Introduction
Depression, particularly treatment resistant
depression (TRD) has become a focus and sensitive topic in
neuropsychiatric research. Depression is a chronic and recurrent
disease characterized by persistent low mood, including no interest
in life, lack of pleasure, impaired concentration, loss of memory
and the repeated idea of suicide (1,2).
There have been advancements in the pharmacological treatment of
depression (1,3); however, >30% of depression
therapies remain ineffective, which is termed TRD (4). At present, the treatment strategies
for TRD, involve increasing the dosage and course of
antidepressants, altering or using other antidepressants, adding
synergists and combining with non-drug therapy (5). Despite clinical efforts, ~90%
patients with TRD experience different degrees of depression, which
not only affects their quality of life; however, additionally
becomes the principal cause of suicide (6–8).
Furthermore, TRD significantly increases the incidence of diabetes
mellitus and cardiovascular and cerebrovascular diseases, resulting
in a marked increase in the disability rate and a burden on society
(9).
Previously, accumulating evidence revealed that
inflammation was closely associated with the occurrence,
development and progression of depression (10–12).
Additionally, the expression levels of peripheral inflammatory
cytokines in patients with TRD were significantly higher compared
with patients with curative depression (13,14).
Similarly, patients with depression with high peripheral
inflammatory cytokines expression had a significantly lower
response to therapies compared with patients with low expression of
inflammatory cytokines (15,16).
Previous studies have demonstrated that tumor necrosis factor (TNF)
antagonism may improve depressive symptoms in patients with TRD
with high baseline inflammatory biomarkers (17,18).
These studies suggested that inflammation may participate in the
development and progression of TRD.
MicroRNAs (miRs) act as a characteristic type of
post-transcriptional modulators of gene expression with significant
stabilization in serum (19). It
has been suggested that microRNA-155 (miR-155), an important member
of miRs, serves crucial roles in organism function, involving
differentiation of hematopoietic cells (20), immunization (21), inflammation (22) and cardiovascular diseases (23). In addition, it was demonstrated
that miR-155 serves as an oncogenic gene and overexpresses in
various malignant tumors, including nasopharynx cancer (24), breast cancer (25), hepatocellular carcinoma (26) and gastric carcinoma (27). It has been reported that
hippocampal dysfunction is associated with the occurrence of
depression (28). Nevertheless, to
the best our knowledge, the roles and mechanisms of miR-155 in
inflammation as a result of TRD remains unclear.
In the present study, the associations between
miR-155 and the inflammatory injury in TRD were analyzed.
Furthermore, it was noteworthy to investigate the exact roles and
mechanisms of miR-155 together with the activation of microglial
cells in the inflammatory injury of TRD.
Materials and methods
Cell culture
The mouse BV-2 microglial cell line was obtained
from the Cell Bank of Chinese Academy of Sciences (Beijing, China)
and the mouse HT22 hippocampal neuron cell line obtained from the
BeNa Culture Collection (Beijing, China). Cells were maintained in
Dulbecco's modified Eagle's medium (DMEM) mixed 1:1 with Ham's F-12
(both Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) in a 5% CO2 atmosphere at 37°C.
Preparation of microglial-conditioned
medium (MCM)
BV-2 microglial cells were maintained in
serum/glucose-free DMEM (Gibco; Thermo Fisher Scientific, Inc.) in
an anoxic environment for 1 h at 37°C. The cells were subsequently
transferred into an anoxic incubator and reserved in the serum-free
medium (Gibco; Thermo Fisher Scientific, Inc.; added with 1% B27, 2
mmol/l glutamine and 10 µl/ml penicillin-streptomycin). After 48 h
treatment, the MCM was harvested. Centrifugation (1,000 × g; 1 min;
4°C) was used to purify the obtained conditioned medium. The MCM
was diluted with serum-free medium to 1:1. Subsequently, the MCM
was used as the culture medium for hippocampal neuron cells in the
subsequent experiments.
Cell grouping and transfection
Four treatment groups were prepared in the present
study, including the control group (BV-2 microglial cells or
hippocampal neuron cells), mimic control group [BV-2 microglial
cells or hippocampal neuron cells transfected with control
scrambled sequences (5′-GAGUAUGUGAGAUUAACUGGUGGC-3′; Shanghai
GenePharma Co., Ltd., Shanghai, China)], miR-155 mimics group [BV-2
microglial cells or hippocampal neuronal cells transfected with
miR-155 mimics (5′-UUAAUGCUAAUCGUGAUAGGGGUU-3′; Shanghai GenePharma
Co., Ltd.)], and lipopolysaccharide (LPS) group. BV-2 microglial
cells or hippocampal neuron cells were treated with LPS (10 µg/ml;
dissolved in PBS; Beijing Solarbio Science & Technology Co.
Ltd., Beijing, China) for 24 h at 37°C. Cells were seeded into
6-well plates at a density of 1×104 cells/well. The
cells were starved overnight and then transfected with 75 pmol
mimic control or miR-155 mimic using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The cells were harvested 24 h after
transfection and then used for subsequent experiments.
Overexpression of suppressor of cytokine signaling 1
(SOCS1) was induced by transfecting cells (1×105
cells/ml) for 24 h at 37°C with 2.5 µg pcDNA3.1-SOCS1 plasmid
(Shanghai GenePharma Co., Ltd, Shanghai, China) using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc), according to the manufacturer's protocol.
pcDNA3.1 served as the negative control.
Cell viability analysis
Cell Counting Kit-8 (CCK-8; Beyotime Institute of
Biotechnology, Shanghai, China) was used to determine the cell
viability of BV-2 microglial cells and hippocampal neuron cells
from the four treatment groups described above. A density of
~6×104 cells/ml in the logarithmic phase were seeded
into the wells of 96-well plates, and subsequently incubated in a
5% CO2 atmosphere at 37°C for 12 h. The cells were
maintained for 12, 24 and 48 h, respectively. Subsequently, 10 µl
CCK reagent was supplemented into the wells. Cells were maintained
for 3 h. A microplate reader (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) was used to record the absorbance at 450 nm. Cell
viability was evaluated by the percentage of cell survival compared
with control.
ELISA
Commercially available ELISA kits were used to
detect interleukin-6 (IL-6; cat. no. M6000B; R&D systems),
IL-10 (cat. no. M1000B; R&D systems), TNF-α (cat. no. MTA00B;
R&D systems), TNF-β (cat. no. E-EL-M1210; Elabscience, Wuhan,
China) and indoleamine 2,3-dioxygenase 1 (IDO1; cat. no.
CSB-EL010996MO; CUSABIO TECHNOLOGY LLC, Wuhan, China). Cell culture
supernatants were added into the corresponding wells, and the wells
were sealed using adhesive tape and maintained at 37°C for 90 min.
A total of 100 µl biotinylated antibody fluids were supplemented
into wells. Subsequently, the wells were sealed by adhesive tape
and incubated for 60 min at room temperature. Chromogenic substrate
was added into the wells, excluding for the blank wells. Plates
were incubated for 10–15 min in the dark at 37°C. Stop solution was
added into each well and immediately mixed for 10 min at room
temperature. The optical density (OD) 450 value was detected using
a microplate reader (Bio-Rad Laboratories, Inc.).
