Introduction
Maturity-onset diabetes of the young (MODY) is an
dominant, autosomal genetic disease characterized by islet function
defects, insufficient insulin secretion and islet-associated
antibody negativity. MODY is the most common type of monogenic
hereditary diabetes (1,2). Due to its similar age at onset and
clinical manifestations, MODY is often misdiagnosed as type I or
atypical type II diabetes. However, different types of diabetes are
characterized by different prognoses and treatment, and MODY
exhibits clear family heredity. Accurate diagnosis is crucial for
patients with MODY and their relatives (3).
At present, there is a lack of effective methods for
the diagnosis or treatment of most hereditary diseases. With the
continuous iterative update of sequencing technology and the
gradual improvement in cost-effectiveness, whole-exome sequencing
(WES) has become more widespread for genetically analyzing and
identifying potential genetic variations that lead to disease
development (4–7). In brief, WES involves the use of
hybrid capture technology to obtain a DNA sequence encoding all
exon regions in the genome that encode the protein, followed by
high-throughput sequencing. There are as many as 180,000 exons in
the human genome, accounting for ~1% of the entire genome. Exome
sequencing by high-throughput sequencing technology is an effective
method for identifying disease-causing genetic variations (8–11).
Therefore, WES may be used to identify novel susceptibility genes
for the MODY-X family of unknown genes.
In the present study, WES was performed on a family
of three (two members with MODY and one healthy member), and a
novel mutation site on the pancreatic and duodenal homeobox 1
(PDX1) gene was identified, which was subsequently verified by
sequencing in relatives of the individuals.
Materials and methods
Research subjects
Proband
The patient was female, aged 23 years, with normal
development and moderate nutritional status, who had been consuming
carbonated drinks since childhood. Two years prior, the patient
exhibited no hunger or an obvious incentive for eating. The
patient's fasting blood glucose level was 19.0 mmol/l 2 days prior
to admission to the hospital. Immediately prior to admission to the
Shanxi Provincial People's Hospital on August 30, 2017, the fasting
blood glucose level was 18.0 mmol/l. The patient reported no
polydipsia, weight loss or blurred vision, and there was no
numbness of the limbs, abdominal pain, diarrhea, fatigue or
discomfort.
Family history
There was no history of diabetes on the maternal
side of the family, whereas there were three diabetic patients
among paternal relatives (grandfather, father and uncle).
Physical examination
On physical examination, the patient's height was
162 cm, with a weight of 56 kg and a body mass index (BMI) of 21.3
kg/m2; blood pressure was 122/67 mmHg. There were no
obvious abnormalities of the heart, lung or abdomen, and there was
no edema in either of the lower limbs.
Clinical data collection
The medical histories of the proband and her family
members were taken into account, and their heights and weights were
measured. In addition, BMI was calculated, laboratory blood glucose
tests were performed, C-peptide and glycosylated hemoglobin (HbA1c)
levels were measured, and liver and kidney function, blood lipids,
urine microalbumin and type I diabetes-related antibodies, namely
anti-islet antibody, anti-insulin autoantibody and anti-glutamate
decarboxylase antibody (ICA, IAA and GAD, respectively), were
assessed. Informed written consent was obtained from the patient
for publication of this case report and any accompanying images,
and the study was approved by the Ethics Committee of the Shanxi
Provincial People's Hospital (Shanxi, China).
DNA extraction
A total of 2 ml venous blood was collected from
three family members (two members with MODY and one healthy
member). The heparin anticoagulation and SE Blood DNA kits (Omega
Bio-Tek, Inc.) were used to extract peripheral blood leukocyte
genomic DNA, which was then sent to Mingma Biotechnology Company
for WES (12).
Bioinformatics analysis of exome
sequence
The original sequence was quality-controlled by
FastQC (version 0.11.8) (13), and
BWA (version r1188) (14) software
was used to align the reads to the human reference genome (hg19).
The Samblaster (version 0.1.24) program (15) was used to remove duplicate reads,
and GATK (version 3.8) realigner (16) was used to realign indel and Base
Quality Score recalibration. To ensure the accuracy of the
identification variation, five software programs (GATK, SAMtools
(version 1.8) (17), FreeBayes
(version 1.1.0) (18), Platypus
(version 0.8.1.2) (19) and
VarScan2 (version 2.4.0) (20)
were used for mutation analysis and dbSNP, 1000Genomes, dbNSFP,
ClinVar were used for filter mutation (4).
