Introduction
Dysfunction of ubiquitin-protein ligase E3A (encoded
by UBE3A gene) is the major cause of Angelman syndrome (AS),
which is a congenital neurodevelopmental disorder presenting a
series of clinical characteristics associated with neurological
damage, including cognitive disability, cognitive delay,
unconscious limb tremor or seizures, as well as inappropriate
emotional response and behavior (1). UBE3A is located at chromosome
15, encoding an enzyme that participates in the protein degradation
process within cells. UBE3A exhibits imprinted gene
expression, that is, only maternally inherited UBE3A can
express functional proteins in the brain (2,3).
This functional defect results in the accumulation of several
proteins in the brain that interfere with neuron development
(4). It has been reported that
single nucleotide variations (SNVs), maternal deletion of
chromosome 15q11.2-q13 region or paternal chromosome 15 uniparental
disomy (UPD) can affect the expression or function of UBE3A
gene and further lead to AS (5).
The majority of reported AS cases are caused by
maternal deletion, whereas few cases are due to paternal UPD, where
both copies of a chromosome are inherited from the father. Studies
have demonstrated that approximately 2–5% of the AS patients were
diagnosed because they had the only paternal allele of the
UBE3A gene, which is specifically silenced in the developing
brain (6,7). Heterodisomy and isodisomy are the two
types of UPD. The former usually occurs when the diploid from one
parent fails to separate into chromatids during meiosis I, while
the latter occurs when the chromatids fail to separate during
meiosis II (8). In consequence,
isodisomy is when both chromosomes are from one parent and
heterodisomy is where the two chromosomes are different copies of
the same chromosome. UPD due to a parental Robertsonian
translocation (ROB) is rare, with a risk of 0.6–0.8% for
non-homologous ROBs (9). However,
isodisomy is a more frequent situation that can result from meiotic
or mitotic duplication of one parental chromosome and may involve
isochromosomes (10).
Several genetic tests have been used for the
screening of UPD, such as microsatellite analysis, single
nucleotide polymorphism (SNP)-based array comparative genomic
hybridization and multiplex ligation-dependent probe amplification
(MLPA). Recently, whole exome sequencing (WES) has been reported to
be another feasible approach to determine UPD and assist the
analysis of disease-causing variants (11–13).
Exons are protein-coding regions consisting of only 1–2% of the
genome, but contributing to almost 85% of reported disease-causing
genes. Isodisomy can cause homozygosity in the autosomal recessive
traits, which is detectable by homozygosity mapping tools, such as
homozygous stretch identifier (HomSI) (14). Furthermore, the primary diagnosis
of paternal isodisomy can be further validated by Sanger-based
segregation analysis (11).
In the present study, the identification of two
patients with an abnormal karyotype of balanced translocations on
chromosome 15 is reported. For patient 1, variants were
investigated using WES, and a higher homozygous rate was observed
on chromosome 15 as compared with all chromosomes (92.69 vs.
42.84%), indicating a high potential UPD. Chromosomes were mapped
by HomSI (14) to further identify
the homozygous regions on chromosome 15. Next, Sanger sequencing
validation indicated that two homozygous variants (STARD9,
NM_020759.2:c.4169C>T; and TTBK2,
NM_173500.3:c.2926C>T) with low frequency in Exome Aggregation
Consortium (ExAC) databases were inherited from the father, which
strongly suggests the paternal isodisomy type of UPD. In addition,
AS was diagnosed in patient 2 by MS-MLPA, who exhibited paternal
UPD on the 15q11-13 region.
Patients and methods
Ethical considerations
The present study was approved by the Ethics
Committees of the Children's Hospital Affiliated to Zhengzhou
University (Zhengzhou, China). The two cases involved patients
admitted at the Children's Hospital Affiliated to Zhengzhou
University, and written informed consents were obtained from all
participating individuals. The parents of the studied patients
declared that they were non-consanguineous and had no family
history of congenital anomalies.