Apoptosis assay
Cell apoptosis was evaluated by flow cytometry
(FCM). Following washing with PBS, cells were trypsinized using
0.25% trypsin (Beyotime Institute of Biotechnology). Following
centrifugation (1,000 × g; 1 min; 4°C), the supernatant was removed
and the cells for assessment were suspended in the incubation
buffer at a density of 1×106 cells/ml. Cells were
incubated with Annexin V-fluorescein isothiocyanate and propidium
iodide (Xilong Scientific, Co., Ltd., Guangdong, China) at room
temperature for 15 min in the dark. Cell apoptosis was subsequently
assessed using a FACSCalibur flow cytometer (BD Biosciences,
Franklin Lakes NJ, USA) and ModFit LT software version 2.0 (Verity
Software House, Topsham, ME, USA).
Western blot analysis
Total protein was isolated from cells using NP40
lysis buffer (Beyotime Institute of Biotechnology). Protein
concentration was measured using Bradford Protein Assay kit
(Bio-Rad Laboratories, Inc.). Proteins (30 µg/lane) were separated
by 12% SDS-PAGE. The separated products were transferred to
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% skimmed milk at room
temperature for 2 h. Blotting was performed with specific primary
antibodies at 4°C overnight: Anti-TNF-α (1:1,000; Abcam, Cambridge,
UK; cat. no. ab6671; rabbit anti-mouse); anti-TNF-β (1:500; Abcam;
cat. no. ab106353; rabbit anti-mouse); anti-IDO1 (1:50; Abcam; cat.
no. ab106134; rabbit anti-mouse); anti-apoptosis regulator Bax
(Bax; 1:1,000; Abcam; cat. no. ab32503; rabbit anti-mouse);
anti-apoptosis regulator Bcl2 (Bcl-2; 1:500; Abcam; cat. no.
ab59348; rabbit anti-mouse); anti-pro-caspase-3 (1:10,000; Abcam;
cat. no. ab32499; rabbit anti-mouse); anti-cleaved-caspase-3
(1:1,000; Abcam; cat. no. ab2302; rabbit anti-mouse); anti-SOCS1
(1:200; Abcam; cat. no. ab3691; rabbit anti-mouse) and anti-actin
(1:5,000; Abcam; cat. no. ab179467; rabbit anti-mouse). Horseradish
peroxidase-conjugated secondary antibodies (1:5,000; Abcam; cat.
no. ab205718; goat anti-rabbit) were added and incubated at room
temperature for 1 h. Enhanced chemiluminescent (ECL) reagents (EMD
Millipore) with an ECL system (GE Healthcare, Chicago, IL, USA)
were used for the evaluation of results. Quantity One Analysis
software version 4.6.2 (Bio-Rad Laboratories, Inc.) was used for
densitometric analysis of the blots.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from cultured cells using
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.). RNA was reverse transcribed to cDNA using the BeyoRT™ First
Strand cDNA Synthesis kit (cat. no. D7166; Beyotime Institute of
Biotechnology), according to the manufacturer's instructions. The
protocol for RT was as follows: 37°C for 60 min, 85°C for 5 min and
then hold at 4°C. RT-qPCR analysis was performed using a
SYBR® Green PCR Master Mix kit (Takara Bio, Inc., Otsu,
Japan) with an ABI 7500 Thermocycler (Applied Biosystems, Foster
City, CA, USA). PCR cycles were as follows: 10 min pretreatment at
95°C, 93°C for 15 sec, 67°C for 45 sec (45 cycles), 93°C for 15
sec, 67°C for 1 min, 95°C for 15 sec, a final extension at 75°C for
10 min and held at 4°C. The primers were designed by Invitrogen
(Thermo Fisher Scientific, Inc.): miR-155, forward:
5′-CTGTTAATGCTAATCGTGAT-3′ and reverse: 5′-AACTGACTCCTACATATTAG-3′
(product 215 bp); IL-6, forward: 5′-TTCTTGGGACTGATGCTGGT-3′ and
reverse: 5′-CAAGTGCATCATCGTTGTTCA-3′ (product 211 bp); IL-10,
forward: 5′-AGTACAGCCGGGAAGACAAT-3′ and reverse:
5′-TTTCTGGGCCATGCTTCTCT-3′ (product 249 bp); TNF-α, forward:
5′-CAGAAAGCATGATCCGCGAC-3′ and reverse: 5′-GGTCTGGGCCATAGAACTGA-3′
(product 224 bp); TNF-β, forward: 5′-CATCCTGAAACCTGCTGCTC-3′ and
reverse: 5′-GGAGGAAAAGAGCTGGACCT-3′ (product: 244 bp); IDO1,
forward: 5′-GACTGTGTCCTGGCAAACTG-3′ and reverse:
5′-GTAGCTATGTCGTGCAGTGC-3′ (product 233 bp); and actin, forward:
5′-TCTGAACTCCAACGATGCCT-3′ and reverse: 5′-TCTTGTCCTTAAGCCTGGGG-3′
(product 221 bp). Actin was used as the control of the input RNA
level. Relative gene expression was determined using the
2−ΔΔCq method (29).
Statistical analysis
All experiments were repeated at least three times.
The results in the present study are presented as the mean ±
standard error. All of the experimental data was analyzed by
one-way analysis of variance following Dunnett's t-test method.
P<0.05 was considered to indicate a statistically significant
difference.