Genetic filtering
According to the patient's family history and
clinical manifestations, MODY was suspected. Therefore, 14 known
mutations in the MODY gene and Mendelian inheritance were analyzed,
and a novel mutation site was identified on the PDX1 gene (6,21–23).
Polymerase chain reaction (PCR)
verification
A fragment of the first exon of PDX1 was amplified
by PCR and the primers were designed via the NCBI website using
Primer Blast (version 3.0; http://www.ncbi.nlm.nih.gov/tools/primer-blast/).
PCR was performed using the Bio-Rad T100 system (Bio-Rad
Laboratories, Inc.). The PCR reaction system consisted of 1 µl DNA
template [extracted using the Omega SE Blood DNA kits (Omega
Bio-Tek, Inc.)], 1 µl forward (5′-CGCAGCTTTACAAGGACCCAT-3′) and
reverse primers (3′-GGTGAGAACCGGAAAGGAGA-5′), 0.5 µl Taq enzyme
(Takara Biotechnology Co., Ltd.), 5 µl dNTP and 4 µl 10X Ex Taq
Buffer (Mg2+ free), supplemented with double-distilled water to a
total volume of 50 µl. The reaction conditions were as follows:
Pre-denaturation at 94°C for 3 min; denaturation at 94°C for 30
sec; annealing at 58°C for 30 sec; extension at 72°C for 45 sec, 30
cycles following 72°C extension for 5 min and, finally, the PCR
product was stored at 4°C. Following the reaction, 1% agarose gel
electrophoresis was performed, followed by ethidium bromide
staining. The strip was observed under a UV lamp. The PCR product
was sent to Shanghai Shenggong Biological Company for
sequencing.
Results
Family diagram, clinical characteristics
of family members and laboratory test results
Family diagram
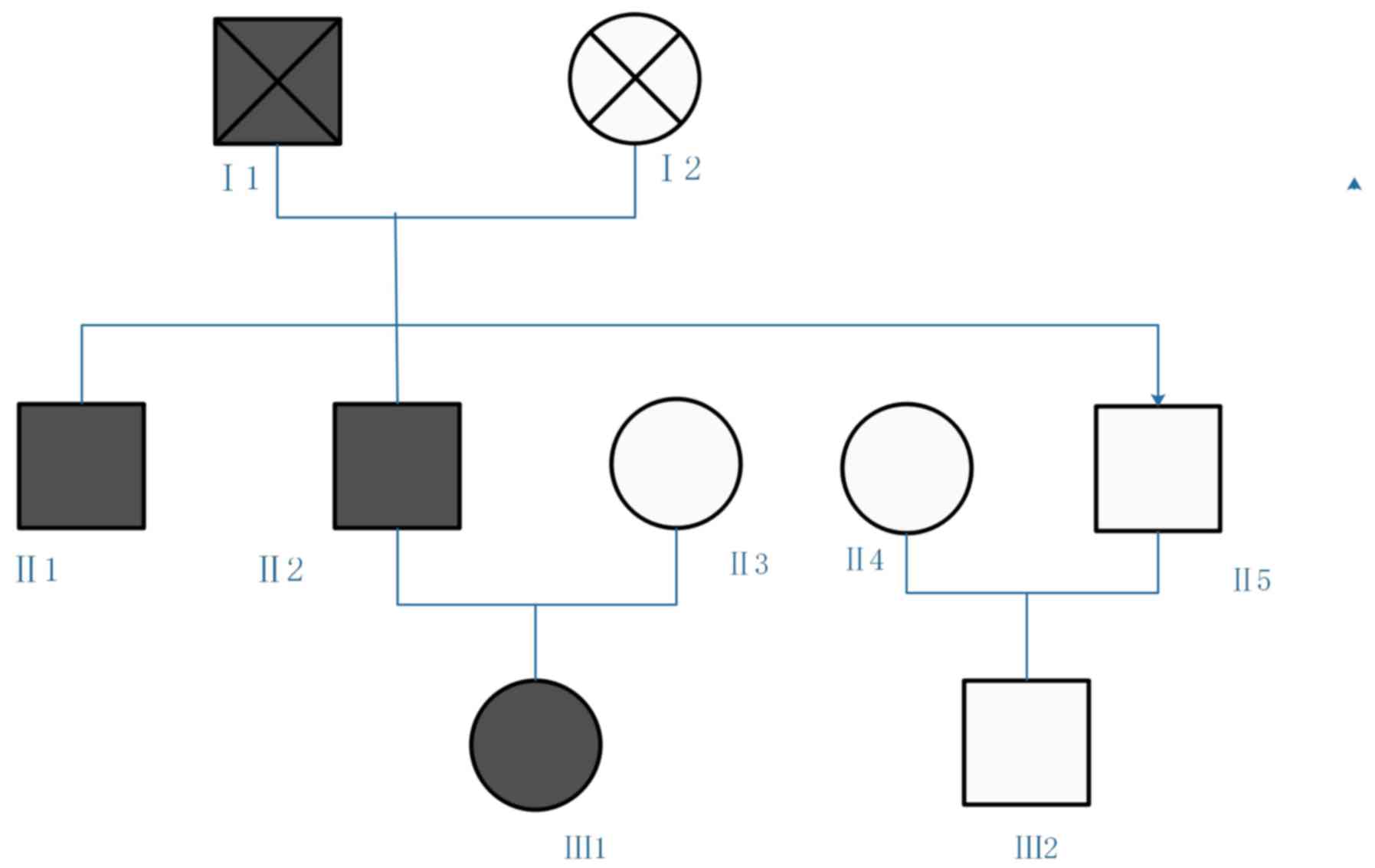
The family included three generations. Blood samples
were collected from five family members: II1, II2, II3, II4 and
III1. The three diabetic patients and the other family members
without clinical symptoms are presented in Fig. 1. A detailed laboratory assessment
was performed on the proband (III1), and the remaining family
members underwent a simple check.
Laboratory data and clinical
characteristics
The proband's HbA1c level was 12.9%, urine sugar was
4+ and urine was negative for ketone bodies. ICA, IAA and GAD were
all negative. An oral glucose tolerance test was performed