Patient details
Patient 1
Patient 1 was a 3-year-old female with complex
developmental issues. The patient had non-consanguineous parents
and a healthy 12-year-old brother, and was born at 37 weeks of
gestation by Caesarian section with a birth weight of 2.7 kg. She
sat at 12 months and stood unsupported for 1 minute or walk a few
steps at 32 months. Absence of speech was reported, and the patient
was only able to say ‘mama’. The patient was shy, but always had a
smiling face, and was able to signal ‘goodbye’ with gestures and
recognize common objects. A seizure was first reported at the age
of 2.5 years, and the anticonvulsant magnesium valproate was
administered thereafter. In addition, the patient exhibited
microcephaly, with a head circumference of 44 cm. Genetic detection
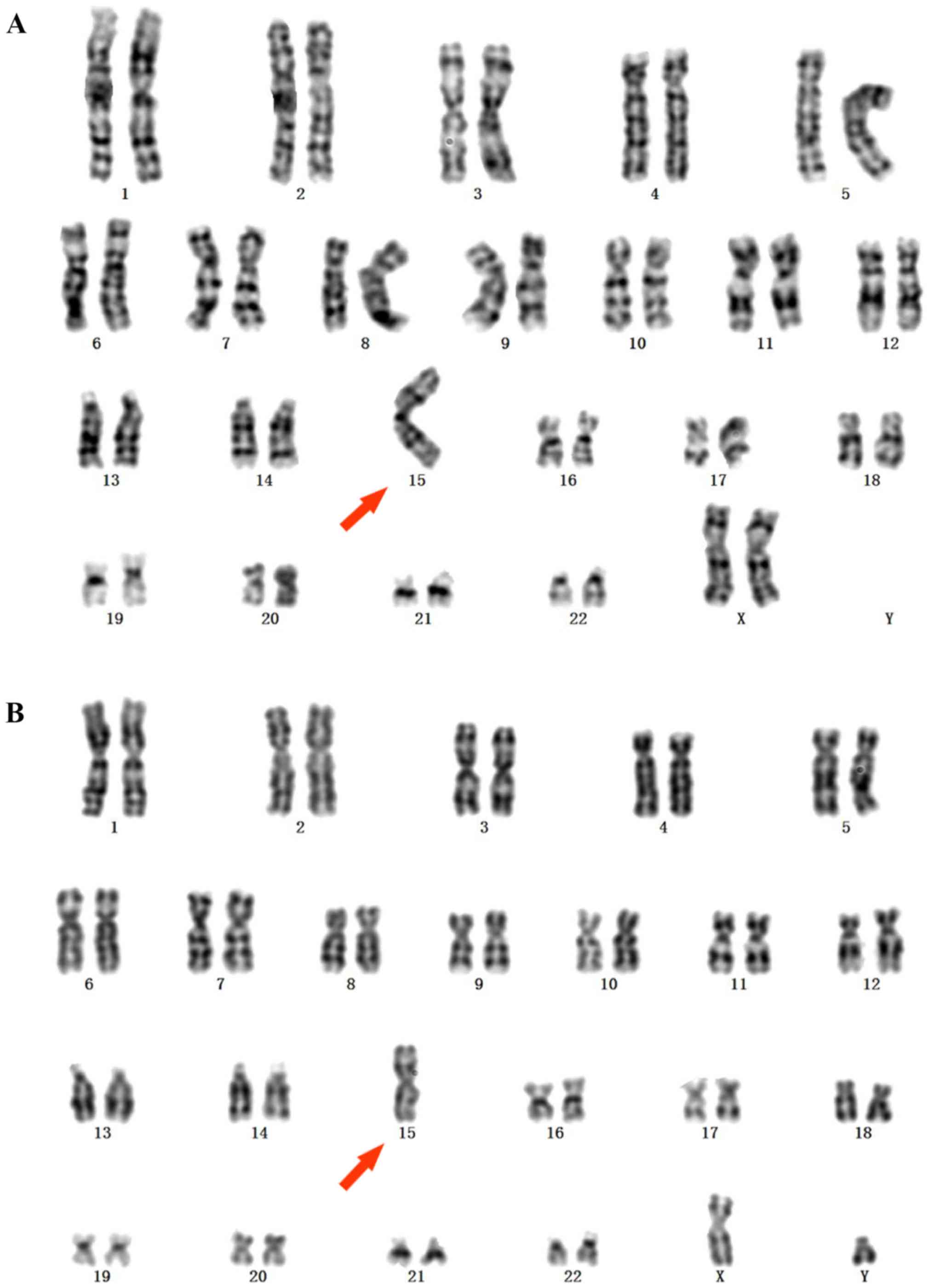
indicated a 45,XX,der(15;15)(q10;q10) karyotype (Fig. 1A). An electroencephalogram (EEG)
indicated a moderate diffuse abnormality, while brain magnetic
resonance imaging (MRI) detected a Rathke's cleft cyst. The blood
acylcarnitine and amino acid profiles of the patient were normal.
Elevated valproic acid was determined in the patient's urine, which
may be the result of the anticonvulsant treatment.
Patient 2
Patient 2 was a 3.5-year-old male with a karyotype
of 45,XY,der(15;15)(q10;q10) (Fig.
1B). The patient was born at 37 weeks of gestation by Caesarian
section due to the umbilical cord being wrapped around the neck for
two weeks, and weighed 3.7 kg at birth. His mother was 36 years old
and the patient was her fifth pregnancy. The patient sat and began
crawling at 8 months, and walked at the age of 1 year and 8 months.
At 3 years old, the patient was able to run, go up and down stairs
independently, and jump with both feet, but was unable to stand on
a single leg. The subject was unable to speak, with the exception
of unconsciously articulating ‘mama’ and ‘nine’. He was able to
understand certain simple commands and follow the instructions to
finish specific actions. The patient had a happy smiling face and
quick temper, as well as spaced teeth, bilateral inverted nipples
and a micropenis. There was no history of seizures, and normal head
development with a head circumference of 50 cm was observed. A
brain MRI scan was normal. Blood acylcarnitine and amino acid
profiles indicated a low level of serum acylcarnitine, which was
treated with L-carnitine. Urinary organic acid levels were found to
be normal.
DNA extraction and library
construction
Genomic DNA was extracted from a 300-µl peripheral
blood sample using a RelaxGene Blood DNA system (Tiangen Biotech
Co., Ltd., Beijing China). Its purity and quantity were determined
with a Nanodrop 2000 spectrophotometer and an Invitrogen
Qubit® 3.0 fluorometer (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The hybrid selection of genomic DNA was
performed using Agilent SureSelect Human All Exon Kit v6 (Agilent
Technologies, Inc., Santa Clara, CA, USA). DNA libraries were
purified by magnetic beads (AMPrure XP beads; Beckman Coulter,
Inc., Brea, CA, USA). Coding regions and intron/exon boundaries
were enriched, and sample quality control was conducted by an
Agilent 2100 Bioanalyzer system (Agilent Technologies, Inc.). The
extracted DNA was stored at −20°C and prepared for sequencing.
