck Surg. 147:826–831. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yamanaka T, Sasa M, Amano T, Miyahara H
and Matsunaga T: Role of glucocorticoid in vestibular compensation
in relation to activation of vestibular nucleus neurons. Acta
Otolaryngol Suppl. 519:168–172. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhi-Yuan LI: An animal model of
experimental vestibular dysfunction by intratympanically injecting
arsanilic acid in mice. J Pharm Pract. 26:427–429. 2008.
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonny C, Borsello T and Zine A: Targeting
the JNK pathway as a therapeutic protective strategy for nervous
system diseases. Rev Neurosci. 16:57–67. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Felszeghy K, Banisadr G, Nyakas C and
Haour F: Dexamethasone downregulates chemokine receptor CXCR4 and
exerts neuroprotection against Hypoxia/ischemia-induced brain
injury in neonatal rats. Neuroimmunomodulation. 11:404–413. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang Z, Zhang Z, Artelt M, Burnet M and
Schluesener HJ: Dexamethasone attenuates early expression of three
molecules associated with microglia/macrophages activation
following rat traumatic brain injury. Acta Neuropathol.
113:675–682. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Du XD, Li H and Liao L: Experimental study
on protective effects of dexamethasone on traumatic brain injury in
rats. Sichuan Da Xue Xue Bao Yi Xue Ban (Chinese). 40:486–489.
2009.
|
|
22
|
Zhang Z, Fauser U and Schluesener HJ:
Early attenuation of lesional interleukin-16 up-regulation by
dexamethasone and FTY720 in experimental traumatic brain injury.
Neuropathol Appl Neurobiol. 34:330–339. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Breivik T, Thrane PS, Gjermo P and Opstad
PK: Glucocorticoid receptor antagonist RU 486 treatment reduces
periodontitis in Fischer 344 rats. J Periodontal Res. 35:285–290.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ning Q, Yang H, Li E, Li C and Xu C:
Effect of corticosterone on the activity and gene expression of
ornithine decarboxylase in rat regenerating hepatocytes. Dong Wu
Xue Bao. 50:778–783. 2004.
|
|
25
|
Rivera C, Li HCJ, Lahtinen H, Viitanen T,
Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K and
Saarma M: BDNF-induced TrkB activation down-regulates the K+-Cl-
cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol.
159:747–752. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mcbride CB, Mcphail LT and Steeves JD:
Emerging therapeutic targets in caspase-dependent disease. Expert
Ther Targets. 3:391–411. 1999.
|
|
27
|
Repici M and Borsello T: JNK pathway as
therapeutic target to prevent degeneration in the central nervous
system. Adv Exp Med Biol. 588:145–155. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shuto M, Higuchi K, Sugiyama C, Yoneyama
M, Kuramoto N, Nagashima R, Kawada K and Ogita K: Endogenous and
exogenous glucocorticoids prevent trimethyltin from causing
neuronal degeneration of the mouse brain in vivo: Involvement of
oxidative stress pathways. J Pharmacol Sci. 110:424–436. 2009.
View Article : Google Scholar : PubMed/NCBI
|