simultaneously with insulin and C-peptide release experiments
(Table I). There were no
abnormalities in the thyroid function tests, blood lipid profile,
urine microalbumin or on fundus examination.
 | Table I.Blood glucose, insulin and C-peptide
levels of diabetic patients in the family. |
Table I.
Blood glucose, insulin and C-peptide
levels of diabetic patients in the family.
|
| Blood sugar
(mmol/l) | Insulin (uIU/ml) | C-peptide
(ng/ml) |
|---|
|
|
|
|
|
|---|
| Time (min) | III1 | II2 | II1 | III1 | II2 | II1 | III1 | II2 | II1 |
|---|
| 0 | 13.7 | 7.9 | 7.3 | 8.45 | 7.86 | 9.34 | 0.79 | 1.56 | 1.78 |
| 30 | 15.4 | 11.6 | 10.8 | 9.12 | 10.39 | 11.45 | 0.98 | 2.09 | 2.36 |
| 60 | 18.2 | 12.9 | 13.7 | 12.79 | 30.76 | 35.89 | 1.03 | 5.88 | 6.02 |
| 120 | 23.6 | 13.9 | 11.8 | 19.07 | 39.54 | 50.07 | 1.24 | 7.64 | 8.19 |
| 180 | 21.4 | 10.5 | 9.8 | 13.39 | 18.67 | 20.34 | 1.16 | 3.83 | 3.95 |
Family
The proband's grandfather and mother had died, but
their age and treatment for diabetes were unknown. The proband's
father and uncle (II2 and II1) were diagnosed with diabetes between
35 and 40 years of age (specific age unknown), and received
intermittent treatment with metformin with regular monitoring of
blood glucose. In II2 and II1, the HbA1c levels were 9.5 and 9.0%,
respectively. ICA, IAA and GAD were negative. The islet function
test results are shown in Table
I.
Treatment and follow-up
The proband was treated with continuous subcutaneous
injection of insulin aspart using an insulin pump for 2 weeks (mean
daily dose, 40 units), which was then changed to hypoglycemic
treatment with subcutaneous insulin (insulin + insulin glargine)
four times per day. At 3 months following discharge from the
hospital, the HbA1c value was 8.8%. Injecting insulin before meals
was then replaced with insulin glargine injection (14 units at
bedtime) and oral agarose. At 6 months following discharge from
hospital, the HbA1c level was 6.8%; insulin glargine was then
replaced with oral metformin (250 mg orally three times/day) and
acarbose (50 mg orally three times/day). At 9 months following
discharge from hospital, the HbA1c level was 7.0%. At present, the
patient's blood sugar is controlled and stable.