WES and data analysis
Samples were sequenced using the Illumina HiSeq X
Ten platform (Illumina, Inc., San Diego, CA, USA). The results were
analyzed and annotated by an in-house pipeline. Briefly, raw reads
were preprocessed to remove reads with low quality or adaptors.
Read alignment was performed using the Burrows-Wheeler Aligner tool
(version 0.7.17) with default parameters against the human genome
assembly hg19 (GRCh37) as previously described (15). The generated bam file was sorted by
SAMtools (16). Genome Analysis
Toolkit (GATK; http://software.broadinstitute.org/gatk/) was
performed to detect SNVs and indels (<50 bp), and CNVkit
(17) was applied to detect the
copy number variations (CNVs). Next, the Variant Effect Predictor
(18) was employed to annotate
SNVs, indels and CNVs. With regard to possible effects on protein
function, variants were evaluated by two widely used prediction
tools, including Sorting Intolerant from Tolerant (SIFT) (19) and Polymorphism Phenotyping version
2 (PolyPhen-2) (20). For
calculating the homozygous ratio with SNV data, the following rules
were adopted: i) SNPs with a depth of <20× were removed; ii)
only SNPs with a variant allele frequency between 0.2 and 0.8 were
considered to be heterozygous.
Prioritization of candidate variants
associated with disease phenotypes
Based on the aforementioned variant annotations, a
series of prioritization strategies were applied to select
candidate variants associated with clinical phenotypes. Detailed
steps were as follows: i) Variants with minor allele frequency
(MAF) of >0.05 according to public databases were excluded; ii)
variants with no damaging results presented in any protein function
predicted by SIFT and Polyphen2 were excluded; and iii) variants
described as benign or likely benign in ClinVar (18), and not as disease-causing in HGMD
(21) were excluded. A candidate
list of ~100 variants was obtained by implementing the
aforementioned three-step prioritization strategy.
Homozygosity mapping
Homozygosity mapping was conducted with HomSI from
next-generation sequencing data. In order to generate input files
compatible with HomSI, VCFtools (22) was used to merge the variant call
format (vcf) files of patient 1 and three healthy control
individuals. Three individuals were all healthy individuals.
Control 1 and 3 were male, while control 2 was a female.
Paternal isodisomy confirmation
Two homozygous variants with high quality (read
depth, ≥10; genotype quality, ≥75) and rarity (1000 Genomes Project
MAF≤0.001 (http://browser.1000genomes.org) and ExAC≤0.001
(http://exac.broadinstitute.org) were
selected among the possible UPD sites (11). The genomic DNA were extracted from
the peripheral blood sample of Patient1 parents, which used
as a template for Polymerase chain reaction (PCR). PCR was
performed using a Bio-Rad thermal cycler (Bio-Rad Laboratories,
Inc., Hercules, CA, USA). The forward and reverse primers for gene
STARD9 (NM_020759.2:c.4169C>T) were
5′-CCATTGTGAGCAGGCTGAAT-3′ and 5′-ACGCTAGAATTGTGGGATGC-3′,
respectively. The forward and reverse primers for gene TTBK2
(NM_173500.3:c.2926C>T) were 5′-GAGAATGAACATGGTGCCCC-3′ and
5′-AAGGGAGCAGGAACAGTAGC-3′, respectively. The PCR conditions were:
Denaturation at 95°C for 5 min, followed by 10 cycles of 95°C for
30 sec, 61°C for 30 sec and 72°C for 30 sec, 32 cycles of 95°C for
30 sec, 58°C for 30 sec and 72°C for 30 sec, and a final elongation
step at 72°C for 5 min. PCR products were separated by 1% agarose
gel electrophoresis and then purified by a QIAquick Gel Extraction
kit (Qiagen, Inc., Valencia, CA, USA). Subsequently, the products
were sequenced by the 3730×l DNA Analyzer Sequencing Standards,
BigDye™ Terminator version 3.1 (Applied Biosystems; Thermo Fisher
Scientific, Inc.) and 3730×l DNA Analyzer (Applied Biosystems;
Thermo Fisher Scientific, Inc.).
MS-MLPA
MS-MLPA was conducted using a commercial MLPA probe
kit, namely the SALSA MS-MLPA ME028-B1 Prader-Willi Syndrome
(PWS)/AS probemix, following the protocols of the manufacturer
(MRC-Holland, Amsterdam, Netherlands). The probemix contains 32
MLPA probes specific for sequences in or near the PWS/AS critical
region of chromosome 15q11, which can be applied to detect the CNVs
in this region. Five of these probes are designed for specific
imprinted gene sequences and contain a recognition site for the
methylation-sensitive HhaI enzyme. In addition, six probes
are designed to detect indels and variations in UBE3A. In
order to analyze the results, 14 reference probes were located
outside the targeted region. Amplicons were identified by capillary
electrophoresis, and the results were analyzed using Coffalyser.Net
(MRC-Holland; http://support.mlpa.com/kb/coffalyser-net).
Results
Variant analysis by WES of patient
1
Exome sequencing yielded a total of 184,605,772
reads with 91.52% above the Q30 score. Variants were annotated for
population frequency and functional predictions. Across all
chromosomes, 42.84% of the detected variants exhibited
homozygosity, which was notably up to be 92.69% on chromosome 15
homozygotic variants (the remaining non-homozygous variants were
mostly in intergenic regions; Table
I).
 | Table I.Results of variants identified by
whole exome sequencing. |
Table I.