Results
miR-155 decreases the cell viability
of BV-2 microglial cells
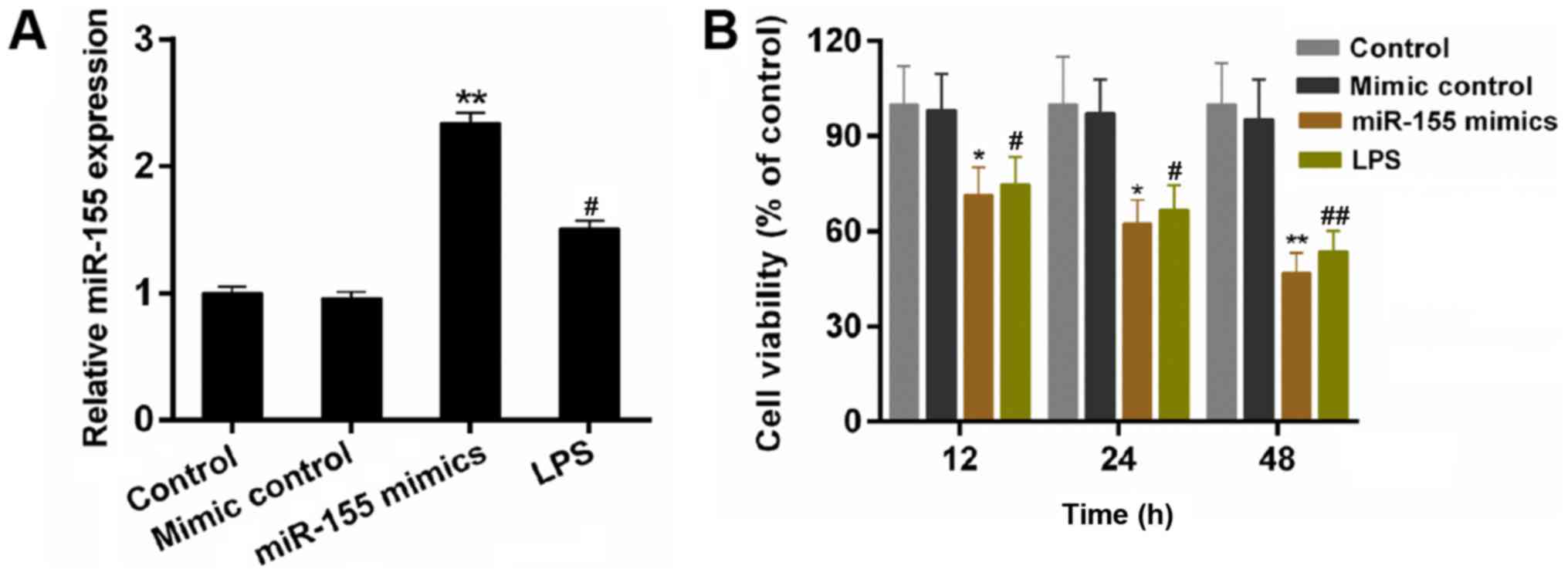
The expression of miR-155 in BV-2 cells was
investigated. The results demonstrated that the expression of
miR-155 was upregulated by LPS stimulation (Fig. 1A; P<0.05). LPS is able to induce
inflammation injury (30). It was
suggested that miR-155 was closely associated with inflammation
response of microglial cells. In the present study, the miR-155
mimics were transfected into BV-2 microglial cells. According to
the RT-qPCR data, the expression level of miR-155 in BV-2
microglial cells was significantly increased by transfecting with
miR-155 mimics compared with the mimic control (Fig. 1A; P<0.01). Furthermore, a CCK-8
assay was performed to measure the cell viability of BV-2
microglial cells in the treatment groups. As demonstrated in the
results, compared with other groups, miR-155 mimics significantly
decreased the cell viability of BV-2 microglial cells, particularly
at 48 h (Fig. 1B; P<0.05). The
results indicated that the miR-155 reduces the cell viability of
BV-2 microglial cells.
miR-155 increases the expression
levels of pro-inflammatory cytokines in BV-2 microglial cells
In order to investigate the associations between
miR-155 and inflammatory injury, the expression levels of
interleukin-6 (IL-6), IL-10, TNF-α, TNF-β and IDO1 were assessed in
BV-2 microglial cells from each treatment group. On the basis of
the ELISA data, significant increases were observed in the protein
levels of IL-6, IL-10, TNF-α, TNF-β and IDO1 in BV-2 microglial
cells transfected with miR-155 mimics (Fig. 2A; P<0.01). Additionally, the
RT-qPCR results indicated that the IL-6, IL-10, TNF-α, TNF-β and
IDO1 mRNA expression in BV-2 microglial cells was significantly
upregulated by transfection with miR-155 mimics (Fig. 2B; P<0.01). Additionally, the
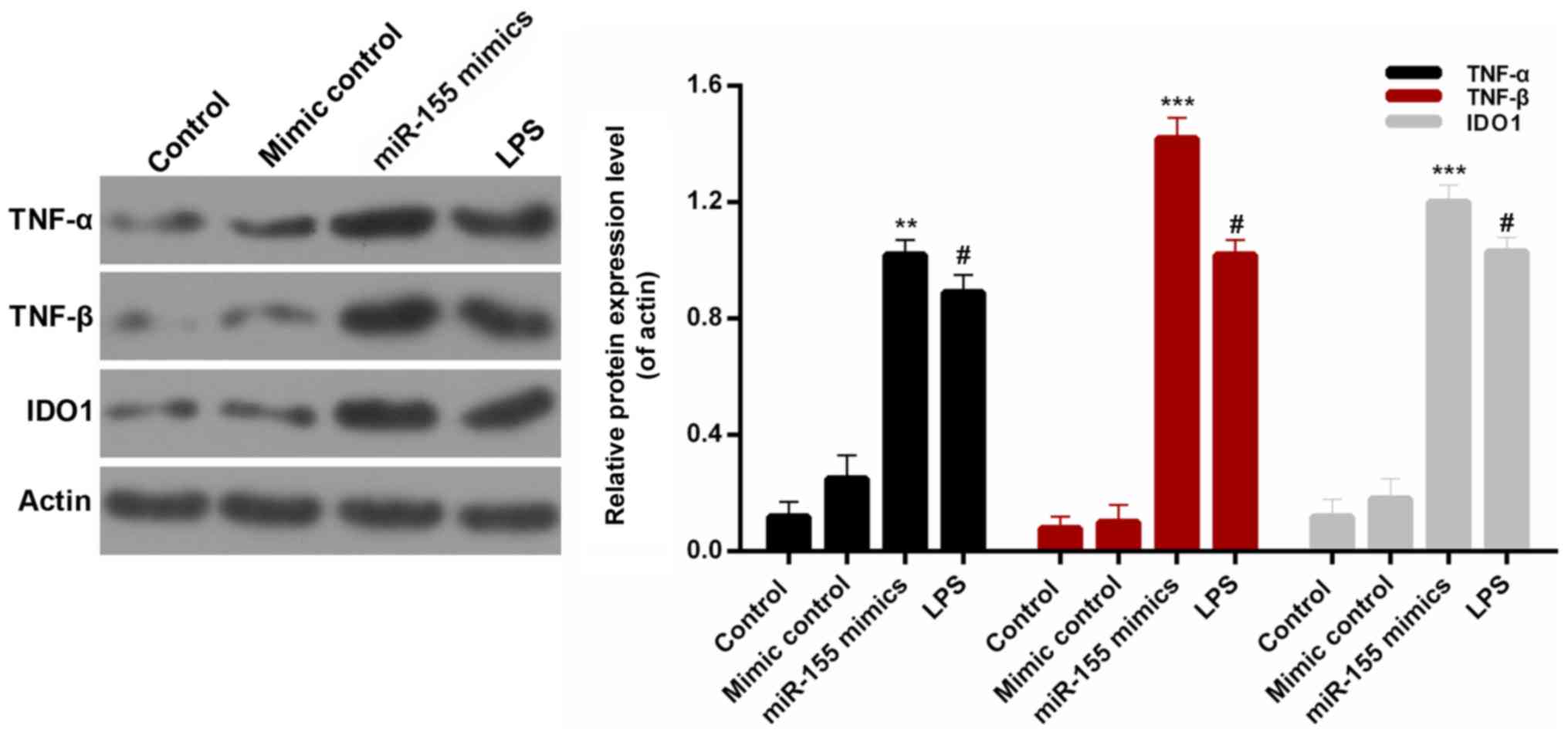
protein expression levels of pro-inflammatory cytokines, TNF-α,
TNF-β and IDO1, were measured by western blotting (Fig. 3). It was revealed that miR-155
significantly increased the expression levels of TNF-α, TNF-β and
IDO1 in BV-2 microglial cells compared with mimic controls
(P<0.01). These results confirmed that miR-155 increased the
expression levels of pro-inflammatory cytokines in BV-2 microglial
cells.
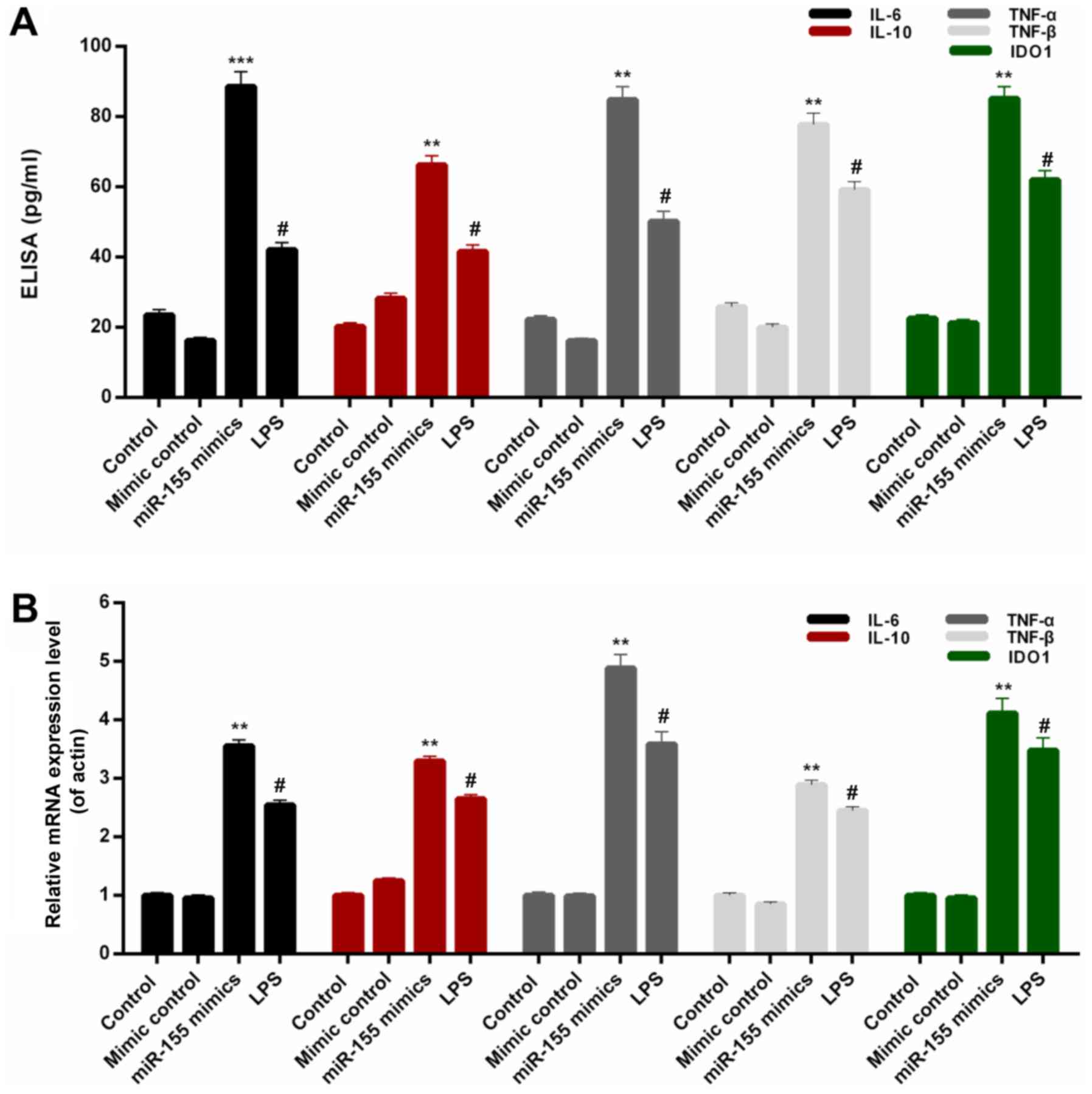 | Figure 2.miR-155 increases the
pro-inflammatory cytokines expression in BV-2 microglial cells.
BV-2 microglial cells were transfected with mimic control, miR-155
mimics or treated with LPS. (A) ELISA and (B) reverse
transcription-quantitative polymerase chain reaction assays were
performed to measure the expression levels of IL-6, IL-10, TNF-α,
TNF-β and IDO1 in BV-2 microglial cells. #P<0.05 vs.
control; **P<0.01, ***P<0.001 vs. mimic control. miR,
microRNA; LPS, lipopolysaccharide; IL, interleukin; TNF, tumor
necrosis factor; IDO1, indoleamine 2,3-dioxygenase 1. |
miR-155 contributes to
microglial-induced injury of hippocampal neuron cells
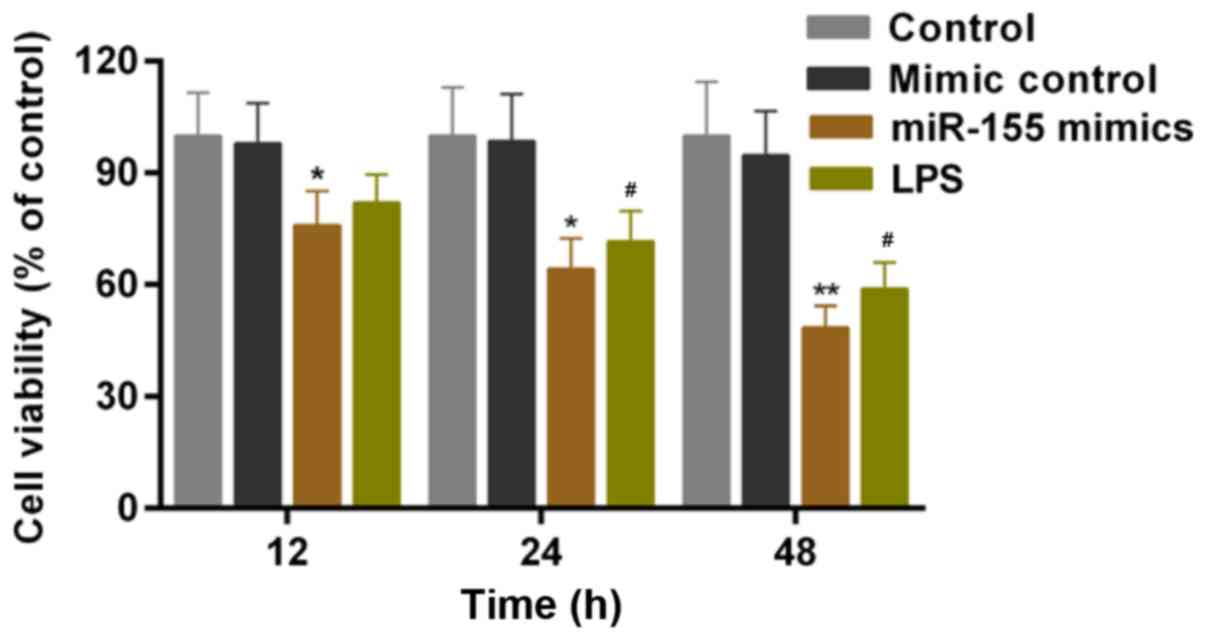
Hippocampal neuron cells were cultured with MCM
in vitro. A CCK-8 assay was conducted to assess the cell
viability of hippocampal neuron cells following culture in the MCM
of microglial cells that received the treatment/transfection
described above. Based on the results, LPS-stimulation of microglia
significantly decreased the cell viability of hippocampal neuron
cells. According to the CCK-8 results, it was additionally
identified that the cell viability of hippocampal neuron cells was
significantly decreased by miR-155-stimulation of microglia
(Fig. 4; P<0.05).
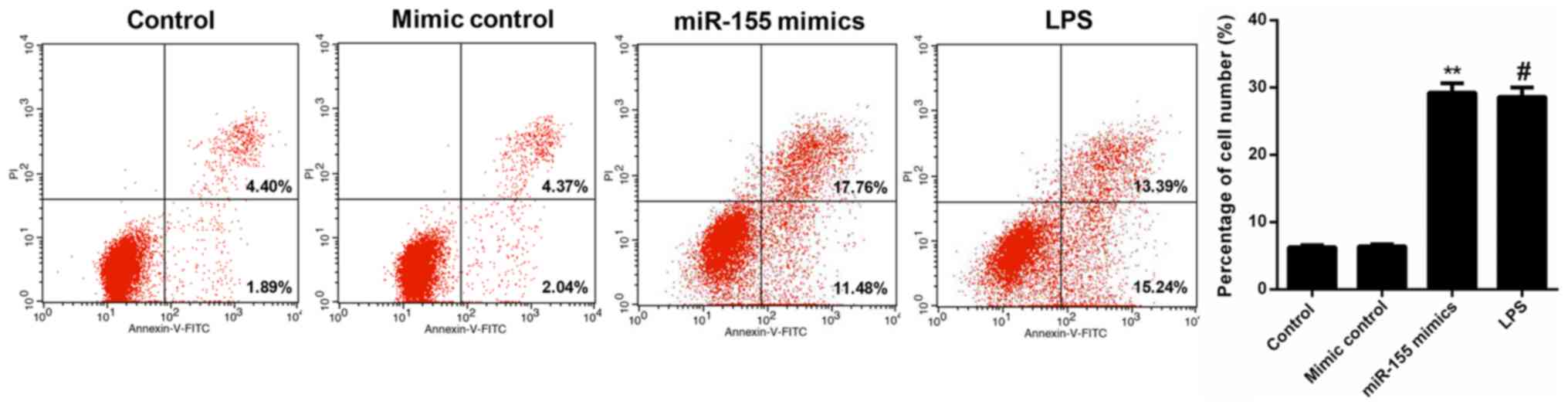
miR-155 contributes to apoptosis of
hippocampal neuron cells
FCM data revealed that the percentage of apoptotic
cells in the miR-155 mimic-transfected hippocampal neuron group was
29.24%, which was significantly increased compared with the control
and mimic control groups (6.29 and 6.41%, respectively; P<0.01).
Furthermore, following treatment with MCM from the LPS-treated
microglia, the apoptosis rate of hippocampal neuron cells was
increased to 28.63%. These data indicated that miR-155-stimulation
of microglia significantly increased the apoptosis of hippocampal
neuron cells, which was even more marked compared with the positive
control (LPS; Fig. 5). In order to
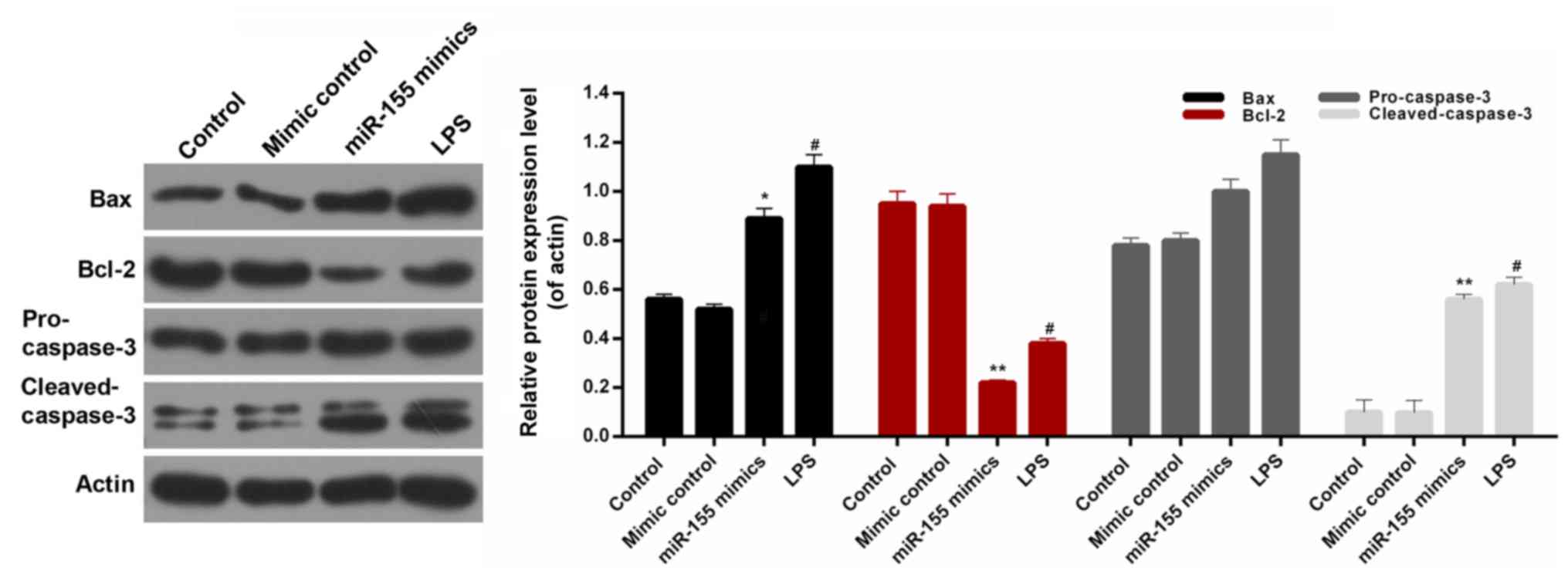
further investigate the associated apoptosis mechanisms in
hippocampal neuron cells, the expression levels of several
apoptosis-associated proteins were assessed, including Bax, Bcl-2,
pro-caspase-3 and cleaved-caspase-3 in the hippocampal neuronal
cells. It was observed that the expression level of Bcl-2 in
hippocampal neuronal cells was significantly decreased in the
miR-155 mimics group (Fig. 6;
P<0.01). However, there was no significant difference in the
expression level of pro-caspase-3 in hippocampal neuronal cells
among the treatment groups. The western blot analysis additionally
revealed similar trends in Bax, Bcl-2, pro-cleaved-3 and
cleaved-caspase-3 protein expression in hippocampal neuron cells
from each treatment group (Fig.
6). Based on these results, it was suggested that
miR-155-stimulation of microglia modulates the expression levels of
Bax, Bcl-2, pro-caspase-3 and cleaved-caspase-3 in hippocampal
neuron cells. Therefore, it was concluded that miR-155 contributed
to increasing the apoptosis of hippocampal neuronal cells by
modulating the levels of Bax, Bcl-2, and cleaved-caspase-3.
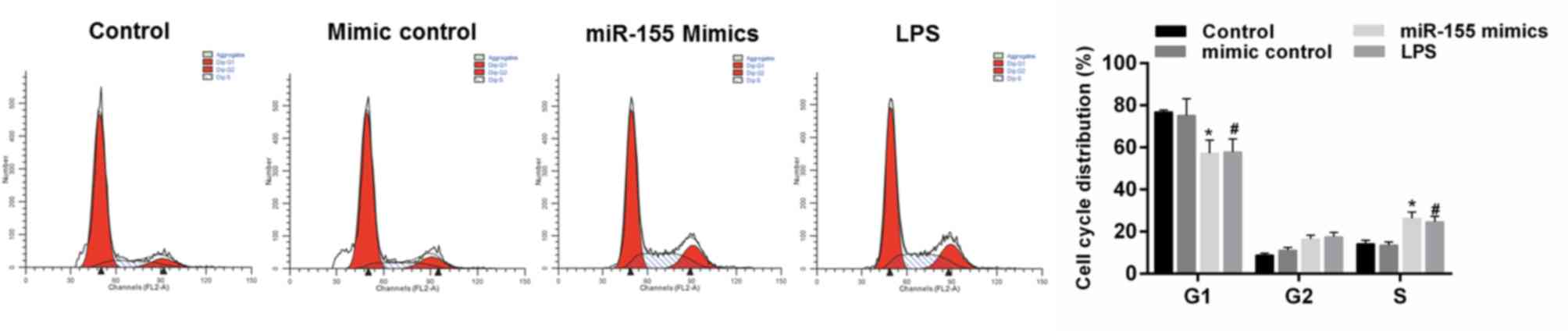
miR-155 promotes the cell cycle arrest
of hippocampal neuron cells
The effect of miR-155 on cell cycle progression of
hippocampal neuronal cells was examined. It was demonstrated that
MCM from LPS-treated microglial cells decreased the cell numbers in
the G1 phase and increased the cell numbers in the S phase compared
with the control group. The cell cycle was also arrested by miR-155
group; identified by the decreasing cell numbers in the G1 phase
and increasing cell numbers in the S phase compared with the mimics
control group (Fig. 7;
P<0.05).
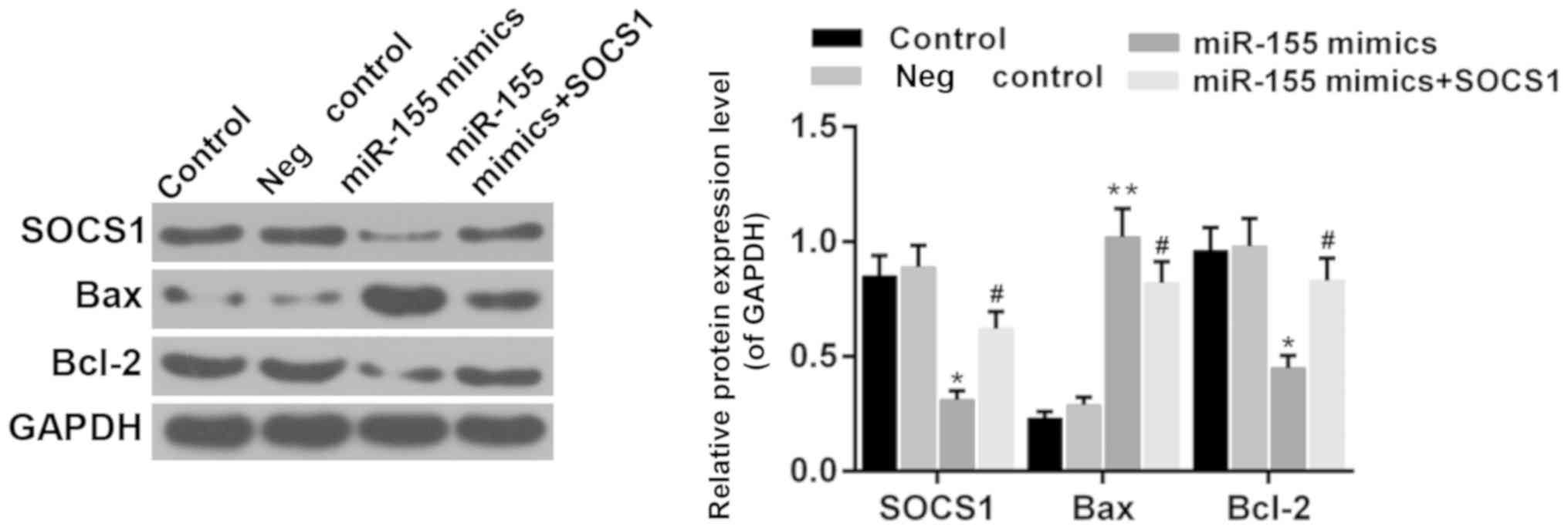
Suppressor of cytokine signaling 1
(SOCS1) is involved in the effect of miR-155
It has been reported that SOCS1 is a direct target
of miR-155 during microglia activation (31,32).
Therefore, the function of SOCS1 was investigated in the present
study. The results demonstrated that the expression of SOCS1 was
reduced in the presence of miR-155 mimics (Fig. 7; P<0.05). Furthermore, the
expression of Bcl-2 and Bax was upregulated and downregulated by
overexpression of SOCS1 in hippocampal cells, respectively,
compared with the miR-155 mimics group (Fig. 8; P<0.05). It was suggested that
miR-155 mediates hippocampal neuron cell injury via SOCS1.
Discussion
A principal limitation in the progression of TRD
research is the lack of a useable model. Microglia are widely
distributed in the central nervous system (CNS), accounting for
5–20% of the total number of neuroglial cells. Microglia are
considered the principal immune effector cells in the CNS, which
act as immune supervisors at rest state (33). Due to the important immune function
and wide distribution, microglia serve extremely important roles in
CNS diseases. When the CNS is subjected to specific noxious
stimuli, including ischemia, hypoxia, infection, trauma and chronic
neuropathy, the microglia will be activated (34). Activated microglia possess can
perform phagocytosis and release specific cytokines, including
pro-inflammatory cytokines and neurotrophic factors (35). Common pro-inflammatory cytokines
secreted by microglia are IL-1, IL-6 and TNF-α, which cause
inflammatory reactions in the nervous system, exacerbate damage to
nerve cells and promote apoptosis of nerve cells (36). Previous studies have demonstrated
that microglia mediate nerve damage in CNS diseases (37). Whereas, more recently, research has
indicated that activated microglia release neurotrophic factors;
however, they also provide neuroprotective effects (38–40).