In terms of the father and uncle of the proband (II2
and II1), improvements in life management were important. This
included control of the intake of starchy foods per meal; according
to work and life conditions, starchy foods in each meal should be
controlled at 100 g. Other instructions included choosing foods
with a lower glycemic index, educating on the importance of
exercising properly following eating for 30 min, to improve
postprandial glycemic control, monitoring changes in body weight,
and ensuring BMI does not exceed 25. At the 3-month follow-up, the
HbA1c levels were 7.8 and 8.1%, respectively; the dose of metformin
was reduced to 1,000 mg/day, and at the 6-month follow-up, the
HbA1c levels were 6.8 and 7.1%, respectively. Metformin was
discontinued, and blood glucose was controlled by diet and regular
exercise. At the 9-month follow-up, the HbA1c levels were 6.9 and
7.0%, respectively (Table
II).
| Table II.Follow-up results of glycosylated
hemoglobin in diabetic patients in the family. |
Table II.
Follow-up results of glycosylated
hemoglobin in diabetic patients in the family.
|
| Glycosylated
hemoglobin (%) |
|---|
|
|
|
|---|
| Time (months) | III1 | II2 | II1 |
|---|
| 0 | 12.9 | 9.5 | 9.0 |
| 3 | 8.8 | 7.8 | 8.1 |
| 6 | 6.8 | 6.8 | 7.1 |
| 9 | 7.0 | 6.9 | 7.0 |
Results of bioinformatics
analysis
The Q30 ratio based on the family was >90%. The
sequencing quality was good, and the comparison rate was >95%.
The repeat sequence was <10%, and the mean sequencing depth was
>120×, which was sufficient to identify the mutation. The
specific comparison information is shown in Table III. The father harbored 641,508
mutations, the mother had 508,998 mutations and their offspring had
490,117 mutations. The specific variation distribution is shown in
Table IV. The proportion of exon
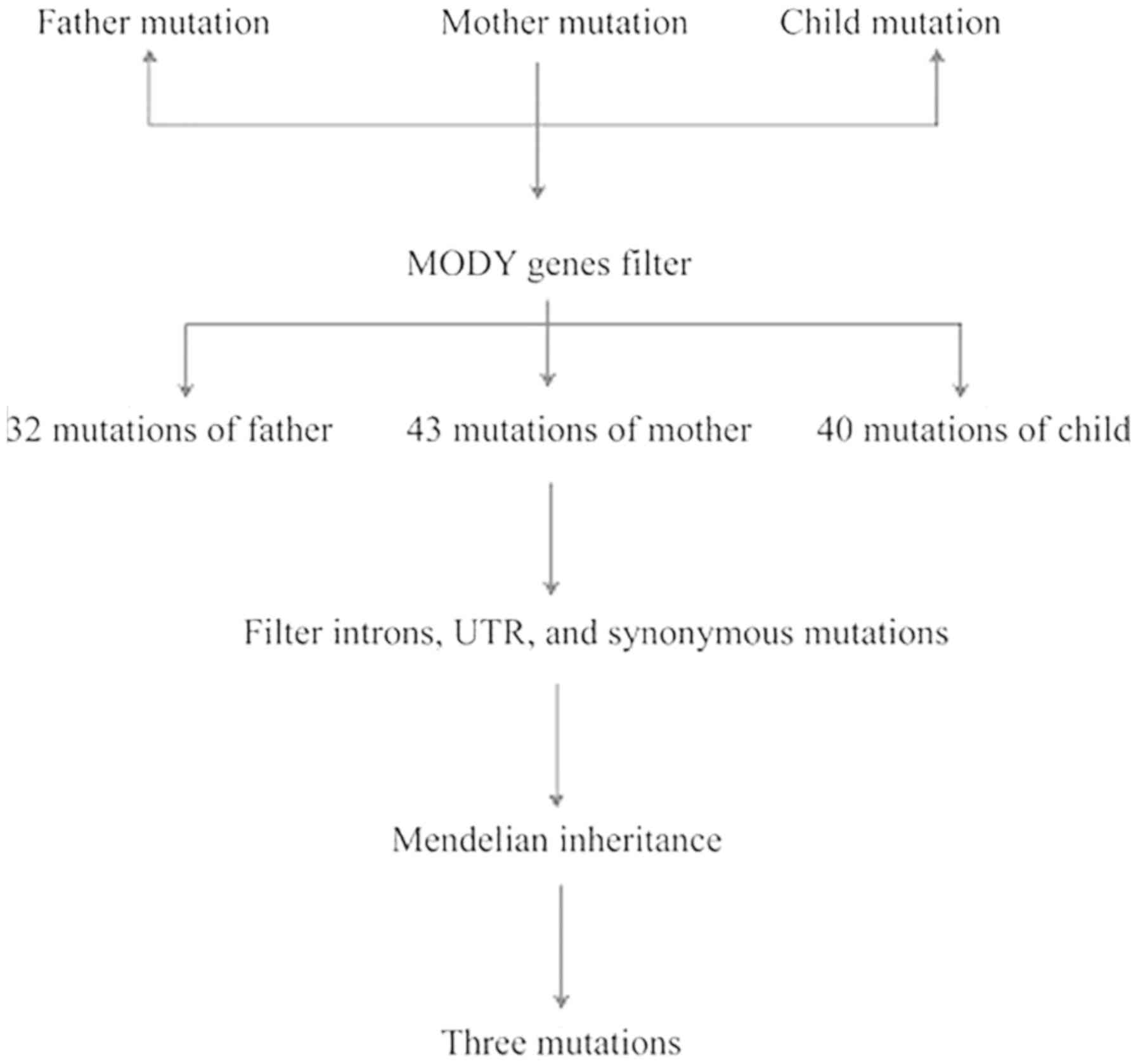
regions accounted for ~20%. When filtered against 14 MODY genes,
the father maintained 32 mutations, the mother maintained 43
mutations and their offspring maintained 40 mutations. Following
the filtering of introns, untranslated regions and synonymous
mutations, the family maintained eight mutations. According to
Mendelian inheritance (filtering the site where the child genotype
is consistent with the parent genotype), the remaining three sites
