Introduction
Leukemia is the most common malignancy among
children between birth and 14 years of age worldwide (1). There are four major types of
leukemia: Acute myeloid leukemia (AML), chronic myeloid leukemia,
acute lymphocytic leukemia (ALL) and chronic lymphocytic leukemia
(2). Multiple factors have been
implicated in the etiology of the disease, such as ionizing
radiation, electromagnetic fields, infection, chemotherapy agents
and certain inherited genetic disorders (3). The treatment of leukemia has been
improved significantly through chemotherapy, interferon therapy,
radiation therapy, stem cell transplantation and surgery, however,
survival rates remain poor (4,5). It
is therefore necessary to investigate novel therapeutic targets or
strategies for the treatment of leukemia.
ENDOCAN, also known as endothelial cell-specific
molecule-1 (ESM-1), was initially cloned from a cDNA library of
human endothelial cells (6). Serum
ENDOCAN levels are increased in essential hypertension, and are
positively correlated with carotid intima-media thickness and
high-sensitivity C-reactive protein (7). High levels of ENDOCAN have been shown
to predict poor survival rate in liver cirrhosis (8). Circulating ENDOCAN may serve as an
outcome measure in the development of Behcet's disease (9) and sepsis (10). Previous studies have reported that
the expression levels of ENDOCAN were elevated in a wide range of
tumor types, including non-small cell lung cancer (11), glioblastoma (12) and hepatocellular carcinoma
(13), and was positively
correlated with the severity of the disease. The overexpression of
ENDOCAN can induce tumor formation (14). By contrast, ENDOCAN knockdown can
inhibit tumor cell proliferation and promote tumor cell apoptosis
(15). Xu et al (16) and Hatfield et al (17) identified elevated levels of ENDOCAN
in patients with untreated AML and ALL. However, the functional
role of ENDOCAN in myeloid leukemia cells remains to be fully
elucidated.
In the present study, the expression of ENDOCAN in
four myeloid leukemia cell lines (THP-1, U937, HL-60 and K562) was
determined, following which ENDOCAN was silenced in two
ENDOCAN-elevated cell lines (U937 and K562) via lentiviral
infection. Subsequently, the effects of ENDOCAN knockdown on cell
proliferation, cell cycle distribution, cell apoptosis and NF-κB
activity were investigated.
Materials and methods
Cells and cell culture
The THP-1 cells (derived from a patient with
monocytic leukemia) and HL-60 cells (derived from a patient with
promyelocytic leukemia) were purchased from the Shanghai Cell Bank
of the Chinese Academy of Sciences (Shanghai, China). The K562
cells (derived from a patient with chronic myelogenous leukemia)
were obtained from CHI Scientific (Jiangyin, China). The U937 cells
(derived from a patient with histiocytic lymphoma) were purchased
from the Institute of Basic Medical Sciences, Chinese Academy of
Medical Sciences (Beijing, China). The HL-60 cells were cultured in
Iscove's modified Dulbecco's medium (Thermo Fisher Scientific,
Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum
(FBS; Biological Industries, Beit Haemek, Israel), 100 U/ml
penicillin and 0.1 mg/ml streptomycin. The THP-1, K562, and U937
cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific,
Inc.) containing 10% FBS (Biological Industries), 100 U/ml
penicillin and 0.1 mg/ml streptomycin. All cell lines were
maintained in an incubator (Shanghai Lishen, Shanghai, China) with
5% CO2 at 37°C.
Lentivirus generation
Lentiviruses containing two short hairpin RNA
(shRNA) sequences specific to ENDOCAN and a negative control (NC)
were generated by a lentiviral packaging system from Wanleibio Co.,
Ltd. (Shenyang, China). The sequences were as follows: ENDOCAN
shRNA1,
5′-TGGTGAAGAGTTTGGTATCTTTCAAGAGAAGATACCAAACTCTTCACCTTTTTTC-3′;
ENDOCAN shRNA2,
5′-TCATCTGGAGATGGCAATATTTCAAGAGAATATTGCCATCTCCAGATGTTTTTTC-3′; NC
shRNA,
5′-TTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTC-3′. The
shRNA sequences were cloned into the AgeI/EcoRI sites
of the lentiviral vector (LV-shRNA). The recombinant lentiviral
vector and packaging plasmids were co-transfected into 293T cells
(Procell Life Science and Technology Co., Ltd., Wuhan, China) with
Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) and the
supernatant containing the virus was collected to infect target
cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from cells using an RNApure
High-purity Total RNA Rapid Extraction kit (BioTeke Corporation,
Beijing, China) and reverse transcribed into cDNA using Super M-MLV
reverse transcriptase (BioTeke Corporation). The cDNA was amplified
by RT-qPCR using Exicycler™ 96 (Bioneer Corporation, Daejeon,
Korea) with the following primers: ENDOCAN forward
5′-CTGGGAAACATGAAGAGCG-3′ and 5′-GCCTGAGACTGTGCGGTAG-3′ reverse;
β-actin forward 5′-CTTAGTTGCGTTACACCCTTTCTTG-3′ and
5′-CTGTCACCTTCACCGTTCCAGTTT-3′ reverse. The reactions were
performed in a 20-µl reaction mixture containing 1 µl template
cDNA, 10 µl SYBR GREEN mastermix (2X), 0.5 µl forward and reverse
primers and 8 µl ddH2O. For the negative control, no
primers were added. The thermocycling steps were as follows:
Initial denaturation at 94°C for 5 min, followed by 40 cycles of
denaturation at 94°C for 10 sec, annealing at 60°C for 20 sec and
elongation at 72°C for 30 sec, and a final extension at 72°C for 2
min 30 sec. Gene expression was analyzed using the
2−ΔΔCq method (18).
Western blot analysis
Total protein was separated from cells using
radioimmunoprecipitation assay lysis buffer (Beyotime Institute of
Biotechnology, Shanghai, China) supplemented with 1%
phenylmethylsulfonyl fluoride. The nuclear protein was extracted
using a nuclear protein extraction kit (Beyotime Institute of
Biotechnology). The protein concentration was determined using a
BCA protein quantitative kit (Beyotime Institute of Biotechnology).