Results of variants identified by
whole exome sequencing.
|
| All variants | MAF<0.01 |
|---|
|
|
|
|
|---|
| Variant type | Whole exome | Chr15 | Whole exome | Chr15 |
|---|
| Total variants | 63,953 | 1,476 | 18,176 | 470 |
| Homozygous | 42.84% | 92.69% | 39.66% | 88.15% |
| Synonymous | 11,664 | 254 | 386 | 3 |
| Missense | 11,150 | 236 | 753 | 21 |
| Splice site | 2,845 | 69 | 197 | 3 |
| Frameshift | 384 | 6 | 69 | 2 |
| In-frame coding
indel | 390 | 11 | 68 | 1 |
| Stop-gained | 123 | 4 | 31 | 2 |
| Stop-lost | 42 | 0 | 5 | 0 |
| Start-lost | 35 | 1 | 6 | 0 |
| Intron | 32,927 | 725 | 10,011 | 210 |
| Other
non-coding | 4,393 | 170 | 6,650 | 228 |
With an MAF cut-off at 0.01, a total of 18,176
variants were identified in whole exome regions, 470 of which were
on chromosome 15. Among all variants on chromosome 15, there were
228 non-coding variants, 21 missense variants, 3 synonymous
variants, 3 splice site variants, 1 indels, 2 frameshifts and 2
stop gained variants, whereas no stop-lost and start-lost variants
were detected. Similar results with that of all variants were
detected for the homozygous variant ratio, with a value of 39.66%
observed for the whole exome and 88.15% for chromosome 15
(P<2.2×10−16; Table I). The homozygous variant ratio of
chromosome 15 was much higher than the whole exome, which indicate
runs of homozygosity for this chromosome.
Homozygosity mapping on chromosome 15
for patient 1
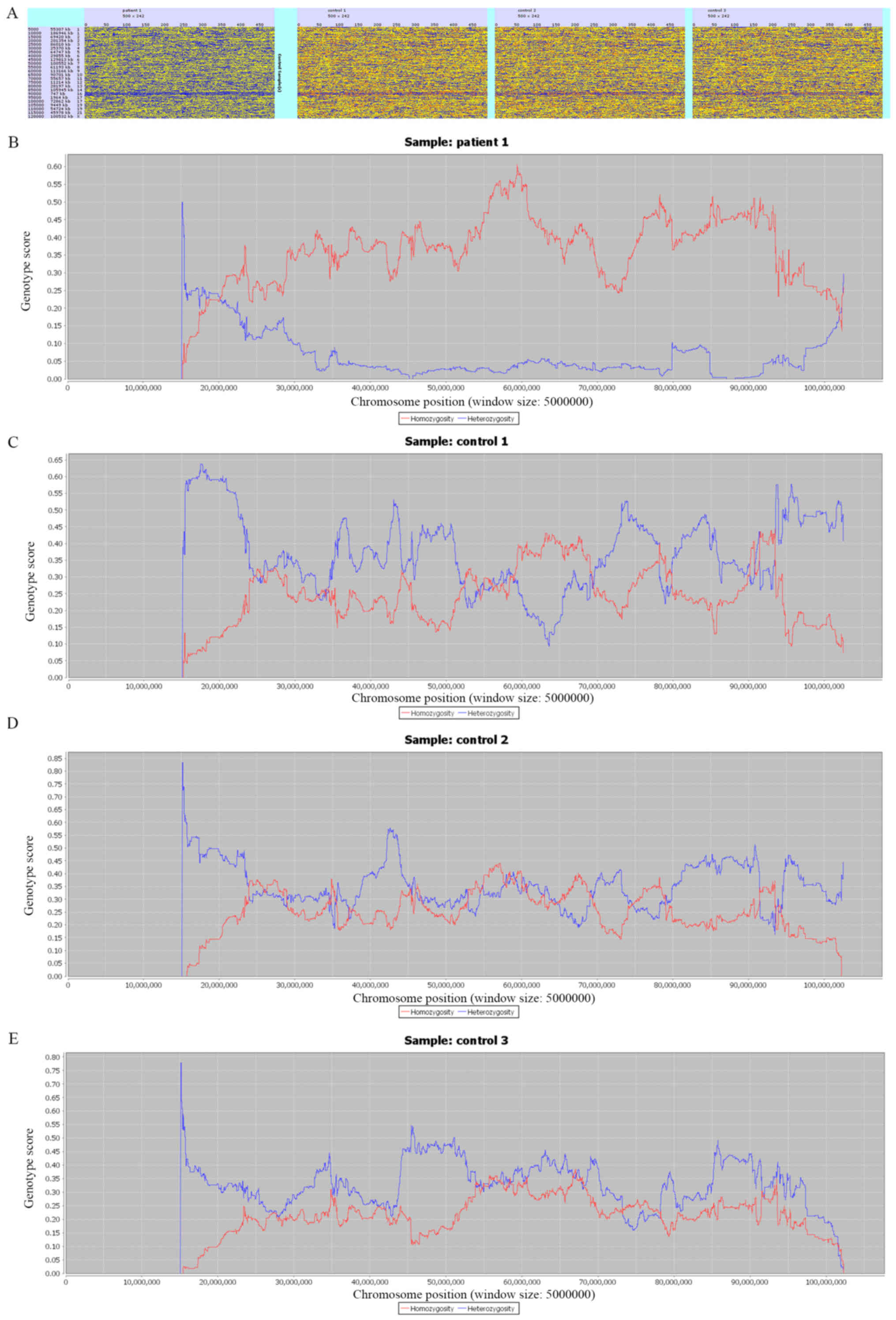
Homozygosity mapping was displayed with HomSI
(Fig. 2). Using *.vcf files as an
input file, HomSI can identify the majority of homozygous regions.