were filtered. The entire filtration process is shown in Fig. 2 and the results are shown in
Table V. The mutation rates of the
ATP binding cassette subfamily C member 8 and the potassium
voltage-gated channel subfamily J member 9 genes are >1% in the
population. However, the frequency of mutations in the PDX1 gene
has not been reported. In addition, Mutation Taster and LRT
predicted that this mutation site is deleterious, the mutation site
was located in the first exon, there were two exons in this gene
and that this mutated amino acid was located in last
transactivation domain of the protein coded by this gene. This
transactivation domain contained other protein binding sites,
including transcription coregulators; therefore, mutation of the
transactivation domain may affect the function of the gene and may
be the pathogenic factor in this family. To the best of our
knowledge, that site has not been previously reported in the
literature.
| Table III.Specific comparison information. |
Table III.
Specific comparison information.
| Quality control
term | Father | Mother | Child |
|---|
| Total_reads | 97,368,897 | 98,766,436 | 104,464,226 |
| Q30 | 0.9053 | 0.9234 | 0.9353 |
|
Mapped_reads_percent | 0.9689 | 0.9789 | 0.9823 |
|
Duplicated_reads_percent | 0.0521 | 0.0773 | 0.0601 |
|
Mapped_average_MAQ | 59.0869 | 59.0744 | 60.2534 |
| Error_rate | 0.0019 | 0.002 | 0.0021 |
|
On_target_region_reads_percent | 0.7049 | 0.7053 | 0.7068 |
|
20X_coverage_bases_percent | 0.9013 | 0.9235 | 0.9145 |
|
Average_coverage | 122× | 128× | 132× |
| Table IV.Specific variation distribution. |
Table IV.
Specific variation distribution.
| Mutation type | Child, number of
mutations | Father, number of
mutations | Mother, number of
mutations |
|---|
| DOWNSTREAM | 86,953 | 105,783 | 89,108 |
| EXON | 118,407 | 112,863 | 119,841 |
| INTERGENIC | 661 | 42,163 | 775 |
| INTRON | 178,572 | 258,482 | 188,825 |
| MOTIF | 227 | 241 | 236 |
|
SPLICE_SITE_ACCEPTOR | 484 | 321 | 405 |
|
SPLICE_SITE_DONOR | 395 | 221 | 385 |
|
SPLICE_SITE_REGION | 13,974 | 11,858 | 14,055 |
| TRANSCRIPT | 3,057 | 2,869 | 3,096 |
| UPSTREAM | 66,565 | 86,377 | 70,492 |
| UTR_3_PRIME | 12,999 | 13,770 | 13,252 |
| UTR_5_PRIME | 7,823 | 6,560 | 8,528 |
| TOTAL | 490,117 | 641,508 | 508,998 |
| Table V.Candidate maturity-onset diabetes of
the young mutation sites. |
Table V.