Protein samples (20–40 µg/lane) were resolved by 8–15% SDS-PAGE,
blotted onto polyvinylidene fluoride membranes, blocked with 5%
skim milk and incubated with primary antibodies against ENDOCAN
(1:300; cat. no. PAC463Hu01; Cloud-Clone Corp., Wuhan, China),
proliferating cell nuclear antigen (PCNA; 1:500; cat. no.
10205-2-AP; Wuhan Sanying Biotechnology, Wuhan, China), cyclin D1
(1:500; cat. no. AP2612d; Abgent, Inc., San Diego, CA, USA), B-cell
lymphoma 2 (BCL2; 1:500; cat. no. D160117; Sangon Biotech Co.,
Ltd., Shanghai, China), BCL-2-associated X protein (BAX; 1:1,000;
D120073; Sangon Biotech Co., Ltd.), cleaved caspase 3 (1:1,000;
cat. no. ab2302; Abcam, Cambridge, MA, USA), cleaved caspase 9
(1:1,000; cat. no. 9501; CST Biological Reagents Co., Ltd.,
Shanghai, China), cleaved poly (ADP-ribose) polymerase (PARP;
1:500; cat. no. ab32561; Abcam), NF-κB, P65 (1:1,000; cat. no.
10745-1-AP; Wuhan Sanying Biotechnology), phosphorylated (p-)P65
(1:400; cat. no. BM3994; Boster Biological Technology, Pleasanton,
CA, USA), NF-κB inhibitor α (IκBα) (1:500; cat. no. bs-1287R;
BIOSS, Beijing, China), p-IκBα (1:500; cat. no. bs-2513R; BIOSS),
β-actin (1:500; bsm-33139M; BIOSS), and histone H3 (1:5,000; cat.
no. GTX122148; GeneTex, Inc., Irvine, CA, USA) overnight at 4°C.
Subsequently, the membranes were treated with horseradish
peroxidase (HRP)-labeled goat anti-rabbit antibodies (1:5,000; cat.
no. A0208; Beyotime Institute of Biotechnology) at 37°C for 45 min.
The protein was visualized using enhanced chemiluminescence (ECL;
Beyotime Institute of Biotechnology) with a gel imaging system
(Beijing Liuyi Instrument Factory, Beijing, China). The protein
expression levels were evaluated by densitometric analysis with
Gel-Pro Analyzer 3.1 (Meyer Instruments, Inc., Houston, TX, USA)
and each target protein was normalized to the corresponding β-actin
or histone H3 band.
Cell counting kit-8 (CCK-8) assay
Cell proliferation ability was assessed using a
CCK-8 assay (Wanleibio, Shenyang, China), based on the activity of
mitochondrial dehydrogenases (19). Briefly, the cells were seeded into
96-well plates (3×103 cells/well). The cells were
cultured for 0, 24, 48, 72 and 96 h at 37°C with 5% CO2.
The supernatant was removed at each time point and replaced with
100 µl of complete medium, and 10 µl CCK-8 solution was added into
each well for 1 h. The absorbance was measured at 450 nm using a
microplate reader (BioTek Instruments, Inc., Winooski, VT,
USA).
Cell cycle analysis
The cell cycle distribution was detected using a
propidium iodide (PI)-based cell cycle analysis kit (Beyotime
Institute of Biotechnology), as previously described (20). The principle of this method is that
the nuclei of dead cells can be stained by PI. Briefly, the cells
were collected and fixed in pre-cooled 70% ethanol for 2 h. The
cells were then suspended in 500 µl of binding buffer containing 25
µl PI and 10 µl ribonuclease A (which can degrade RNA and prevent
the PI staining of RNA) for 30 min at 37°C in the dark. The cell
cycle was analyzed using a flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA), based on forward light scatter, side light scatter
and PI (FL-2 channel, wavelength 575 nm) fluorescence.
Cell apoptosis
Cell apoptosis was evaluated using an Annexin
V-fluorescein isothiocyanate (FITC)/PI apoptosis detection kit
(Wanleibio Co., Ltd.), as previously described (21). Annexin V binds to the externalized
phosphatidylserine of cells undergoing programmed cell death, and
the nuclei of dead cells can be stained by PI. Briefly, the cells
were collected and resuspended in 500 µl binding buffer.
Subsequently, 5 µl Annexin V-FITC and 10 µl PI were added and mixed
well for 15 min at room temperature away from light. The cells were
analyzed using a flow cytometer (BD Biosciences), based on forward
light scatter, side light scatter, and FITC (FL-1 channel,
wavelength 530 nm) and PI (FL-2 channel, wavelength 575 nm)
fluorescence.
Electrophoretic mobility shift assay
(EMSA)
The principle of EMSA is that DNA-protein complexes
migrate at a slower rate than unbound DNA in a gel. The DNA binding
activity of NF-κB was determined using the EMSA kit (Viagene
Biotech, Ningbo, China), according to the manufacturer's protocol
(22). Briefly, nuclear protein
was extracted using a nuclear extraction kit (Beyotime Institute of
Biotechnology). Protein concentration was determined using the BCA
method. The nuclear protein (15 µg) was mixed with a biotin-labeled
NF-κB probe for 20 min at room temperature. Subsequently, the
DNA-protein complex was separated using a 6.5% polyacrylamide gel
at 180 V for 80 min, transferred onto a nylon membrane at 360 mA
for 1 h and crosslinked to the nylon membrane under UV light for 30
min. The bands were visualized using streptavidin-HRP and an ECL
kit. The sequence of the probe used in EMSA was
5′-AGTTGAGGGGACTTTCCCAGGC-3′.
Statistical analysis
The results are presented as the mean ± standard
deviation. Each experiment was repeated three times. Statistical
differences were analyzed using Student's t-test, one-way or
two-way analysis of variance or a Kruskal-Wallis test using
GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Downregulation of ENDOCAN in U937 and
K562 cells
To investigate the function of ENDOCAN in leukemia
cells, its expression was determined in four myeloid leukemia cell
lines. The results of the RT-qPCR and western blot analyses showed
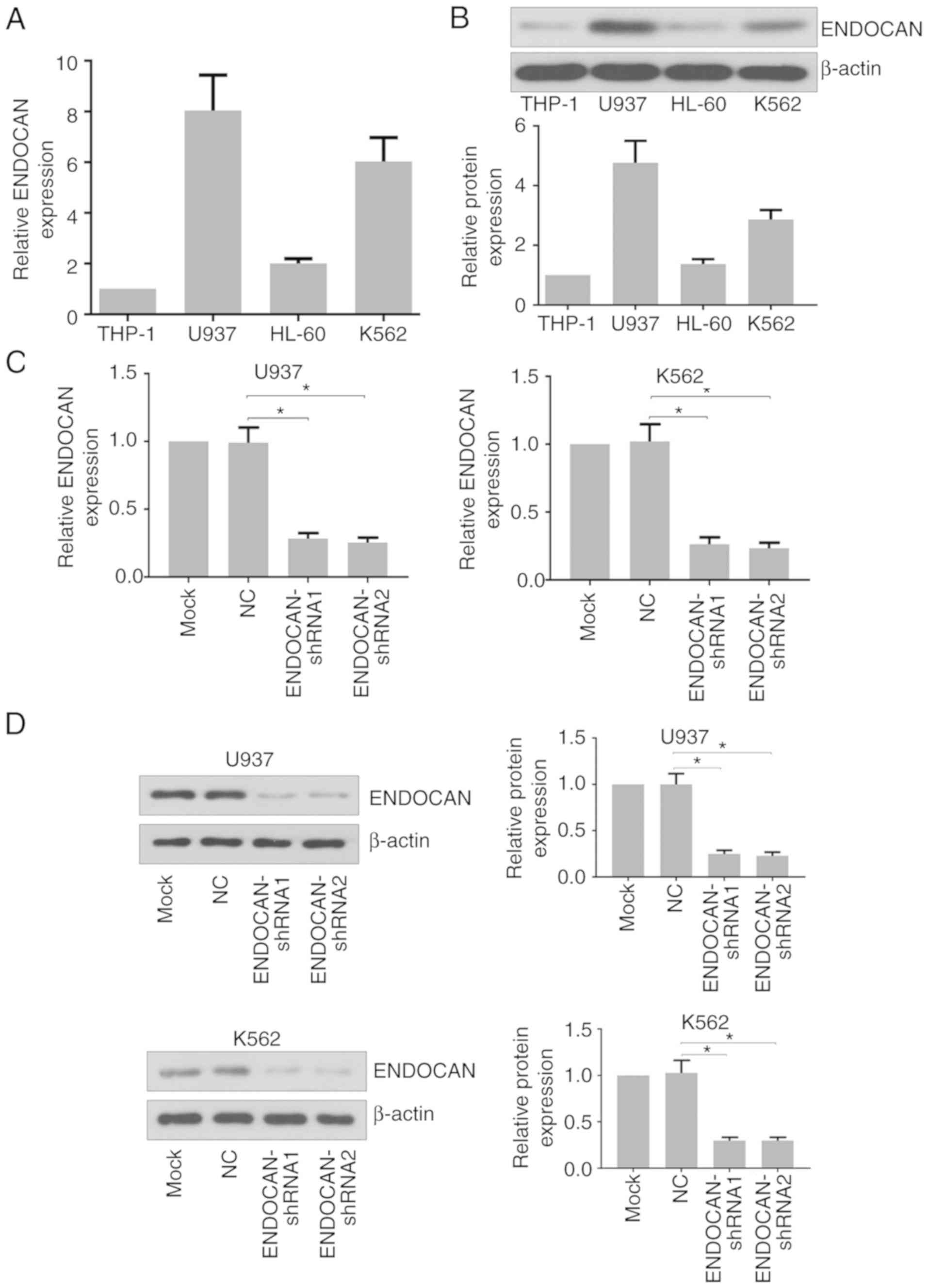
that the expression of ENDOCAN was higher in the U937 and K562
cells than in the THP-1 and HL-60 cells (Fig. 1A and B). The U937 and K562 cells
were then infected with lentivirus carrying ENDOCAN shRNA-1, −2 or
scramble shRNA for 48 h. The mRNA and protein expression levels of
ENDOCAN were evaluated by RT-qPCR and western blot analyses. As
shown in Fig. 1C and D, its
expression was significantly reduced in these cells in response to
the ENDOCAN shRNAs.
Effect of ENDOCAN silencing on the
proliferation of myeloid leukemia cells
Subsequently, experiments were performed to
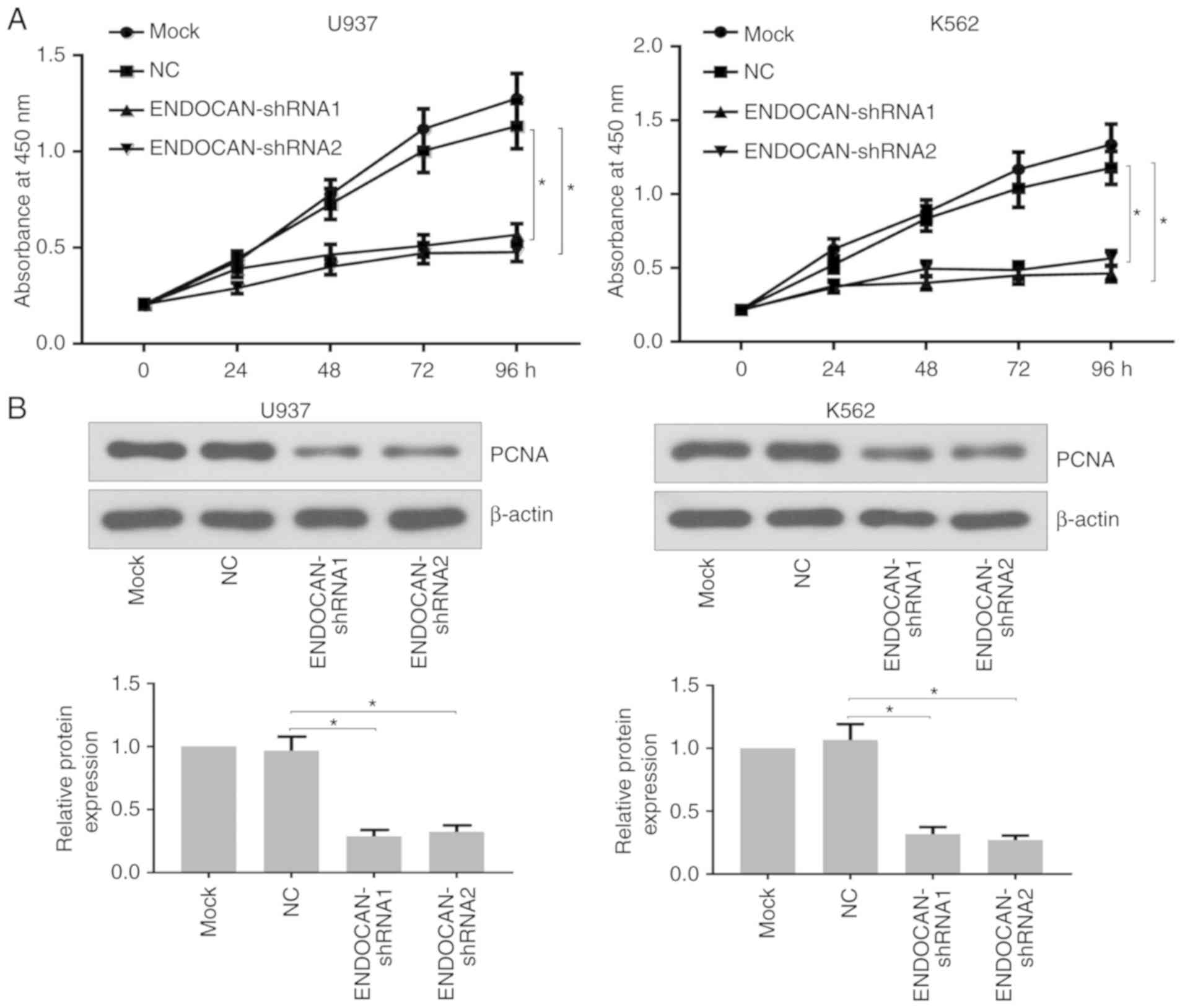
determine whether ENDOCAN knockdown affected cell proliferation.
The U937 and K562 cells were treated with lentivirus harboring
ENDOCAN shRNA-1, −2 or scramble shRNA for 0, 24, 48, 72 and 96 h. A
CCK-8 assay was then used to evaluate the proliferation ability of
the U937 and K562 cells. As shown in Fig. 2A, the proliferation potential of
the ENDOCAN-silenced cells was distinctly inhibited. Furthermore,
reduced expression of PCNA was identified in these cells following
ENDOCAN silencing for 48 h, as determined by western blot analysis
(Fig. 2B). In addition, following
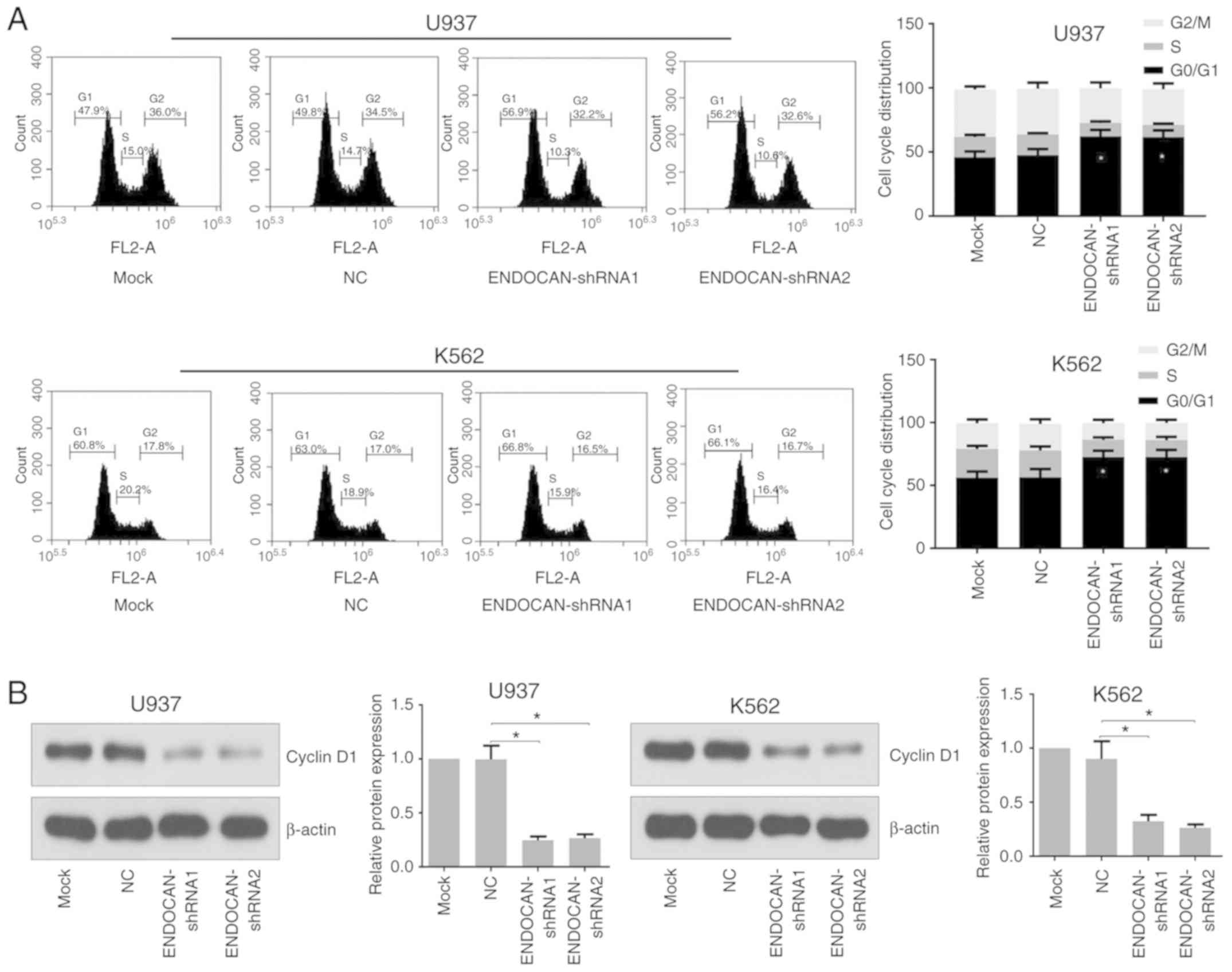
infection for 48 h, the flow cytometry results showed that ENDOCAN
knockdown led to G0/G1 cell cycle arrest in the U937 and K562 cells
(Fig. 3A). The western blot
analysis results showed that the expression level of cyclin D1 was
decreased in the ENDOCAN-silenced cells (Fig. 3B).
Effect of ENDOCAN silencing on the
apoptosis of myeloid leukemia cells
The apoptotic rates of ENDOCAN-silenced myeloid
leukemia cells were then evaluated by flow cytometry using Annexin
V/PI staining. Following infection of the U937 and K562 cells with
lentivirus harboring ENDOCAN shRNA-1, −2 or scramble shRNA for 72
h, the apoptotic rate was significantly increased in the cells with
ENDOCAN knockdown (Fig. 4A). In
addition, the expression of apoptosis-related genes was detected by
western blot analysis following infection for 72 h. As shown in
Fig. 4B, the expression of BCL2
was decreased, whereas the levels of BAX, cleaved caspases 3 and 9,
and cleaved PARP were increased in the ENDOCAN-silenced myeloid
leukemia cells.
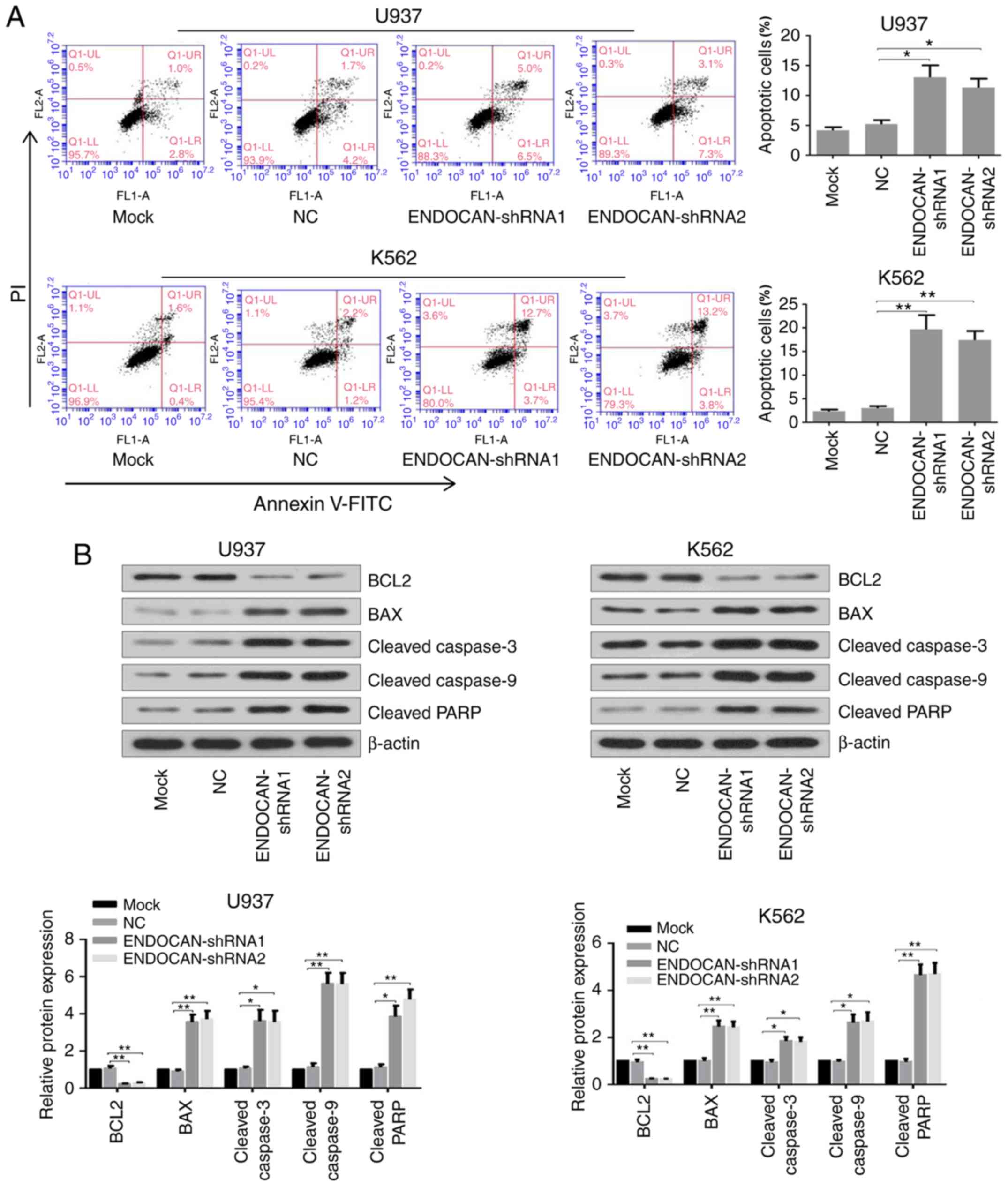 | Figure 4.ENDOCAN knockdown promotes apoptosis
in myeloid leukemia cells. (A) Apoptosis of U937 and K562 cells
with or without ENDOCAN silencing was determined by flow cytometry
following Annexin V-FITC/PI staining. (B) Expression levels of
BCL2, BAX, cleaved caspases 3 and 9, and PARP in ENDOCAN-knocked
down U937 and K562 cells were detected by western blot analysis.
The results are expressed as the mean ± standard deviation.
*P<0.05, **P<0.01. FITC, fluorescein isothiocyanate; PI,
propidium iodide; BCL2, B-cell lymphoma 2; BAX, BCL2-associated X
protein; PARP, cleaved poly (ADP-ribose) polymerase; NC, negative
control; shRNA, short hairpin RNA. |
Effect of ENDOCAN silencing on NF-κB
activity in myeloid leukemia cells
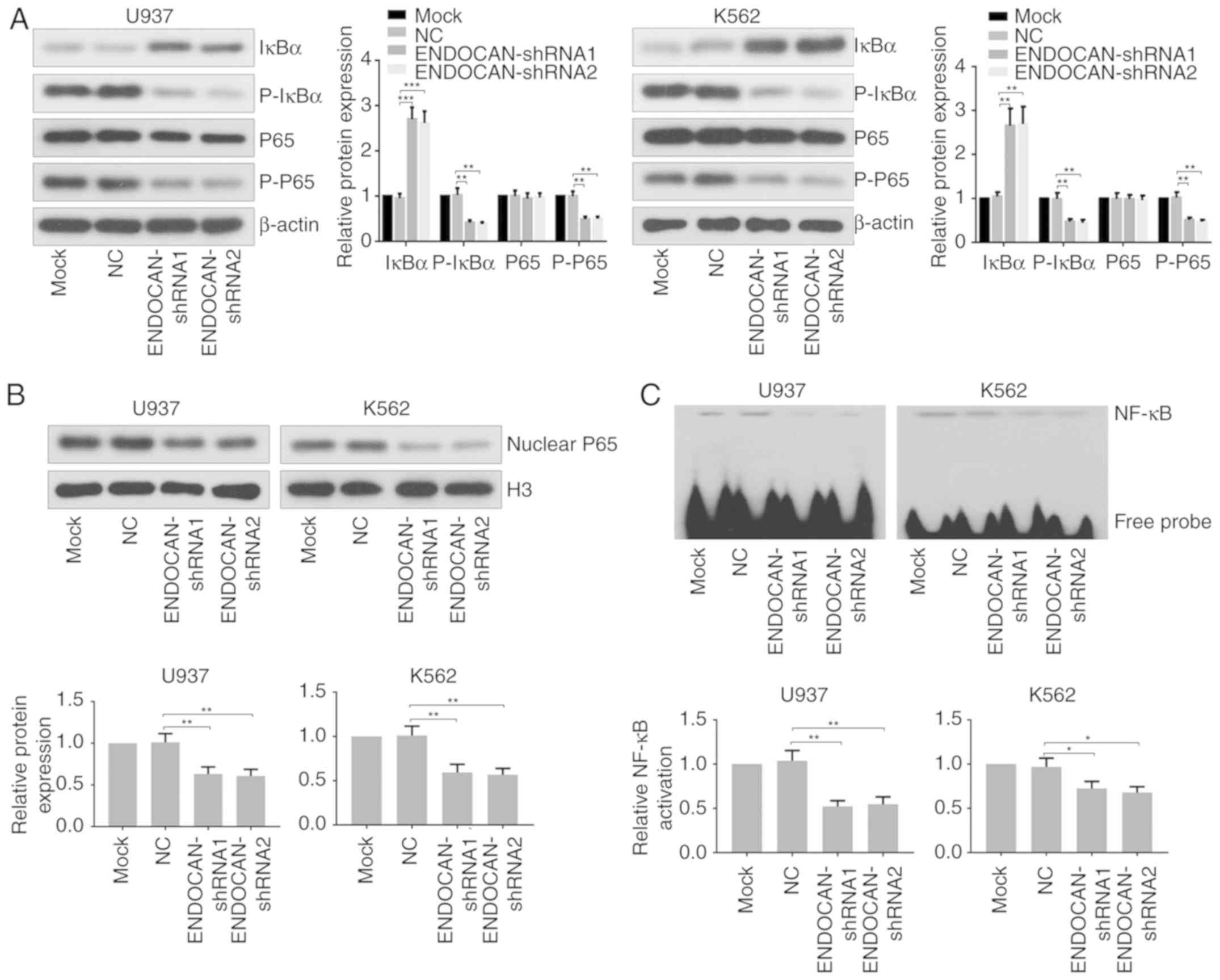
Finally, the effect of ENDOCAN knockdown on the
activity of NF-κB was assessed in U937 and K562 cells infected with
lentivirus carrying ENDOCAN shRNA-1, −2 or scramble shRNA for 48 h.
As shown in Fig. 5A, the results
of the western blot analysis showed that the expression of IκBα was
increased, whereas the expression levels of p-IκBα and p-p65 were
decreased following ENDOCAN knockdown in U937 and K562 cells. In
addition, the expression of nuclear p65 was found to be reduced in
the ENDOCAN-silenced myeloid leukemia cells, as determined by
western blot analysis (Fig. 5B).
The EMSA assay revealed that the DNA binding activity of NF-κB was
attenuated in the myeloid leukemia cells following ENDOCAN
silencing (Fig. 5C).
Discussion
The anticancer effect of ENDOCAN knockdown has been
investigated in several types of cancer, including hepatocarcinoma
(15), and prostate (23), head and neck (24), and gastric cancer (25). ENDOCAN has been found to be
elevated in the serum and bone marrow blasts of patients with acute
leukemia (16,17). In the present study, it was
demonstrated that ENDOCAN silencing effectively suppressed cell
proliferation, induced G0/G1 cell cycle arrest, stimulated cell
apoptosis and attenuated NF-κB activity in myeloid leukemia cells
in vitro.
Uncontrolled proliferation and suppression of
apoptosis is a hallmark of tumor cells. PCNA has been widely used
as a marker in the assessment of tumor cell proliferation (26). The inhibition of cyclin D1, a key
cell cycle regulatory protein, can induce cell-cycle arrest at the
G0/G1 phase (27). Members of the
BCL2 family are pivotal in the regulation of apoptosis (28). For example, reducing the ratio of
BCL2 to BCX can trigger the release of cytochrome c from
mitochondria, leading to an increase in cleaved caspases-3 and −9
and PARP, which contributes to cell apoptosis (29). By detecting the above factors, the
antiproliferation and pro-apoptotic effects of ENDOCAN silencing on
myeloid leukemia cells were confirmed in the present study.
The NF-κB family consists of five members, NF-κB1
(p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB and c-Rel, which
form homo- or heterodimers. IκB sequesters NF-κB in the cytoplasm
and prevents its translocation to the nucleus. Upon the appropriate
stimulus, IκB is phosphorylated and degraded, resulting in the
translocation of NF-κB into the nucleus, where it can bind to
specific DNA sequences in the promoters of target genes and
stimulate their transcription (30,31).
Previous studies have shown that NF-κB is constitutively activated
in several types of cancer, including thyroid (32) and breast cancer (33), melanoma (34) and leukemia (35). It has been reported that the
inhibition of NF-κB can restrain the proliferation and migration,
and promote the apoptosis of leukemia cells (36). The function of ENDOCAN in
regulating the activity of NF-κB has been demonstrated in
colorectal cancer and hepatocarcinoma (15,37).
Kang et al (37) revealed
that ENDOCAN can interact with NF-κB and activate NF-κB promoter.
These reports, in combination with the present findings, suggest
that the antitumor effect of ENDOCAN silencing may, at least in
part, be attributed to a reduction in the activity of NF-κB in
myeloid leukemia cells. Based on the present data, ENDOCAN
inhibitors offer potential to be developed as therapeutic agents
for leukemia.
Furthermore, NF-κB has been demonstrated to be an
important mediator of autophagy in cancer (38). Autophagy is the process through
which cellular contents are engulfed by autophagosomes and
delivered to lysosomes for degradation, and has emerged as a key
pathway in cancer development and therapy (39). A previous study has shown that the
suppression of autophagy can assist in overcoming chemoresistance
in leukemia (40). Wang et
al (41) found that
bardoxolone methyl, a potent NF-κB inhibitor, induced cell cycle
arrest, apoptosis and autophagy in leukemia cells. Therefore,
ENDOCAN may be involved in the autophagy of leukemia cells. Other
tumor growth-related pathways, including p53, epidermal growth
factor receptor (EGFR), vascular EGFR type 2, phosphoinositide
3-kinase, Akt and mammalian target of rapamycin are also regulated
by ENDOCAN (42,43). These pathways may also be involved
in ENDOCAN-mediated proliferation and apoptosis.
In conclusion, the results of the present study
showed that the knockdown of ENDOCAN inhibited cell growth and
induced cell apoptosis, which was accompanied by the inhibition of
NF-κB activity in myeloid leukemia cells, indicating that ENDOCAN
is a promising therapeutic target in leukemia.
Acknowledgements
Not applicable.
Funding
This work was supported by the Nature Science
Foundation of Liaoning Province (grant no. 20170541015).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
WY and LS conceived, designed and prepared the
manuscript. LS performed the experiments, analyzed and interpreted
the data. CS and JS performed part of the experiments and data
collection. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Miller KD, Siegel RL, Lin CC, Mariotto AB,
Kramer JL, Rowland JH, Stein KD, Alteri R and Jemal A: Cancer
treatment and survivorship statistics, 2016. CA Cancer J Clin.
66:271–289. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang D, Hong Q, Shen Y, Xu Y, Zhu H, Li
Y, Xu C, Ouyang G and Duan S: The diagnostic value of DNA
methylation in leukemia: A systematic review and meta-analysis.
PLoS One. 9:e968222014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Weng HH, Tsai SS, Chen CC, Chiu HF, Wu TN
and Yang CY: Childhood leukemia development and correlation with
traffic air pollution in Taiwan using nitrogen dioxide as an air
pollutant marker. J Toxicol Environ Health A. 71:434–438. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guo J, Cahill MR, McKenna SL and
O'Driscoll CM: Biomimetic nanoparticles for siRNA delivery in the
treatment of leukaemia. Biotechnol Adv. 32:1396–1409. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tsai TC, Huang HP, Chang KT, Wang CJ and
Chang YC: Anthocyanins from roselle extract arrest cell cycle G2/M
phase transition via ATM/Chk pathway in p53-deficient leukemia
HL-60 cells. Environ Toxicol. 32:1290–1304. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lassalle P, Molet S, Janin A, Heyden JV,
Tavernier J, Fiers W, Devos R and Tonnel AB: ESM-1 is a novel human
endothelial cell-specific molecule expressed in lung and regulated
by cytokines. J Biol Chem. 271:20458–20464. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balta S, Mikhailidis DP, Demirkol S,
Ozturk C, Kurtoglu E, Demir M, Celik T, Turker T and Iyisoy A:
Endocan-A novel inflammatory indicator in newly diagnosed patients
with hypertension: A pilot study. Angiology. 65:773–777. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toshikuni N, Ozaki K, George J and
Tsutsumi M: Serum endocan as a survival predictor for patients with
liver cirrhosis. Can J Gastroenterol Hepatol. 29:427–430. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Balta I, Balta S, Koryurek OM, Demirkol S,
Mikhailidis DP, Celik T, Cakar M, Kucuk U, Eksioglu M and Kurt YG:
Serum endocan levels as a marker of disease activity in patients
with Behcet disease. J Am Acad Dermatol. 70:291–296. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mihajlovic DM, Lendak DF, Brkic SV,
Draskovic BG, Mitic GP, Novakov Mikic AS and Cebovic TN: Endocan is
useful biomarker of survival and severity in sepsis. Microvasc Res.
93:92–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Grigoriu BD, Depontieu F, Scherpereel A,
Gourcerol D, Devos P, Ouatas T, Lafitte JJ, Copin MC, Tonnel AB and
Lassalle P: Endocan expression and relationship with survival in
human non-small cell lung cancer. Clin Cancer Res. 12:4575–4582.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maurage CA, Adam E, Mineo JF, Sarrazin S,
Debunne M, Siminski RM, Baroncini M, Lassalle P, Blond S and
Delehedde M: Endocan expression and localization in human
glioblastomas. J Neuropathol Exp Neurol. 68:633–641. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ozaki K, Toshikuni N, George J, Minato T,
Matsue Y, Arisawa T and Tsutsumi M: Serum endocan as a novel
prognostic biomarker in patients with hepatocellular carcinoma. J
Cancer. 5:221–230. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scherpereel A, Gentina T, Grigoriu B,
Senechal S, Janin A, Tsicopoulos A, Plenat F, Bechard D, Tonnel AB
and Lassalle P: Overexpression of endocan induces tumor formation.
Cancer Res. 63:6084–6089. 2003.PubMed/NCBI
|
|
15
|
Yang J, Sheng S, Yang Q, Li L, Qin S, Yu S
and Zhang X: Endocan silencing induces programmed cell death in
hepatocarcinoma. Oncol Lett. 14:5333–5339. 2017.PubMed/NCBI
|
|
16
|
Xu Z, Zhang S, Zhou Q, Wang Y and Xia R:
Endocan, a potential prognostic and diagnostic biomarker of acute
leukemia. Mol Cell Biochem. 395:117–123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hatfield KJ, Lassalle P, Leiva RA, Lindas
R, Wendelboe O and Bruserud O: Serum levels of endothelium-derived
endocan are increased in patients with untreated acute myeloid
leukemia. Hematology. 16:351–356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-ΔΔCT method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang X, Zhou Q, Yu Z, Wu X, Chen X, Li J,
Li C, Yan M, Zhu Z, Liu B and Su L: Cancer-associated
fibroblast-derived Lumican promotes gastric cancer progression via
the integrin beta1-FAK signaling pathway. Int J Cancer.
141:998–1010. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo L, Ou S, Ma X, Zhang S and Lai Y:
MACC1 silencing inhibits cell proliferation and induces cell
apoptosis of lung adenocarcinoma cells through the beta-catenin
pathway. Neoplasma. 65:552–560. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu X, Song S, Wang Q, Yuan T and He J: A
mutation in beta-amyloid precursor protein renders SHSY5Y cells
vulnerable to isoflurane toxicity: The role of inositol 1,4,5
trisphosphate receptors. Mol Med Rep. 14:5435–5442. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qin M, Luo Y, Lu S, Sun J, Yang K, Sun G
and Sun X: Ginsenoside F1 ameliorates endothelial cell inflammatory
injury and prevents atherosclerosis in mice through A20-mediated
suppression of NF-κB signaling. Front Pharmacol. 8:9532017.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rebollo J, Geliebter J and Reyes N: ESM-1
siRNA knockdown decreased migration and expression of CXCL3 in
prostate cancer cells. Int J Biomed Sci. 13:35–42. 2017.PubMed/NCBI
|
|
24
|
Bender O, Gunduz M, Cigdem S, Hatipoglu
OF, Acar M, Kaya M, Grenman R, Gunduz E and Ugur KS: Functional
analysis of ESM1 by siRNA knockdown in primary and metastatic head
and neck cancer cells. J Oral Pathol Med. 47:40–47. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao W, Sun M, Li S, Wang Y and Liu J:
Biological and clinical implications of endocan in gastric cancer.
Tumour Biol. 35:10043–10049. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Furuta C, Li C, Taneda S, Suzuki AK,
Kamata K, Watanabe G and Taya K: Immunohistological study for
estrogenic activities of nitrophenols in diesel exhaust particles.
Endocrine. 27:33–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ye D, Luo H, Lai Z, Zou L, Zhu L, Mao J,
Jacob T, Ye W, Wang L and Chen L: ClC-3 chloride channel proteins
regulate the cell cycle by Up-regulating cyclin D1-CDK4/6 through
suppressing p21/p27 expression in nasopharyngeal carcinoma cells.
Sci Rep. 6:302762016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hata AN, Engelman JA and Faber AC: The
BCL2 family: Key mediators of the apoptotic response to targeted
anticancer therapeutics. Cancer Discov. 5:475–487. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qin J, Xie LP, Zheng XY, Wang YB, Bai Y,
Shen HF, Li LC and Dahiya R: A component of green tea,
(−)-epigallocatechin-3-gallate, promotes apoptosis in T24 human
bladder cancer cells via modulation of the PI3K/Akt pathway and
Bcl-2 family proteins. Biochem Biophys Res Commun. 354:852–857.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dolcet X, Llobet D, Pallares J and
Matias-Guiu X: NF-κB in development and progression of human
cancer. Virchows Arch. 446:475–482. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pacifico F and Leonardi A: Role of
NF-kappaB in thyroid cancer. Mol Cell Endocrinol. 321:29–35. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang W, Nag SA and Zhang R: Targeting the
NFkappaB signaling pathways for breast cancer prevention and
therapy. Curr Med Chem. 22:264–289. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Madonna G, Ullman CD, Gentilcore G,
Palmieri G and Ascierto PA: NF-kappaB as potential target in the
treatment of melanoma. J Transl Med. 10:532012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lopez-Guerra M and Colomer D: NF-kappaB as
a therapeutic target in chronic lymphocytic leukemia. Expert Opin
Ther Targets. 14:275–288. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cilloni D, Martinelli G, Messa F,
Baccarani M and Saglio G: Nuclear factor kB as a target for new
drug development in myeloid malignancies. Haematologica.
92:1224–1229. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang YH, Ji NY, Han SR, Lee CI, Kim JW,
Yeom YI, Kim YH, Chun HK, Kim JW, Chung JW, et al: ESM-1 regulates
cell growth and metastatic process through activation of NF-kappaB
in colorectal cancer. Cell Signal. 24:1940–1949. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Baldwin AS: Regulation of cell death and
autophagy by IKK and NF-kappaB: Critical mechanisms in immune
function and cancer. Immunol Rev. 246:327–345. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kondo Y, Kanzawa T, Sawaya R and Kondo S:
The role of autophagy in cancer development and response to
therapy. Nat Rev Cancer. 5:726–734. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Auberger P and Puissant A: Autophagy, a
key mechanism of oncogenesis and resistance in leukemia. Blood.
129:547–552. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang XY, Zhang XH, Peng L, Liu Z, Yang YX,
He ZX, Dang HW and Zhou SF: Bardoxolone methyl (CDDO-Me or RTA402)
induces cell cycle arrest, apoptosis and autophagy via
PI3K/Akt/mTOR and p38 MAPK/Erk1/2 signaling pathways in K562 cells.
Am J Transl Res. 9:4652–4672. 2017.PubMed/NCBI
|
|
42
|
Sumei Z, Shaolong C, Xiang W, Yinliang Q,
Qing Z and Yuan W: Endocan reduces the malign grade of gastric
cancer cells by regulating associated protein expression. Tumour
Biol. 37:14915–14921. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cai L, Leng ZG, Guo YH, Lin SJ, Wu ZR, Su
ZP, Lu JL, Wei LF, Zhuge QC and Jin Kand Wu ZB: Dopamine agonist
resistance-related endocan promotes angiogenesis and cells
viability of prolactinomas. Endocrine. 52:641–651. 2016. View Article : Google Scholar : PubMed/NCBI
|