Fig. 2A shows the homozygosity
maps for the whole genome, with a continuous blue line observed in
patient 1 (first panel), but not in the control individuals (three
panels on the right). Fig. 2B-E
show the Hom (red) and Het (blue) signal for three control
individuals and patient 1, which was consistent with the
homozygosity variant ratio mentioned earlier. In patient 1, the Hom
signal was much stronger than the Het signal across the entire
chromosome. This indicated the presence of UPD for chromosome
15.
Paternal isodisomy confirmation of
patient 1
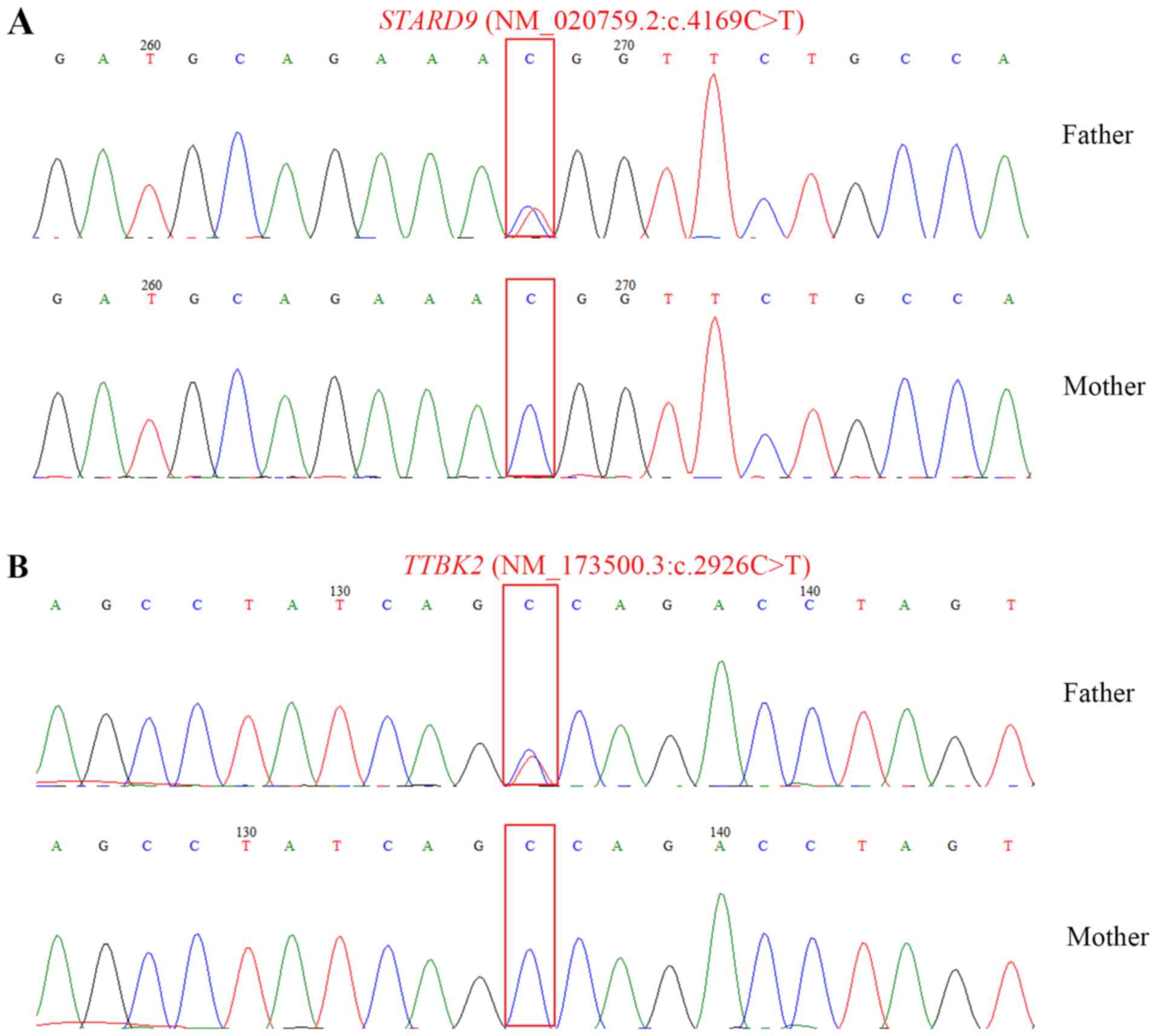
In order to validate the putative isodisomy, two
homozygous variants within chromosome 15 were selected by
recommended approaches (11) for
Sanger sequencing. The selected variants, namely STARD9
(NM_020759.2:c.4169C>T) and TTBK2
(NM_173500.3:c.2926C>T), were of high quality (read depth, ≥10;
genotype quality, ≥75) and rarity (1000 Genomes Project; MAF≤0.001
and ExAC≤0.001) (11). The
sequencing results demonstrated that the patient's father was
heterozygous for c.4169C>T in gene STARD9 and
heterozygous for c.2926C>T in gene TTBK2, whereas both
sites on the patient's mother were wild-type with C genotype
(Fig. 3). These results suggested
that the patient exhibited paternal UPD.
AS identified by MS-MLPA in patient
2
Patient 2 demonstrated similar karyotype and
phenotypes to patient 1. Suspected AS for patient 2 was validated
by another type of efficient genetic test, namely MS-MLPA, which is
able to identify CNVs on related regions and the methylation status
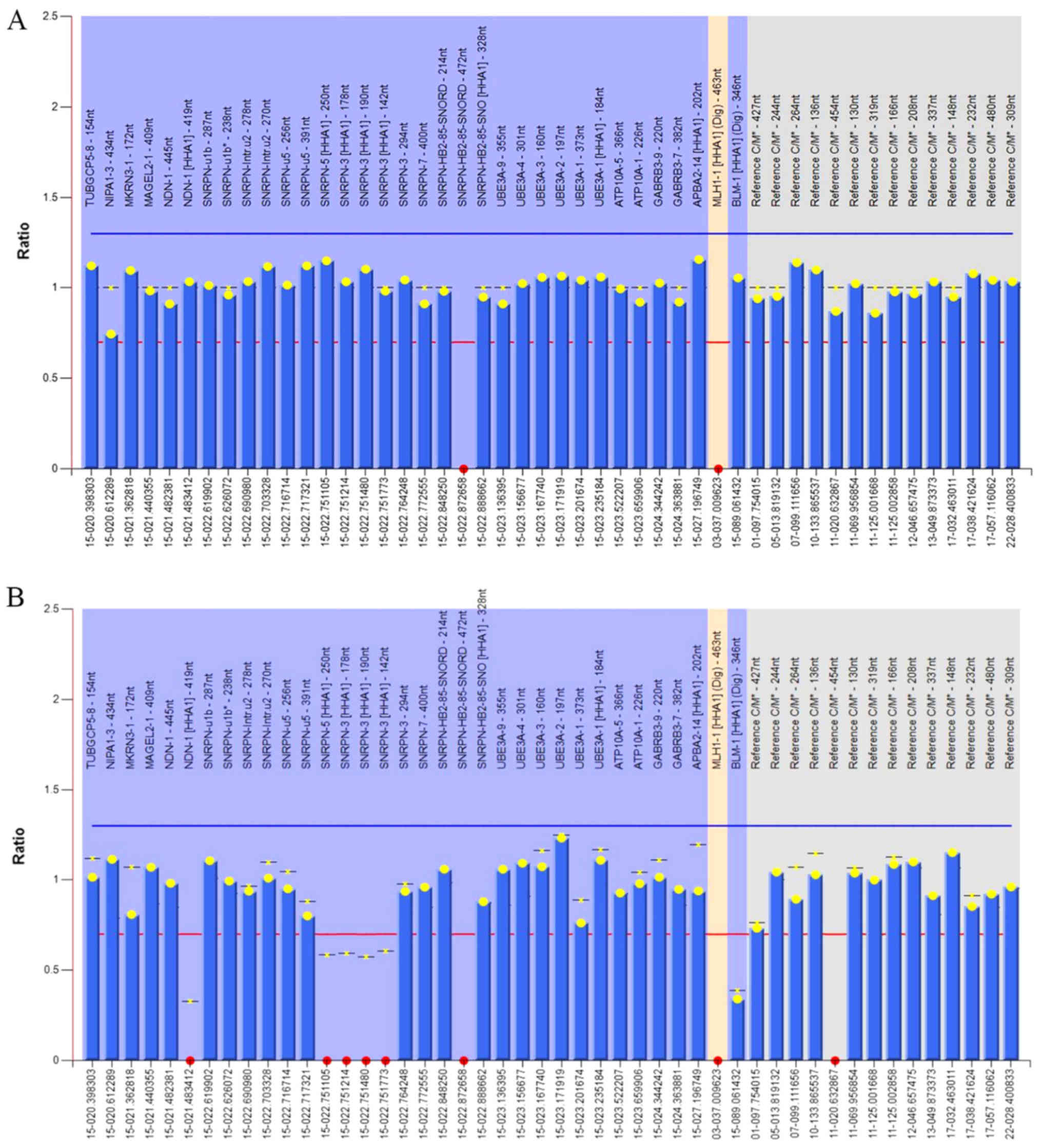
of imprinted genes. Two ratio charts were created using Coffalyser.
Net, based on the MS-MLPA results (Fig. 4). Red dots represent no detected
signal in the probes. The MLH-1[HHA1] (Dig) served as a digestion
control probe, which exhibited no signal upon digestion. The
relative ratio in the 15q11-13 region was ~1, indicating that no
duplication or deletion occurred in this region (Fig. 4A). Furthermore, probes also
contained the UBE3A gene, which enabled the detection of any
deletions on exon 1, 2, 3, 4 and 9. Similarly, the ratio was ~1,
suggesting that there were no deletions on exon 1, 2, 3, 4 and 9 of
UBE3A (Fig. 4A).
Since the imprinted expression gene small nuclear
ribonucleoprotein polypeptide N (SNRPN) is methylated on the
maternal, but not the paternal allele (23), the methylation status of MS-MLPA
results can be used to discriminate the type of UPD, that is,
whether it is paternal or maternal. Five probes were designed for
detecting the methylation patterns in the imprinted gene region:
NDN-1[HHA1]-419nt, SNRPN-5[HHA1]-250nt, SNRPN-3[HHA1]-178nt,
SNRPN-3[HHA1]-190nt and SNRPN-3[HHA1]-142nt. The results revealed
that no methylation signals were detected on these probes,
indicating that patient 2 is highly possible having a paternal UPD
(Fig. 4B).
Discussion
In the present study, two independent cases of
suspected AS that was detected by two different testing methods
were described. The two patients carried the same karyotype of
Robertsonian-like translocation in chromosome 15. The methods used
were both effective in detecting potential UPD, whereas further
segregation analysis was required to confirm the parental origin of
the disomy. For patient 1, the two selected variants demonstrated
that the patient was homozygous at the loci for which the father
was heterozygous, suggesting that the structural rearrangement was
an isodisomic 15q. A similar approach can be further applied to
patient 2. Since paternal UPD only accounts for 2–3% of the AS
population, similar cases of AS resulting from isodisomic 15q
associated UPD would be very rare. Currently, the majority of
reports on paternal UPD-associated AS cases are heterodisomic
(24–27), and cases due to isodisomy are
limited (10,28,29).
The severity of AS symptoms varies greatly with
mutation types. It has been suggested that AS patients with UPD at
chromosome 15 exhibit milder symptoms compared with those with
other underlying genetic abnormalities, such as deletions (28,30).
As for the two cases reported in the present study, less ataxia and
seizures, and better development were observed, which supports
previous findings. Varela et al (31) and Williams et al (32) suggested that AS patients with UPD
may remain undiagnosed due to their less typical phenotype leading
to an under-diagnosis state. WES is a promising approach for
increasing the diagnosis rate. Currently, WES is widely used in
clinical practice. As compared with whole genome sequencing, exome
sequencing displays efficient and effective applications in
detecting Mendelian diseases caused by genetic defects, and may
further provide therapeutic implications for certain cases
(33,34). WES is able to detect SNVs, indels,
isodisomic UPD and even CNVs (35)
in a single test, which has been widely accepted as a robust and
cost-effective approach. In suspected AS patients, DNA methylation
analysis is typically the first test performed, since individuals
with AS caused by a 5-to 7-Mb deletion of 15q11.2-q13, UPD or an
imprinting defect only have an unmethylated (i.e. ‘paternal’)
contribution.
It should be noted that the two methods used in the
current study have limitations in the diagnosis of AS. MLPA/MS-MLPA
can identify maternal deletions and imprinting defects, including
paternal UPD (both isodisomy and heterodisomy); however, it is
unable to detect point mutations and indels. WES of the proband can
detect small nucleotide pathogenic variants in the UBE3A
gene, uniparental isodisomy and certain large deletions. However,
segregation studies of parental samples are required to determine
the parental origin of the affected chromosomes. Furthermore, WES
is unable to reliably detect deletions, and does not detect
heterodisomy unless parental and grandparental samples are
analyzed. Therefore, WES may not be more suitable than MS-MLPA for
clinical genetic testing of AS. However, WES can identify most
types of genetic abnormalities, which is particularly effective for
cases with mild or atypical symptoms. Thus, with the advance of
sequencing technology, WES would be more suitable and
cost-effective in the near future for clinical genetic testing.
In conclusion, the current study suggested the
application of WES data in analyzing clinical cases, even if the
suspected UPD.
Acknowledgements
The authors would like to thank Dr. Yongchu Liu and
Dr. Yang Liu [Aegicare (Shenzhen) Technology Co., Ltd., Shenzhen,
China] for their technical and scientific advice.
Funding
The present study was supported by The Science and
Technology Development Plan of Zhengzhou, 2014 (International
Science and Technology Cooperation; grant no. 20140962) and the
Science and Technology Development Plan of Zhengzhou, 2014 (grant
no. 141PGJHZ530).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
HL, HY and QS collected the clinical cases and
designed the study. HL and HY performed the analysis and wrote the
manuscript. NL performed the MS-MLPA analysis for patient 2. CM and
JL prepared the figures and revised the manuscript. QS revised the
manuscript and provided instructions for the entire study. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committees of the Children's Hospital Affiliated to Zhengzhou
University. The two cases involved patients admitted at the
Children's Hospital Affiliated to Zhengzhou University.
Patient consent for publication
Informed consent was obtained from both set of
parents for routine and investigative studies.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kishino T, Lalande M and Wagstaff J:
UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 15:70–73.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rougeulle C, Glatt H and Lalande M: The
Angelman syndrome candidate gene, UBE3A/E6-AP, is imprinted in
brain. Nat Genet. 17:14–15. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sato M: Early origin and evolution of the
angelman syndrome ubiquitin ligase gene Ube3a. Front Cell Neurosci.
11:622017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Greer PL, Hanayama R, Bloodgood BL,
Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z,
Maehr R, et al: The angelman syndrome protein Ube3A regulates
synapse development by ubiquitinating arc. Cell. 140:704–716. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Clayton-Smith J and Laan L: Angelman
syndrome: A review of the clinical and genetic aspects. J Med
Genet. 40:87–95. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fridman C and Koiffmann CP: Origin of
uniparental disomy 15 in patients with Prader-Willi or Angelman
syndrome. Am J Med Genet. 94:249–253. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Knoll JH, Glatt KA, Nicholls RD, Malcolm S
and Lalande M: Chromosome 15 uniparental disomy is not frequent in
Angelman syndrome. Am J Hum Genet. 48:16–21. 1991.PubMed/NCBI
|
|
8
|
Siegel DH and Slavotinek A: Uniparental
disomy. Pediatr Dermatol. 22:482–487. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shaffer LG: Risk estimates for uniparental
disomy following prenatal detection of a nonhomologous Robertsonian
translocation. Prenat Diagn. 26:303–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Poyatos D, Guitart M, Gabau E, Brun C,
Mila M, Vaquerizo J and Coll MD: Severe phenotype in Angelman
syndrome resulting from paternal isochromosome 15. J Med Genet.