Candidate maturity-onset diabetes of
the young mutation sites.
| Chromosome | Site | Reference gene | Mutant base | Gene | Protein
variation | Crowd
frequency | Child genotype | Father
genotype | Mother
genotype |
|---|
| chr1 | 160057528 | A | G | KCNJ9 | p.Glu368Gly | 0.04 | 0/1 | 0/1 | 0/0 |
| chr11 | 17452500 | C | T | ABCC8 | p.Val560Met | 0.010883 | 0/1 | 0/1 | 0/0 |
| chr13 | 28494493 | T | C | PDX1 | p.Leu73Pro | – | 0/1 | 0/0 | 0/0 |
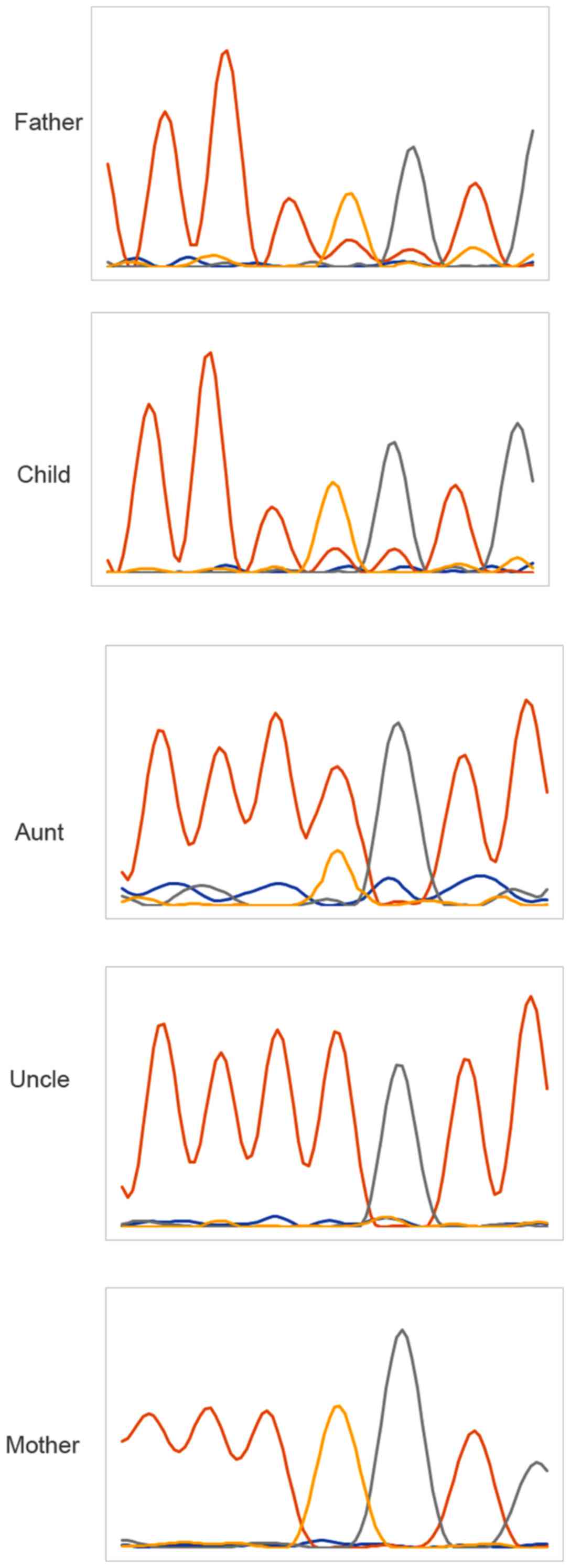
Family verification
In order to verify whether the candidate site is a
true pathogenic site, next-generation sequencing was used to
analyze the information of the family and found that the uncle and
the proband had the same genotype, whereas the genotype of the aunt
was normal (Fig. 3).
Discussion
MODY is a rare monogenic type of diabetes caused by
heterozygous mutations of PDX1, an autosomal dominant gene that is
important for regulating pancreatic function and development. The
proteins encoded by PDX1 are transcriptional activators of insulin,
somatostatin, glucokinase, islet amyloid polypeptide and glucose
transporter 2. The encoded nuclear protein is involved in early
development of the pancreas and the glucose-dependent regulation of
insulin gene expression. Defects in this gene are responsible for
islet hypoplasia, which may lead to early-onset insulin-dependent
diabetes mellitus (IDDM) and MODY type 4 (MODY4) (24,25).