In order to elucidate the exact role and mechanism of activated
microglia in nervous system diseases, it is necessary to conduct an
in-depth study on the function of microglia. On the basis of the
pivotal role of microglia in CNS, microglia were selected as the
research model in the present study.
A previous study has demonstrated that miR-155 was
able to modulate the microglia-mediated immune response (32). The present results demonstrated
that miR-155 was increased in microglial cells by LPS stimulation.
However, the roles of miR-155 in TRD remain unclear. Therefore, the
present study proposed to investigate the accurate functions of
miR-155 in microglia and the associated mechanisms. Initially,
miR-155 mimics were subsequently transfected into the mouse BV-2
microglial cells. RT-qPCR confirmed that the expression levels of
miR-155 in BV-2 microglial cells transfected with miR-155 mimics
were significantly higher compared with the other treatment groups.
Furthermore, the cell viability of BV-2 microglial cells from each
treatment group was measured. The results indicated that miR-155
significantly reduced the cell viability of BV-2 microglial cells,
even more significantly compared with the LPS positive control. The
expression levels of a number of pro-inflammatory cytokines in BV-2
microglial cells from each treatment group were assessed. Based on
ELISA data, miR-155 significantly increased the expression of IL-6,
IL-10, TNF-α, TNF-β and IDO1 in BV-2 microglial cells. RT-qPCR data
additionally demonstrated similar trends of the pro-inflammatory
cytokines in BV-2 microglial cells. Furthermore, the protein
expression levels of TNF-α, TNF-β and IDO1 in BV-2 microglial cells
were assessed. It was observed that miR-155 significantly
upregulated the expression levels of TNF-α, TNF-β and IDO1 in BV-2
microglial cells. Collectively, it was concluded that miR-155
activated an inflammatory response in BV-2 microglial cells and
increased the release of pro-inflammatory cytokines. A previous
study has reported that knockdown of miR-155 may protect microglia
against LPS-induced inflammatory injury by downregulating the
expression of IL-6/8 and TNF-α (41). Therefore, the data from the
previous study and the present study indicate that miR-155 is
closely associated with the inflammatory response in microglia.
To further investigate the role of miR-155 in the
CNS, the cell viability of mouse hippocampal neuronal cells
cultured with MCM from each treatment group was measured. The
results revealed that miR-155 significantly reduced the cell
viability of hippocampal neuron cells. Furthermore, the cell
apoptosis of hippocampal neuron cells was detected by FCM. On the
basis of FCM data, it was observed that miR-155 significantly
increased the apoptosis of hippocampal neuronal cells. The
associated mechanisms of apoptosis were additionally examined. The
expression levels of a number of apoptosis-associated proteins in
hippocampal neuronal cells were evaluated from each treatment
group. The results indicated that miR-155 significantly upregulated
the expression levels of Bax and cleaved-caspase-3, and reduced the
Bcl-2 expression in hippocampal neuron cells. These results
confirmed that miR-155 promoted the apoptosis of hippocampal neuron
cells by modulating the expression levels of Bax, Bcl-2,
pro-caspase-3 and cleaved-caspase-3. Furthermore, cell cycle arrest
was induced in the miR-155 group compared with the mimic control
group.
SOCS1 regulates the immune response to cytokines or
to other inflammatory stimuli (42). Previous studies have demonstrated
that SOCS1 is a direct target of miR-155 in microglia activation
(5,31). Therefore, it was hypothesized that
SOCS1 is associated with the microglial-induced hippocampal neuron
cell injury. The present results revealed that SOCS1 was
downregulated by miR-155 in microglial cells. Furthermore, the
expression of Bcl-2 and Bax in hippocampal neuron cells were
reversed by SOCS1 overexpression compared with the miR-155 mimics
group. It was demonstrated that miR-155 mediated microglia via
SOCS1 and subsequently regulated the hippocampal neuronal cell
injury.
Collectively, the present research demonstrated that
miR-155 mediated inflammatory injury in hippocampal neuron cells
via the activation of microglial cells. The present results provide
novel insight for understanding the pathogenesis of TRD and a
noteworthy potential approach for the therapy of TRD.
Acknowledgements
Not applicable.
Funding
This research was supported by Zhejiang Provincial
Natural Science Foundation of China (grant nos. LQ16H090008 and
LY18H290004) and National Natural Science Foundation of China
(grant no. 81601183).
Availability of data and materials
All data generated and/or analyzed during this study
are included in this published article.
Authors' contributions
XHS and LMY designed the study. XHS, MFS, HDS, YWW
and MJL performed the experiments. MFS, HDS and YWW performed data
analysis. XHS wrote the manuscript. XHS, MFS, HDS and LMY revised
the manuscript. All authors reviewed the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Moussavi S, Chatterji S, Verdes E, Tandon
A, Patel V and Ustun B: Depression, chronic diseases, and
decrements in health: Results from the World Health Surveys.
Lancet. 370:851–858. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nestler EJ, Barrot M, DiLeone RJ, Eisch
AJ, Gold SJ and Monteggia LM: Neurobiology of depression. Neuron.
34:13–25. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fava M: Diagnosis and definition of
treatment-resistant depression. Biol Psychiatry. 53:649–659. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Trivedi MH, Rush AJ, Wisniewski SR,
Nierenberg AA, Warden D, Ritz L, Norquist G, Howland RH, Lebowitz
B, McGrath PJ, et al: Evaluation of outcomes with citalopram for
depression using measurement-based care in STAR*D: Implications for
clinical practice. Am J Psychiatry. 163:28–40. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carvalho AF, Berk M, Hyphantis TN and
McIntyre RS: The integrative management of treatment-resistant
depression: A comprehensive review and perspectives. Psychother
Psychosom. 83:70–88. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Conwell Y and Brent D: Suicide and aging.
I: Patterns of psychiatric diagnosis. Int Psychogeriatr. 7:149–164.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kochanek KD, Murphy SL, Anderson RN and
Scott C: Deaths: Final data for 2002. Natl Vital Stat Rep.
53:1–115. 2004.PubMed/NCBI
|
|
8
|
Weissman MM, Bland RC, Canino GJ,
Greenwald S, Hwu HG, Joyce PR, Karam EG, Lee CK, Lellouch J, Lepine
JP, et al: Prevalence of suicide ideation and suicide attempts in
nine countries. Psychol Med. 29:9–17. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Petersen T, Gordon JA, Kant A, Fava M,
Rosenbaum JF and Nierenberg AA: Treatment resistant depression and
axis I co-morbidity. Psychol Med. 31:1223–1229. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lopresti AL, Maker GL, Hood SD and
Drummond PD: A review of peripheral biomarkers in major depression:
The potential of inflammatory and oxidative stress biomarkers. Prog
Neuropsychopharmacol Biol Psychiatry. 48:102–111. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miller AH and Raison CL: The role of
inflammation in depression: From evolutionary imperative to modern
treatment target. Nat Rev Immunol. 16:22–34. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rethorst CD, Bernstein I and Trivedi MH:
Inflammation, obesity, and metabolic syndrome in depression:
Analysis of the 2009–2010 National health and nutrition examination
survey (NHANES). J Clin Psychiatry. 75:e1428–e1432. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Maes M, Bosmans E, De Jongh R, Kenis G,
Vandoolaeghe E and Neels H: Increased serum IL-6 and IL-1 receptor
antagonist concentrations in major depression and treatment
resistant depression. Cytokine. 9:853–858. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sluzewska A, Sobieska M and Rybakowski JK:
Changes in acute-phase proteins during lithium potentiation of
antidepressants in refractory depression. Neuropsychobiology.
35:123–127. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lanquillon S, Krieg JC, Bening-Abu-Shach U
and Vedder H: Cytokine production and treatment response in major
depressive disorder. Neuropsychopharmacology. 22:370–379. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mikova O, Yakimova R, Bosmans E, Kenis G
and Maes M: Increased serum tumor necrosis factor alpha
concentrations in major depression and multiple sclerosis. Eur
Neuropsychopharmacol. 11:203–208. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Raison CL, Rutherford RE, Woolwine BJ,
Shuo C, Schettler P, Drake DF, Haroon E and Miller AH: A randomized
controlled trial of the tumor necrosis factor antagonist infliximab
for treatment-resistant depression: The role of baseline
inflammatory biomarkers. JAMA Psychiatry. 70:31–41. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weinberger JF, Raison CL, Rye DB, Montague
AR, Woolwine BJ, Felger JC, Haroon E and Miller AH: Inhibition of
tumor necrosis factor improves sleep continuity in patients with
treatment resistant depression and high inflammation. Brain Behav
Immun. 47:193–200. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sochor M, Basova P, Pesta M, Dusilkova N,
Bartos J, Burda P, Pospisil V and Stopka T: Oncogenic microRNAs:
miR-155, miR-19a, miR-181b, and miR-24 enable monitoring of early
breast cancer in serum. BMC Cancer. 14:4482014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Garzon R and Croce CM: MicroRNAs in normal
and malignant hematopoiesis. Curr Opin Hematol. 15:352–358. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Leng RX, Pan HF, Qin WZ, Chen GM and Ye
DQ: Role of microRNA-155 in autoimmunity. Cytokine Growth Factor
Rev. 22:141–147. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Teng G and Papavasiliou FN: Shhh!
Silencing by microRNA-155. Philos Trans R Soc Lond B Biol Sci.
364:631–637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Staszel T, Zapala B, Polus A,
Sadakierska-Chudy A, Kieć-Wilk B, Stepien E, Wybranska I, Chojnacka
M and Dembinska-Kiec A: Role of microRNAs in endothelial cell
pathophysiology. Pol Arch Med Wewn. 121:361–366. 2011.PubMed/NCBI
|
|
24
|
Zhu X, Wang Y, Sun Y, Zheng J and Zhu D:
MiR-155 up-regulation by LMP1 DNA contributes to increased
nasopharyngeal carcinoma cell proliferation and migration. Eur Arch
Otorhinolaryngol. 271:1939–1945. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang CM, Zhao J and Deng HY: MiR-155
promotes proliferation of human breast cancer MCF-7 cells through
targeting tumor protein 53-induced nuclear protein 1. J Biomed Sci.
20:792013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang L, Wang W, Li X, He S, Yao J, Wang
X, Zhang D and Sun X: MicroRNA-155 promotes tumor growth of human
hepatocellular carcinoma by targeting ARID2. Int J Oncol.
48:2425–2434. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu Q, Jin H, Yang Z, Luo G, Lu Y, Li K,
Ren G, Su T, Pan Y, Feng B, et al: MiR-150 promotes gastric cancer
proliferation by negatively regulating the pro-apoptotic gene EGR2.
Biochem Biophys Res Commun. 392:340–345. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tang MM, Lin WJ, Pan YQ, Guan XT and Li
YC: Hippocampal neurogenesis dysfunction linked to depressive-like
behaviors in a neuroinflammation induced model of depression.
Physiol Behav. 161:166–173. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stayte S, Rentsch P, Tröscher AR,
Bamberger M, Li KM and Vissel B: Activin a inhibits MPTP and
LPS-induced increases in inflammatory cell populations and loss of
dopamine neurons in the mouse midbrain in vivo. PLoS One.
12:e01672112017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zheng X, Huang H, Liu J, Li M, Liu M and
Luo T: Propofol attenuates inflammatory response in LPS-activated
microglia by regulating the miR-155/SOCS1 pathway. Inflammation.
41:11–19. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cardoso AL, Guedes JR, Pereira de Almeida
L and Pedroso de Lima MC: miR-155 modulates microglia-mediated
immune response by down-regulating SOCS-1 and promoting cytokine
and nitric oxide production. Immunology. 135:73–88. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wake H and Fields RD: Physiological
function of microglia. Neuron Glia Biol. 7:1–3. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Filipovic R and Zecevic N: Neuroprotective
role of minocycline in co-cultures of human fetal neurons and
microglia. Exp Neurol. 211:41–51. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hanisch UK: Microglia as a source and
target of cytokines. Glia. 40:140–155. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Smith JA, Das A, Ray SK and Banik NL: Role
of pro-inflammatory cytokines released from microglia in
neurodegenerative diseases. Brain Res Bull. 87:10–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kreutzberg GW: Microglia: A sensor for
pathological events in the CNS. Trends Neurosci. 19:312–318. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mizuno T, Doi Y, Mizoguchi H, Jin S, Noda
M, Sonobe Y, Takeuchi H and Suzumura A: Interleukin-34 selectively
enhances the neuroprotective effects of microglia to attenuate
oligomeric amyloid-β neurotoxicity. Am J Pathol. 179:2016–2027.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Noda H, Takeuchi H, Mizuno T and Suzumura
A: Fingolimod phosphate promotes the neuroprotective effects of
microglia. J Neuroimmunol. 256:13–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Polazzi E and Monti B: Microglia and
neuroprotection: From in vitro studies to therapeutic applications.
Prog Neurobiol. 92:293–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yin H, Song S and Pan X: Knockdown of
miR-155 protects microglia against LPS-induced inflammatory injury
via targeting RACK1: A novel research for intracranial infection. J
Inflamm (Lond). 14:172017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yoshimura A, Nishinakamura H, Matsumura Y
and Hanada T: Negative regulation of cytokine signaling and immune
responses by SOCS proteins. Arthritis Res Ther. 7:100–110. 2005.
View Article : Google Scholar : PubMed/NCBI
|