39:E42002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bis DM, Schule R, Reichbauer J, Synofzik
M, Rattay TW, Soehn A, de Jonghe P, Schöls L and Züchner S:
Uniparental disomy determined by whole-exome sequencing in a
spectrum of rare motoneuron diseases and ataxias. Mol Genet Genomic
Med. 5:280–286. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Carmichael H, Shen Y, Nguyen TT,
Hirschhorn JN and Dauber A: Whole exome sequencing in a patient
with uniparental disomy of chromosome 2 and a complex phenotype.
Clin Genet. 84:213–222. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kurth I, Baumgartner M, Schabhuttl M,
Tomni C, Windhager R, Strom TM, Wieland T, Gremel K and
Auer-Grumbach M: Whole exome sequencing in congenital pain
insensitivity identifies a novel causative intronic NTRK1-mutation
due to uniparental disomy. Am J Med Genet B Neuropsychiatr Genet.
171:875–878. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gormez Z, Bakir-Gungor B and Sagiroglu MS:
HomSI: A homozygous stretch identifier from next-generation
sequencing data. Bioinformatics. 30:445–447. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence Alignment/Map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Talevich E, Shain AH, Botton T and Bastian
BC: CNVkit: Genome-Wide copy number detection and visualization
from targeted DNA sequencing. PLoS Comput Biol. 12:e10048732016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17:1222016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stenson PD, Mort M, Ball EV, Shaw K,
Phillips A and Cooper DN: The human gene mutation database:
Building a comprehensive mutation repository for clinical and
molecular genetics, diagnostic testing and personalized genomic
medicine. Hum Genet. 133:1–9. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Danecek P, Auton A, Abecasis G, Albers CA,
Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST,
et al: The variant call format and VCFtools. Bioinformatics.
27:2156–2158. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Glenn CC, Driscoll DJ, Yang TP and
Nicholls RD: Genomic imprinting: Potential function and mechanisms
revealed by the Prader-Willi and Angelman syndromes. Mol Hum
Reprod. 3:321–332. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tonk V, Schultz RA, Christian SL, Kubota
T, Ledbetter DH and Wilson GN: Robertsonian (15q;15q) translocation
in a child with Angelman syndrome: Evidence of uniparental disomy.
Am J Med Genet. 66:426–428. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Freeman SB, May KM, Pettay D, Fernhoff PM
and Hassold TJ: Paternal uniparental disomy in a child with a
balanced 15;15 translocation and Angelman syndrome. Am J Med Genet.
45:625–630. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fridman C, Varela MC, Nicholls RD and
Koiffmann CP: Unusual clinical features in an Angelman syndrome
patient with uniparental disomy due to a translocation 15q15q. Clin
Genet. 54:303–308. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ramsden S, Gaunt L, Seres-Santamaria A and
Clayton-Smith J: A case of Angelman syndrome arising as a result of
a de novo Robertsonian translocation. Acta Genet Med Gemellol
(Roma). 45:255–261. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Horvath E, Horvath Z, Isaszegi D, Gergev
G, Nagy N, Szabó J, Sztriha L, Széll M and Endreffy E: Early
detection of Angelman syndrome resulting from de novo paternal
isodisomic 15q UPD and review of comparable cases. Mol Cytogenet.
6:352013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Robinson WP, Christian SL, Kuchinka BD,
Peñaherrera MS, Das S, Schuffenhauer S, Malcolm S, Schinzel AA,
Hassold TJ and Ledbetter DH: Somatic segregation errors
predominantly contribute to the gain or loss of a paternal
chromosome leading to uniparental disomy for chromosome 15. Clin
Genet. 57:349–358. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Smith A, Robson L and Buchholz B: Normal
growth in Angelman syndrome due to paternal UPD. Clin Genet.
53:223–225. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Varela MC, Kok F, Otto PA and Koiffmann
CP: Phenotypic variability in Angelman syndrome: Comparison among
different deletion classes and between deletion and UPD subjects.
Eur J Hum Genet. 12:987–992. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Williams CA, Beaudet AL, Clayton-Smith J,
Knoll JH, Kyllerman M, Laan LA, Magenis RE, Moncla A, Schinzel AA,
Summers JA and Wagstaff J: Angelman syndrome 2005: Updated
consensus for diagnostic criteria. Am J Med Genet A. 140:413–418.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu
YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D,
et al: Whole-exome sequencing in undiagnosed genetic diseases:
Interpreting 119 trios. Genet Med. 17:774–781. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stranneheim H and Wedell A: Exome and
genome sequencing: A revolution for the discovery and diagnosis of
monogenic disorders. J Intern Med. 279:3–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pfundt R, Del Rosario M, Vissers LELM,
Kwint MP, Janssen IM, de Leeuw N, Yntema HG, Nelen MR, Lugtenberg
D, Kamsteeg EJ, et al: Detection of clinically relevant copy-number
variants by exome sequencing in a large cohort of genetic
disorders. Genet Med. 19:667–675. 2017. View Article : Google Scholar : PubMed/NCBI
|