PDX1 is the main transcription factor that maintains
β-cell function. During the formation of endocrine cells, the
increase in PDX1 levels is crucial for the development and
differentiation of pancreatic β cells. In adulthood, PDX1 is
located in β and δ islet cells, and it may regulate the balance of
glucose in the body by maintaining β-cell function and regulating
insulin, glucose transporter 2 and glucokinase. The inhibition of
PDX1 nuclear localization leads to β-cell dysfunction. Forkhead box
(Fox)a2 and PDX1 are key regulators of β-cell development and
function, and their mutations are associated with susceptibility to
MODY, pancreatic hypoplasia and diabetes. Although Foxa2 has been
shown to directly regulate the expression of PDX1 during mouse
embryonic development, the effect of the regulation of this gene on
postpartum β-cell maturation remains to be elucidated. Elucidating
the gene regulatory network following β-cell maturation may improve
current understanding of the pathological mechanisms involved in
diabetes (26,27).
Previous studies have demonstrated that the genetic
and acquired reduction in the expression of PDX1 may lead to type 2
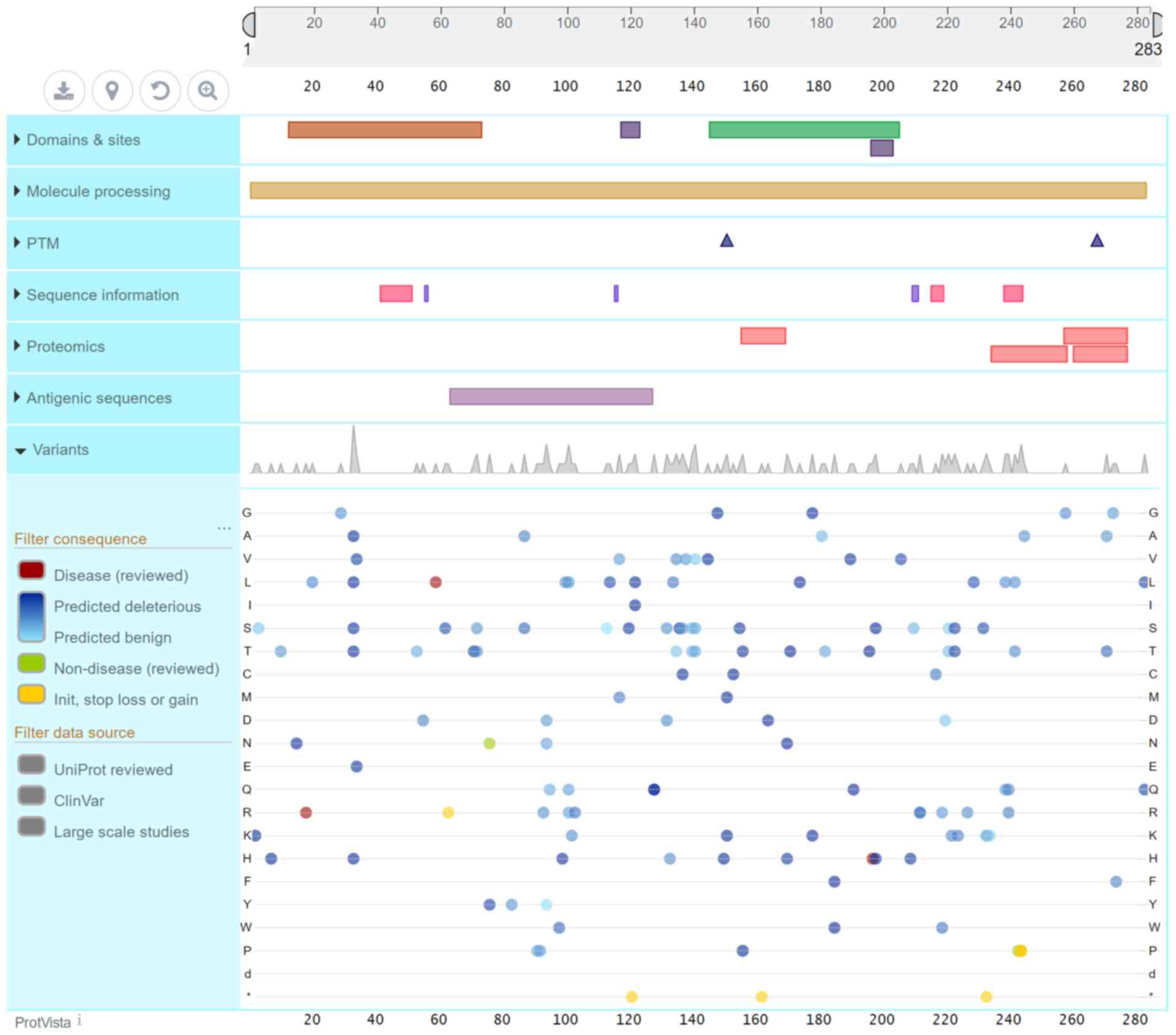
diabetes and β-cell dysfunction. The PDX1 gene encodes a protein
consisting of 283 amino acid residues (Fig. 4). The PDX1 protein sequence is
homologous across different species. It has a transactivation
domain (13–73) at the N-terminus, and the mutation site of the
family in the present study was located at the end of this
transactivation domain, which may affect the function of the
protein. The majority of the 15 mutation sites in the ClinVar
database on the PDX1 gene that resulted in MODY4 were not
identified as pathogenic; however, these sites are likely
pathogenic or associated with conflicting interpretations of
pathogenicity. The early diabetes caused by PDX1 mutations is not
associated with any signs of insulin resistance; thus, its
diagnosis poses a major challenge. In this family, a novel mutation
site was identified, which was considered to be a pathogenic site
based on bioinformatics analysis.
MODY4 is a rare type of diabetes. The number of
reports on MODY caused by gene mutations is currently limited. In
the present study, the clinical manifestations of MODY4 were
different. The patient age at onset was 13–67 years, with a mean
age of 35 years, which was higher compared with other reported MODY
ages at onset. The patients may be obese or non-obese. Insulin
secretion disappeared in phase 1 and significantly decreased in
phase 2 following glucose stimulation. The majority of cases have
been managed with dietary modifications and oral hypoglycemic
agents, with a small number of patients requiring insulin
therapy.
In clinical practice, MODY is similar to type 1
diabetes in that the age of onset overlaps with adolescents, and
hyperglycemia, dry mouth, polydipsia and weight loss occur.
However, a difference is also notable. Patients with type 1
diabetes have high blood sugar, often accompanied by ketosis,
nausea, vomiting and positivity for antibodies (ICA, IAA and GAD)
in laboratory tests, the function of islets is poor and long-term
insulin replacement is required. Patients with MODY generally have
no ketosis. In the present study, no ketosis was present in the
disease course of the proband or family members, they were negative
for autoantibodies, islet function was acceptable and the
hypoglycemic treatment did not require long-term insulin
replacement.
In terms of MODY and type 2 diabetes, the
similarities include overlap in the age of onset and negativity for
antibodies, and treatment generally does not require long-term
insulin; however, differences are also notable, more adolescent
patients with type 2 diabetes with obesity have metabolic syndrome,
and insulin function is mainly caused by insulin resistance; by
contrast, patients with MODY are generally not obese and have no
metabolic syndrome, and islet function defects are mainly caused by
insulin secretion defects. The most marked difference between them
is the genetic background, MODY exhibits multi-generation vertical
inheritance, whereas type 2 diabetes exhibits multi-generation
non-vertical inheritance. Therefore, patients with MODY are young,
have no ketosis, have a family history of three or more generations
of diabetes, and are consistent with autosomal dominant
inheritance. The pathological basis is the primary defect of islet
β-cell function.
In the present study, the age of the proband at
onset was 23 years, and the insulin secretion curve was low,
indicating poor pancreatic islet function, although there was no
tendency for ketosis. In the initial stage, insulin aspart was
administered via an insulin pump, and the patient's blood sugar
levels quickly stabilized and were restored to normal.
Following relief of high-glucose toxicity, the
insulin dose was gradually reduced. Following hospital discharge,
in order to maintain stable blood sugar level, subcutaneous
injection of insulin + insulin glargine was administered as
hypoglycemic therapy four times per day. Following 3 months of
evaluation, the HbA1c level was 8.8%, and the genetic analysis
results confirmed a diagnosis of MODY4 due to a PDX1 mutation.
Based on this result, the patients' subsequent hypoglycemic regimen
was adjusted. Insulin before meals was discontinued, and the
patient was only treated with insulin glargine injection (14 units)
at bedtime, and oral acarbose at mealtimes. At 6 months following
discharge from hospital, the HbA1c level was 6.8%. