Introduction
The incidence rate of type 2 diabetes (T2D) is
rising sharply around the world. T2D is classified as a metabolic
disorder, but increasing evidence has indicated that it is also
strongly associated with inflammation mediated by the innate immune
system (1,2). Unlike type 1 diabetes (T1D), the
inflammatory response in the islets of patients with T2D is ‘low
grade’. It was demonstrated that elevated circulating levels of
interleukin (IL)-1β, IL-6, MCP1 and C-reactive protein are
predictive of T2D; these inflammatory cytokines are known to induce
insulin resistance and impair insulin secretion (1). In particular, IL-1β was found to
regulate pancreatic β-cell death and dysfunction (2). Several studies have described the
involvement of IL-1β in T1D; IL-1β is also an important
inflammatory mediator in T2D (1,2).
Currently, clinical trials using IL-1β-blocking antibodies to treat
patients with T2D have been very encouraging, clearly indicating
that IL-1β plays vital roles in the onset of T2D.
Recently, a large number of studies have clarified
the mechanisms by which biologically active IL-1β is produced. The
secretion of bioactive IL-1β is primarily controlled by the
activation of caspase-1 through the assembly of inflammasomes
formed by NOD-like receptor family pyrin domain-containing protein
3 (NLRP3), the adaptor protein ASC and procaspase-1. The
best-characterized inflammasome protein to date is NLRP3, which is
also recognized as a sensor of metabolic dysregulation, such as the
pathogenic status of T2D (3). In
pancreatic β-cells and macrophages, increased reactive oxygen
species (ROS) triggers the activation of the NLRP3 inflammasome
through a thioredoxin interacting protein (TXNIP)- or islet amyloid
polypeptide-dependent pathway, respectively. In addition,
TXNIP−/− or NLRP3−/− mice showed improved
glucose tolerance and insulin sensitivity (2). These studies encouraged the
hypothesis that the NLRP3 inflammasome mediates IL-1β secretion,
and IL-1β mechanistically regulates the pathogenesis of T2D.
Autophagy has been demonstrated to be necessary to
maintain the structure, mass and function of pancreatic β-cells
(4). In T1D and T2D, chronic
elevation of blood glucose concentration causes an imbalance of
antioxidant activity in cells, leading to
oxidative/nitrosative-mediated stress and injury. Furthermore, a
study has indicated that autophagy serves as a defense mechanism to
clear proteins and organelles damaged by oxidative stress (5). Autophagy is also implicated in the
maintenance of mitochondrial function by facilitating mitochondrial
turnover (6). Mitochondria are the
primary site of ROS generation, and accumulated ROS induce
mitochondria damage and dysfunction. Thus, autophagy has a vital
role in the maintenance of the structural and functional integrity
of mitochondria (7), and impaired
autophagy may result in the accumulation of ROS (8–10).
Therefore, impaired autophagy associated with ROS accumulation may
contribute to insulin resistance and pancreatic β-cell dysfunction.
Autophagy, as a dynamic lysosomal degradation of damaged organelles
and proteins, may be associated with the dysfunction and death of
pancreatic β-cells, which contributes to the onset of T2D. Although
IL-1β is an important inflammatory mediator of T2D (11), the process leading to the increased
expression and secretion of IL-1β in T2D remains unclear. The
current results indicated that enhanced autophagy may induce
mitochondrial damage, inhibit the accumulation of ROS and
subsequently suppress IL-1β production. Therefore, the role of
autophagy was investigated in diabetes-induced hyperlipemic INS-1
cells with lipopolysaccharide (LPS)-induced inflammasome activation
and IL-1β production. The aim of this research was to investigate
the association between the autophagy and inflammation in INS-1
cells, which hoped to provide novel ideas for T2D treatment.
Materials and methods
Cell culture
INS-1 cells were acquired from the Department of
Endocrinology, The Second Affiliated Hospital of Harbin Medical
University (Harbin, China). INS-1 cells were passaged in RPMI-1640
medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% (v/v) fetal bovine serum (FBS; Hangzhou
Sijiqing Biological Engineering Materials Co., Ltd., Hangzhou,
China) in a humidified atmosphere containing 95% air and 5%
CO2 (12). The cells
were treated with LPS (100 ng/ml; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 24 h. N-acetyl-L-cysteine (NAC; 5 µM;
Sigma-Aldrich; Merck KGaA) was used to inhibit ROS (13) and 3-methyladenine (3-MA; 5 mM;
Sigma-Aldrich; Merck KGaA) was used to inhibit autophagy (4).
Cell viability assay
Cell viability was assessed using Cell Counting Kit
8 (CCK8) (Dojindo Molecular Technologies, Inc., Kumamoto, Japan)
according to the manufacturer's protocol (12). Briefly, the cells were treated with
LPS (100 ng/ml), NAC (5 µM) and 3-MA (5 mM) for 24 h, and the
detail treatments for each experiment were displayed in the figure
legends. The INS-1 cells were plated in 96-well plates at a density
of 5×103 per well and treated with CCK8 solution at 37°C
for 1 h. Absorbance was detected using a microplate reader at a
wavelength of 450 nm (12).
Apoptosis assay
Cell apoptosis was detected using flow cytometry.
INS-1 cells were stained with Annexin V and propidium iodide (PI)
using the Annexin-V-FLUOS staining kit (Roche Diagnostics, Basel,
Switzerland) in accordance with the manufacturer's protocol.
Briefly, the INS-1 cells were plated in 24-well plates
(1.2×105 cells/cm2), treated with lysosomal
proteases, and stained with Annexin V (1 µM) and PI (0.1 µM) for 10
min at 25°C in the dark. Images of the apoptotic cells were
analyzed using a fluorescence microscope (Nikon Eclipse TE2000-U;
Nikon Corporation, Tokyo, Japan) with an excitation wavelength in
the range of 450–500 nm and a detection wavelength in the range of
515–565 nm (14).
Intracellular ROS measurement
2′,7′-Dichlorofluorescin diacetate (DCFH-DA) was
used to analyze intracellular ROS by flow cytometry. INS-1 cells
were divided into four groups (control, LPS, LPS + NAC or LPS +
3-MA), seeded in 6-well plates (1×106 cells/ml) and
incubated for 24 h. Then, DCFH-DA (10 µM) was incubated with the
cells for 20 min at 37°C. The medium including DCFH-DA was
aspirated to remove the extracellular dye, washed with serum-free
medium three times and then ice-cold serum-free medium was added to
the cells. The cells were placed on ice in the dark. The
fluorescence was measured using a flow cytometer (BD FACSCalibur;
BD Biosciences, Franklin Lakes, NJ, USA) at an excitation
wavelength of 488 nm and an emission wavelength of 530 nm, and the
data was analyzed by using FACSDiva version 6.1.2 (BD
Biosciences).
Western blotting
Cells were washed with PBS and lysed in lysis buffer
[62.5 mM Tris-HCl (pH 6.8), 2% SDS, 5% 2-Mercaptoethanol (BME), 1%
TritonX-100, 1 mM EDTA, 1 mM EGTA, 10 mM dithiothreitol and 1 mm
Na3VO4]. The lysates were then incubated on
ice for 30 min, followed by centrifugation at 8,000 × g for 5 min
at 4°C. The protein concentrations were determined using a
bicinchoninic acid assay. Equal quantities of protein (50 µg) were
separated by SDS-PAGE on 10–15% gels. Following the transfer of the
proteins to polyvinylidine difluoride membranes, the blots were
blocked with 5% non-fat dry milk at room temperature for 1 h. The
membranes were exposed to specific primary antibodies at 4°C
overnight. The following antibodies were used at a 1:1,000
dilution: Light chain 3 (LC3; Cell Signaling Technology, Inc.,
Danvers, MA, USA; cat. no. 2775), IL-1β (Cell Signaling Technology,
Inc.; cat. no. 2002), caspase-1 (Abcam, Cambridge, UK; cat. no.
2225), NF-kB p65 (Abcam, Cambridge, UK; cat. no. ab16502)and
β-actin (Cell Signaling Technology, Inc.; cat. no. 8475). The
membranes were washed with PBS containing 0.1% of Tween-20 three
times, then incubated with horseradish peroxidase-conjugated
secondary antibodies (AP178P; cat. no. AC111P; 1:5,000;
Sigma-Aldrich; Merck KGaA) at room temperature for 1 h (12). Protein bands were detected using
enhanced chemiluminescence (Beyotime Institute of Biochemistry,
Haimen, China). Immunoblots were quantified by densitometric
analysis using Image Lab software v2.0.1 (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The relative expression level of a
certain protein was normalized to β-actin and experiments were
repeated in triplicate.
Transmission electron microscopy
(TEM)
The INS-1 cells were fixed in 2% paraformaldehyde
and 2.5% glutaraldehyde in phosphate buffer (pH 7.4) at room
temperature for 1 h, followed by 1% OsO4 at room
temperature for 1 h. The cells were dehydrated through a graded
ethanol series (70, 80, 95 and 100%, 5 min each) and then embedded
using the Quetol 651 Embedding Kit (Polysciences, Inc., Warrington,
PA, USA). The embedded cells were sliced into 70 nm sections. Then
the sections were exposed to uranyl acetate (1%) and lead citrate
(1%) at room temperature for 30 min, then wash with
ddH2O three times and examined by TEM (magnification,
×40,000; H-7100; Hitachi, Ltd., Tokyo, Japan) as marked in the
figure legends.
Statistical analysis
All values are reported as the mean ± standard
deviation. Comparisons of a single variable in more than two groups
were evaluated using one-way analysis of variance followed by
Tukey's multiple comparison tests using GraphPad Prism Version 5
(GraphPad Software, Inc., La Jolla, CA, USA). Data were analyzed
using the paired and unpaired t-test between two groups using SPSS
12.0 software (SPSS, Inc., Chicago, IL, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
LPS induces INS-1 cell apoptosis and
autophagy
LPS, an outer membrane component of Gram negative
bacteria, has been linked to obesity and insulin resistance
(15). It has been reported that
high fat-containing diets elevate enterobacterial production and
the translocation of LPS to cells, while LPS administration to
healthy subjects rapidly induced insulin resistance (16). Thus, the effect of LPS (100 ng/ml,
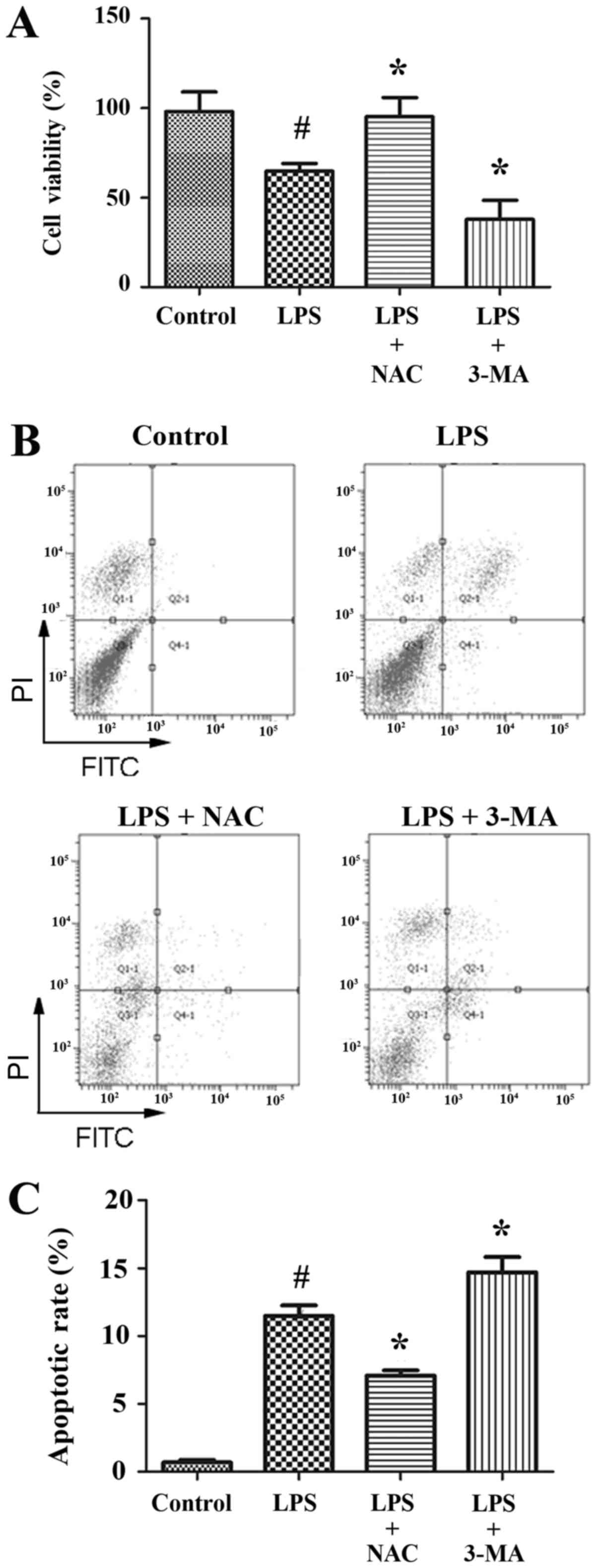
24 h) on INS-1 cells viability was investigated in the current
study. LPS treatment significantly decreased the viability of INS-1
cells (67%) compared with non-treated cells (Fig. 1A). Using an Annexin V-FITC/PI
quantification assay apoptosis was detected in LPS-treated INS-1
cells. LPS dramatically increased the number of apoptotic cells
(11.5%) compared with the control group (0.7%; Fig. 1B and C). The activation of
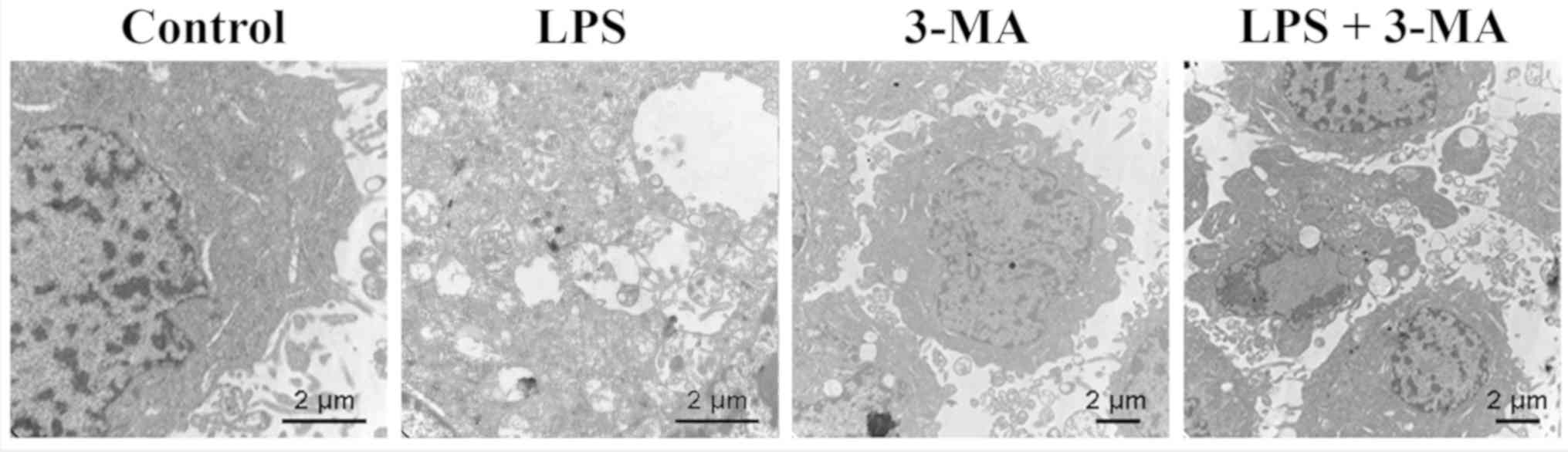
autophagy in INS-1 cells by LPS (100 ng/ml) was detected by
electron microscopy. In INS-1 cells, double-membrane autophagic
vesicles containing cell organelles in the cytoplasm of INS-1 cells
formed an integrated autophagosome (Fig. 2). LPS treatment increased the
formation of autophagic vesicles, and 3-MA treatment ameliorated
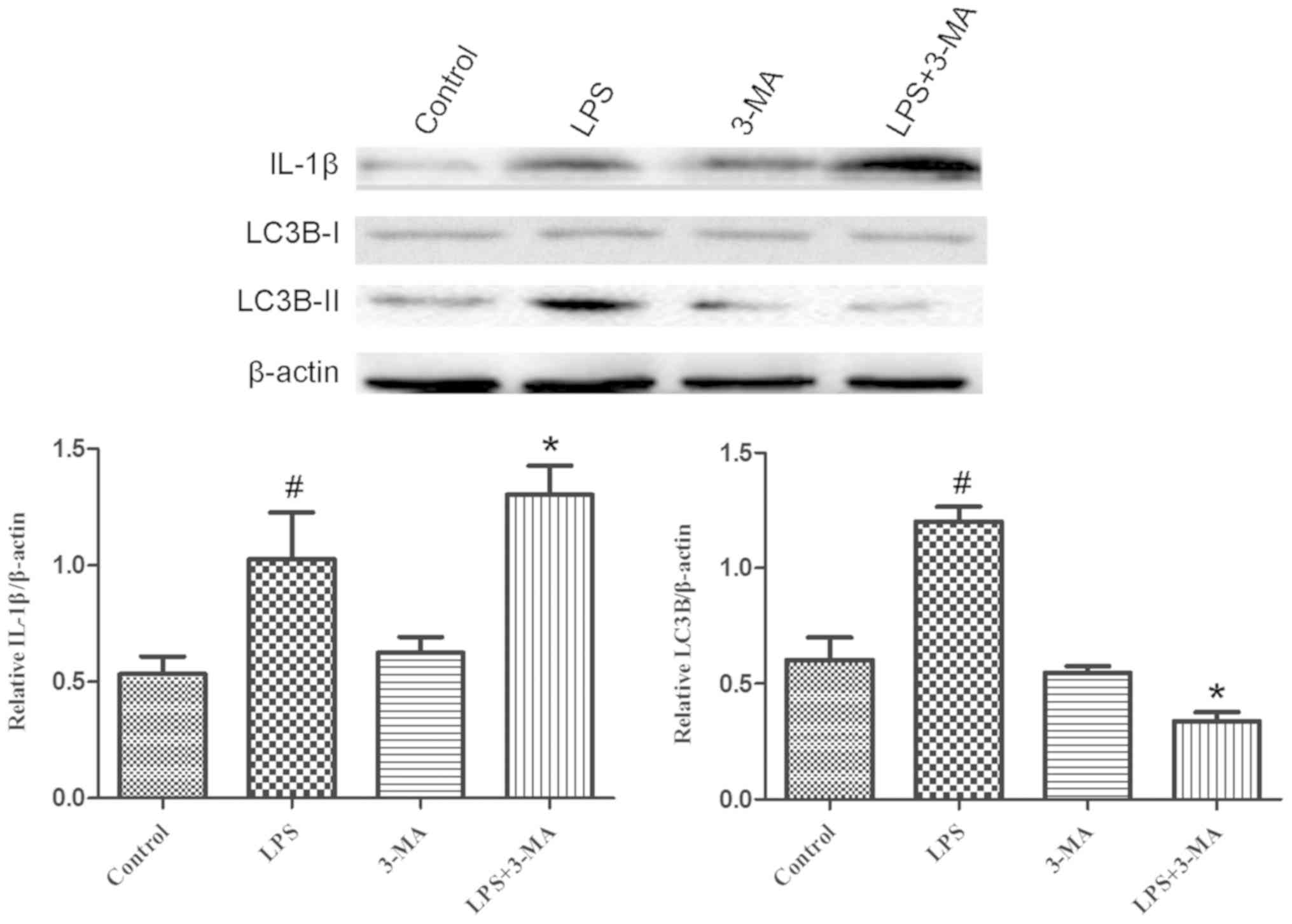
the LPS-induced increase of autophagic vesicles formation (Fig. 2). LC3B protein expression was
detected by western blotting, which further corroborated that LPS
activated autophagy in INS-1 cells. LC3B is a soluble protein
distributed ubiquitously in mammalian cells. Upon the induction of
autophagy, LC3B is cleaved by Atg4B to generate LC3B-II. Therefore
LC3B is considered a specific autophagy marker (17). As expected, the expression of
LC3B-II was remarkably increased in INS-1 cells following LPS
treatment (Fig. 3).
Role of autophagy in the LPS-induced
Caspase 1 cleavage and IL-1β maturation
Previous studies have indicated that autophagy
protects INS-1 cell viability under various challenges, including
palmitate and ROS-induced damage (18,19).
However, the role of autophagy in the inflammation of pancreatic
β-cells (INS-1) has not been well established. To investigate the
role of autophagy in LPS-induced INS-1 cell death, 3-MA, a specific
autophagy inhibitor, was used. Treatment with 3-MA only did not
exhibit a significant effect on cell proliferation and viability in
INS-1 cells (data not shown). Following pre-treatment with 3-MA (5
mM, 24 h), the viability of LPS-treated INS-1 cells was decreased
to 40% (Fig. 1A), which was
significantly lower than that of INS-1 cells only treated with LPS
only (67%). Similarly, apoptosis was also increased to 14.7% in
LPS-treated INS-1 cells pretreated with 3-MA (Fig. 1B and C), which was significantly
higher than that of LPS-treated INS-1 cells. Subsequently, the
expression of IL-1β and LC3B was determined using western blotting.
The expression of LC3B-II was markedly decreased in LPS-treated
INS-1 cells pretreated with 3-MA compared with that of INS-1 cells
treated with LPS only (Fig. 3).
Notably, the expression of IL-1β was significantly enhanced in
LPS-treated INS-1 cells pretreated with 3-MA, which was higher than
that of INS-1 cells treated with LPS only (Fig. 3). These results demonstrated that
autophagy may have protective effects on the LPS-induced
inflammation of INS-1 cells; autophagy may also be necessary for
the maintenance of the normal architecture and function of INS-1
cells.
Inhibition of autophagy augments
inflammation via ROS-mediated Caspase 1 cleavage and IL-1β
maturation
It was also investigated how autophagy regulates
LPS-induced NLRP3 inflammasome activation in INS-1 cells. ROS
activate the NLRP3 inflammasome, amplify the inflammatory response
and promote pro-inflammatory cytokine secretion. Therefore, it was
hypothesized that ROS have an important role in LPS-induced NLRP3
inflammasome activation. Thus, NAC (an ROS inhibitor) was used to
abolish ROS in INS-1 cells (13).
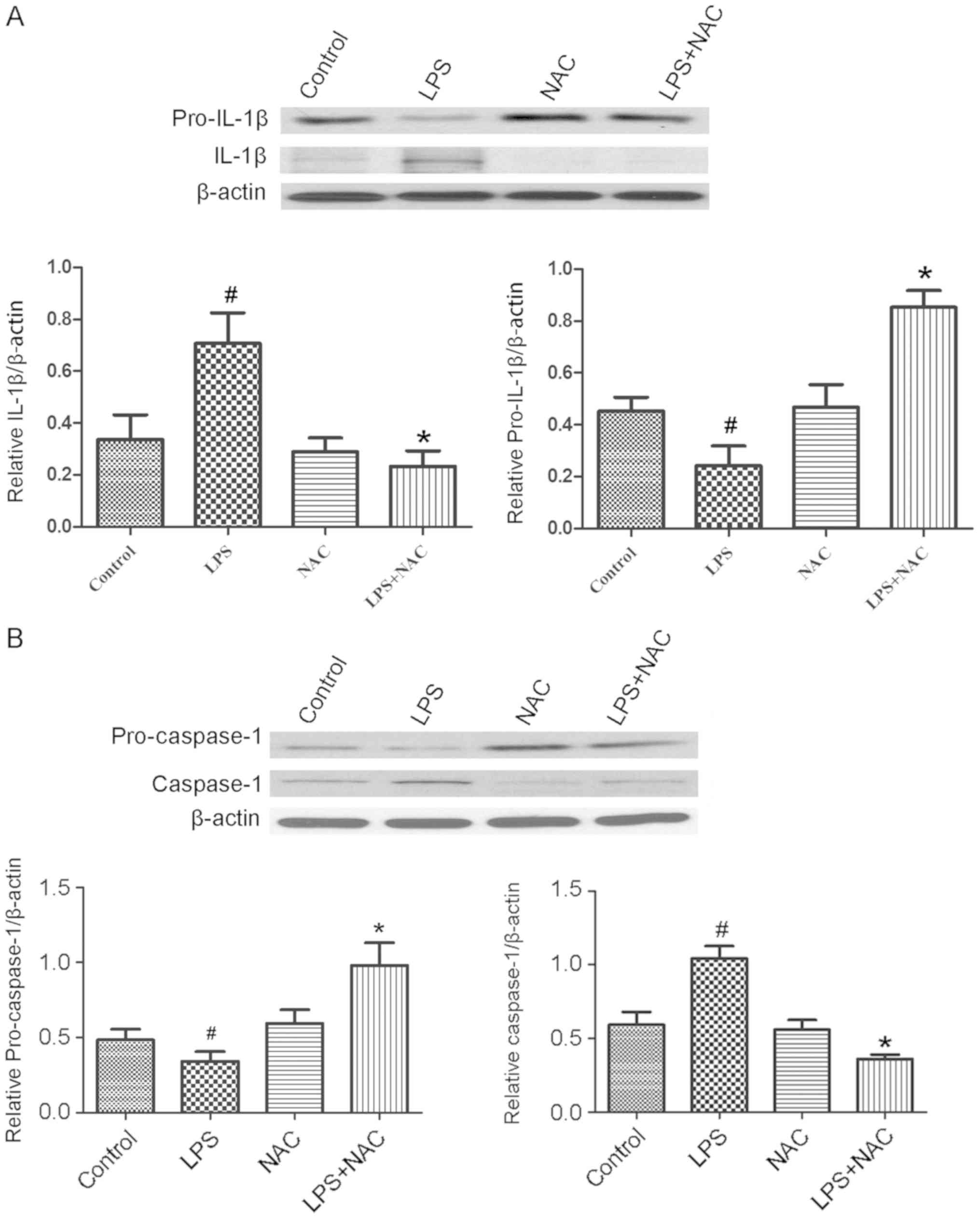
The expression levels of cleaved caspase-1 and secreted IL-1β were
markedly increased in INS-1 cells following LPS treatment (100
ng/ml for 24 h), which may indicate the activation of
inflammasomes, especially for NLRP3 inflammasome (Fig. 4). Additionally, a significant
increase in ROS production was observed in the LPS treated INS-1
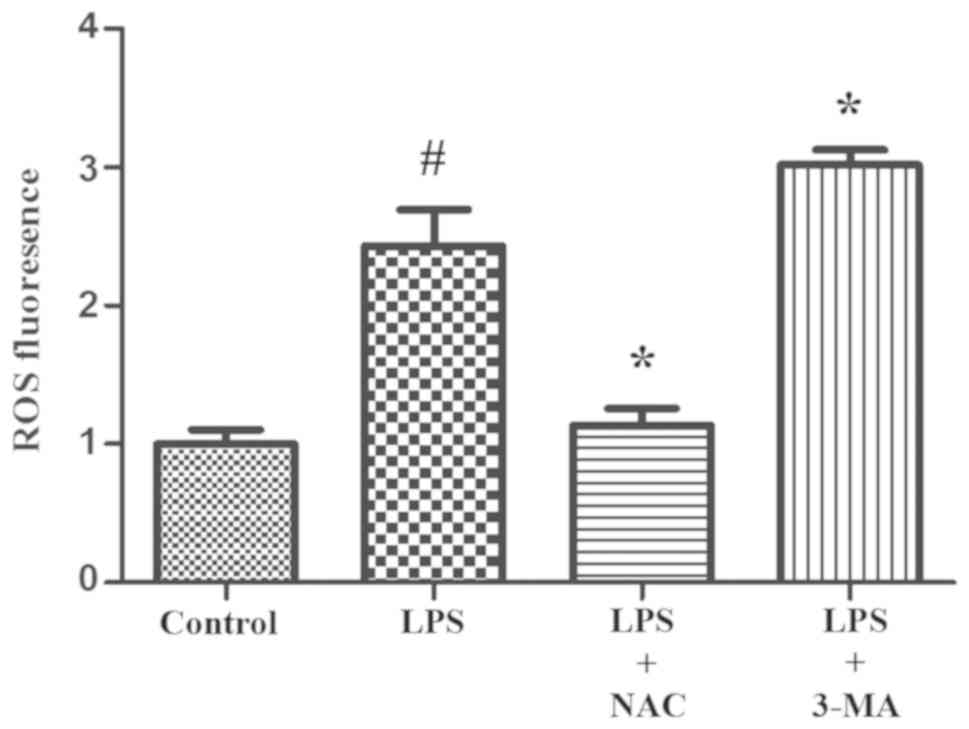
cells compared with the control cells (Fig. 5). On the other hand, when ROS
levels were inhibited by NAC (5 µM), the expression of IL-1β was
decreased, which was accompanied by the significantly enhanced
expression of pro-IL-1β (Fig. 4A).
Similarly, the expression of caspase-1 was significantly decreased
by NAC following LPS stimulation, which was accompanied by the
significantly enhanced expression of pro-caspase-1 (Fig. 4B). These results suggested that the
inhibition of ROS suppressed NLRP3 inflammasome activation and
reduced the maturation of IL-1β.
Whether autophagy protects against NLRP3
inflammasome activation through the inhibition of ROS accumulation
was also investigated. LPS treatment (100 ng/ml, 24 h) elevated ROS
generation in INS-1 cells (Fig.
5). Pretreatment with 3-MA (5 mM, 24 h), an autophagy
inhibitor, significantly increased the ROS level in LPS-treated
INS-1 cells (Fig. 5) and decreased
the expression of LC3B-II (Fig.
3).
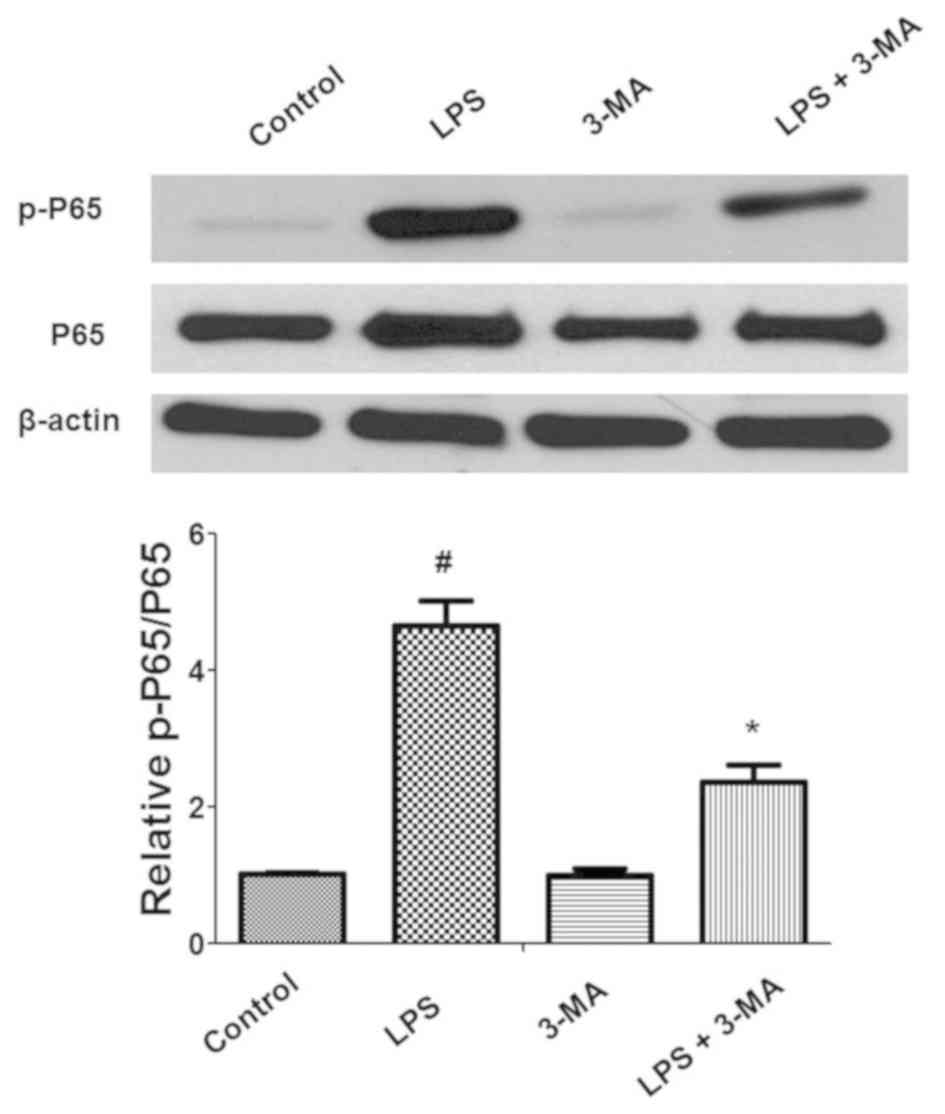
Additionally, the effect of autophagy on nuclear
factor-κB (NF-κB) activation during inflammation was also
determined. LPS significantly induced the phosphorylation of P65,
which promotes the translocation of P65 to the cell nucleus and
activation of the NF-κB pathway (Fig.
6). Pretreatment with 3-MA significantly attenuated LPS-induced
phosphorylation of P65 in INS-1 cells. Collectively, these data
indicated that ROS and NF-κB signaling were associated with
inflammation and autophagy in INS-1 cells.
Discussion
Type 2 diabetes (T2D) is clearly associated with
chronic low-grade inflammation, lipotoxicity and glucotoxicity
(1,20). Autophagy also has a role in T2D
progression by impairing pancreatic β-cell function and promoting
insulin resistance. However, the complete role of autophagy in T2D
remains unclear. To investigate the association between
inflammation and T2D, whether LPS contributed to IL-1β generation
in INS-1 cells was investigated. In the current study, the data
indicated that LPS induced IL-1β production, and that the
inhibition of autophagy by 3-MA reduced cell viability and
increased apoptosis in INS-1 cells. This indicated that autophagy
is required for INS-1 cell survival. Furthermore, it was
demonstrated that autophagy protects against LPS-induced INS-1 cell
death by blocking the ROS-dependent activation of the NLRP3
inflammasome and IL-1β production in INS-1 cells.
Previous studies demonstrated that IL-1β expression
is elevated in human β-cells in response to high concentrations of
glucose (21,22). In β-cells from humans and rats,
IL-1β expression is strongly enhanced by autostimulatory processes
(21,23). Previous investigations using an
immunoaffinity column containing anti-human IL-1β polyclonal
antibodies indicated that IL-1β is toxic to insulin-producing
β-cells (24). Regardless of the
mechanism of action, clinical proof of the importance of IL-1β in
β-cell failure has been obtained in patients with T2D (25,26).
In the present study, LPS induced IL-1β production in INS-1 cells.
The concentration of IL-1β was relatively low because β-cells were
used, rather than macrophages or other inflammatory cells (27). Indeed, even at low concentrations,
IL-1β has been reported to negatively influence β-cell function and
turnover (28).
Autophagy is necessary to maintain the structure,
mass and function of pancreatic β-cells (4). The findings of the current study
indicated that autophagy may be an important factor in the
development of T2D. When autophagy was inhibited with 3-MA, ROS
generation was significantly increased, which was accompanied by
enhanced expression of IL-1β in INS-1 cells. Additionally, the
inhibition of ROS generation by NAC dramatically decreased the
expression of IL-1β in INS-1 cells. The results demonstrated that
the stimulation of autophagic activity may confer beneficial
anti-inflammatory effects through the removal of ROS. Similar
results were observed in aging cells with activated inflammasomes;
these investigations indicated that autophagy is an important
regulator of innate immune responses in the host defense (29). Furthermore, recent studies have
demonstrated that the inhibition of mitophagy may lead to the
accumulation of damaged mitochondria that produce ROS and
consequently activate the inflammasome (30). By contrast, there is now convincing
experimental data to demonstrate that efficient autophagy can
prevent inflammasome activation and inflammatory responses
(31).
The current study further investigated how autophagy
regulates the process of IL-1β secretion in LPS-challenged INS-1
cells. IL-1β is produced as an inactive cytoplasmic precursor,
pro-IL-1β, and requires proteolytic cleavage to become bioactive.
Caspase-1 was initially identified as the main IL-1β-converting
enzyme. It is synthesized as a zymogen, pro-caspase-1, that is
subsequently converted to active caspase-1 through cleavage, and
binds to NLRP3 to form the inflammasome. The findings of the
current study demonstrated that the expression of IL-1β was
significantly enhanced and pro-IL-1β expression was decreased by
treatment with LPS. Additionally, the expression of caspase-1 was
enhanced and the expression of pro-caspase-1 was decreased.
Similarly, a recent study demonstrated that LPS treatment triggered
IL-1β secretion via the NLRP3 inflammasome. It was also
demonstrated that caspase-1 clustering induces the activation of,
and the caspase-1-dependent maturation and secretion of IL-1β
(32). The results suggested that
the inhibition of autophagy may promote ROS generation, NLRP3
inflammasome complex formation and the activation of caspase-1,
which results in the cleavage of pro-IL-1β to form mature
IL-1β.
There are many shortcomings to this study, for
example more experiments are needed to investigate the
interrelationship between autophagy and inflammation, and the
effect of them on the T2D progression and potential molecular
mechanism. A previous study demonstrated the inhibition of
autophagy may lead to the accumulation of damaged mitochondria that
produce ROS and consequently activate inflammation, which induces
pancreatic β-cell death and dysfunction (33). However, the molecular mechanism of
β-cell death in this context is not well understood and, therefore,
requires further research.
In conclusion, the findings of the current study
demonstrated that autophagy has a protective role in the
LPS-induced inflammatory response and cell death. Whether
LPS-induced NLRP3/caspase-1 activation triggered IL-1β generation
through the promotion of ROS accumulation was also investigated.
Furthermore, ROS generation suppressed by autophagy may be a key
mechanism for the inhibition of IL-1β expression in INS-1 cells.
These results suggest that autophagy may be a potential therapeutic
target to delay the inflammatory response in T2D.
Acknowledgements
Not applicable.
Funding
The present work was supported by two grants from
the National Natural Science Foundation of China (grant nos.
81370929 and 81770820), a grant from the Novo Nordisk China
Diabetes Young Scientific Talent Research Funding-2016 and a
clinical medicine grant from the Chinese Medical Association (grant
no. 13040670452).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
LZ and MC performed the cell experiments. JW and YS
performed intracellular ROS measurements and western blot analysis,
WJ and GL performed transmission electron microscopy. YL
contributed to the study design and to the writing of the
manuscript.
Ethics approval and consent to
participate
The present study was approved by Ethic Committee of
First Affiliated Hospital of Harbin Medical University
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of
interest
Glossary
Abbreviations
Abbreviations:
|
LPS
|
lipopolysaccharide
|
|
NAC
|
N-acetyl-L-cysteine
|
|
IL-1β
|
interleukin-1β
|
|
DCFH-DA
|
2′,7′-dichlorofluorescein
diacetate
|
|
LC3
|
light chain 3
|
|
NLRP3
|
NOD-like receptor family pyrin
domain-containing protein 3
|
|
T2D
|
type 2 diabetes
|
|
CCK8
|
Cell Counting Kit-8
|
|
3-MA
|
3-methyladenine
|
|
T1D
|
type 1 diabetes
|
|
ROS
|
reactive oxygen species
|
|
TEM
|
transmission electron microscopy
|
|
SD
|
standard deviation
|
References
|
1
|
Donath MY and Shoelson SE: Type 2 diabetes
as an inflammatory disease. Nat Rev Immunol. 11:98–107. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Eguchi K and Manabe I: Macrophages and
islet inflammation in type 2 diabetes. Diabetes Obes Metab. 3
(Suppl 15):S152–S158. 2013. View Article : Google Scholar
|
|
3
|
Yang CM, Shin DM and Jo EK: The role of
NLR-related protein3 inflammasome in host defense and inflammatory
diseases. Int Neurourol J. 16:2–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Masini M, Bugliani M, Lupi R, del Guerra
S, Boggi U, Filipponi F, Marselli L, Masiello P and Marchetti P:
Autophagy in human type 2 diabetes pancreatic beta cells.
Diabetologia. 52:1083–1086. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gomes LC, Di Benedetto G and Scorrano L:
During autophagy mitochondria elongate, are spared from degradation
and sustain cell viability. Nat Cell Biol. 13:589–598. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldman SJ, Taylor R, Zhang Y and Jin S:
Autophagy and the degradation of mitochondria. Mitochondrion.
10:309–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tal MC, Sasai M, Lee HK, Yordy B, Shadel
GS and Lwasaki A: Absence of autophagy results in reactive oxygen
species-dependent amplification of RLR signaling. Proc Natl Acad
Sci USA. 106:2770–2775. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Petersen KF, Dufour S, Befroy D, Garcia R
and Shulman GI: Impaired mitochondrial activity in the
insulin-resistant offspring of patients with type 2 diabetes. N Eng
J Med. 350:664–671. 2004. View Article : Google Scholar
|
|
11
|
Masters SL, Dunne A, Subramanian SL, Hull
RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen
Z, et al: Activation of the Nlrp3 inflammasome by islet amyloid
polypeptide provides a mechanism for enhanced IL-1β in type 2
diabetes. Nat Immunol. 11:897–904. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jing Yin J, Bo Li Y, Ming Cao M and Wang
Y: Liraglutide improves the survival of INS-1 cells by promoting
macroau-tophagy. Int J Endocrinol Metab. 11:184–190. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wen H, Gris D, Lei Y, Jha S, Zhang L,
Huang MT, Brickey WJ and Ting JP: Fatty acid-induced NLRP3ASC
inflammasome activation interferes with insulin signaling. Nat
Immunol. 12:408–415. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chan JY, Cooney GJ, Biden TJ and Laybutt
DR: Differential regulation of adaptive and apoptotic unfolded
protein response signalling by cytokine-induced nitric oxide
production in mouse pancreatic beta cells. Diabetologia.
54:1766–1776. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cani PD, Amar J, Iglesias MA, Poggi M,
Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et
al: Metabolic endotoxemia initiates obesity and insulin resistance.
Diabetes. 56:1761–1772. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Van der Crabben SN, Blümer RM, Stegenga
ME, Ackermans MT, Endert E, Tanck MW, Serlie MJ, van der Poll T and
Sauerwein HP: Early endotoxemia increases peripheral and hepatic
insulin sensitivity in healthy humans. J Clin Endocrinol Metab.
94:463–468. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi SE, Lee SM, Lee YJ, Li LJ, Lee SJ,
Lee JH, Kim Y, Jun HS, Lee KW and Kang Y: Protective role of
autophagy in palmitate-induced INS-1 beta-cell death.
Endocrinology. 150:126–134. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xia G, Zhu T, Li X, Jin Y, Zhou J and Xiao
J: ROS-mediated autophagy through the AMPK signaling pathway
protects INS-1 cells from human islet amyloid polypeptide-induced
cytotoxicity. Mol Med Rep. 18:2744–2752. 2018.PubMed/NCBI
|
|
20
|
Ashcroft FM, Rohm M, Clark A and Brereton
MF: Is type 2 diabetes a glycogen storage disease of pancreatic β
Cells? Cell Metab. 26:17–23. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Böni-Schnetzler M, Thorne J, Parnaud G,
Marselli L, Ehses JA, Kerr-Conte J, Pattou F, Halban PA, Weir GC
and Donath MY: Increased interleukin (IL)-1β messenger ribonucleic
acid expression in beta-cells of individuals with type 2 diabetes
and regulation of IL-1beta in human islets by glucose and
autostimulation. J Clin Endocrinol Metab. 93:4065–4074. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Maedler K, Sergeev P, Ris F, Oberholzer J,
Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA and Donath MY:
Glucose-induced beta-cell production of interleukin-1beta
contributes to glucotoxicity in human pancreatic islets. J Clin
Invest. 110:851–860. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ribaux P, Ehses JA, Lin-Marq N, Carrozzino
F, Böni-Schnetzler M, Hammar E, Irminger JC, Donath MY and Halban
PA: Induction of CXCL1 by extracellular matrix and autocrine
enhancement by interleukin-1 in rat pancreatic beta-cells.
Endocrinology. 148:5582–5590. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mandrup-Poulsen T, Bendtzen K, Nerup J,
Dinarello CA, Svenson M and Nielsen JH: Affinity-purified human
interleukin I is cytotoxic to isolated islets of langerhans.
Diabetologia. 29:63–67. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Donath MY, Schumann DM, Faulenbach M,
Ellingsgaard H, Perren A and Ehses JA: Islet inflammation in Type 2
diabetes: from metabolic stress to therapy. Diabetes Care. 2 (Suppl
31):S161–S164. 2008. View Article : Google Scholar
|
|
26
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhao G, Dharmadhikari G, Maedler K and
Meyer-Hermann M: Possible role of interleukin-1β in type 2 diabetes
onset and implications for anti-inflammatory therapy strategies.
PLoS Comput Biol. 10:e10037982014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Maedler K, Schumann DM, Sauter N,
Ellingsgaard H, Bosco D, Baertschiger R, Iwakura Y, Oberholzer J,
Wollheim CB, Gauthier BR and Donath MY: Low concentration of
interleukin-1beta induces FLICE-inhibitory protein-mediated β-cell
proliferation in human pancreatic islets. Diabetes. 55:2713–2722.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Green DR, Galluzzi L and Kroemer G:
Mitochondria and the autophagy-inflammation-cell death axis in
organismal aging. Science. 333:1109–1112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kroemer G and Levine B: Autophagic cell
death: The story of a misnomer. Nat Rev Mol Cell Biol. 9:1004–1010.
2008. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-in-teracting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lohitesh K, Saini H, Srivastava A,
Mukherjee S, Roy A and Chowdhury R: Autophagy inhibition
potentiates SAHA-mediated apoptosis in glioblastoma cells by
accumulation of damaged mitochondria. Oncol Rep. 39:2787–2796.
2018.PubMed/NCBI
|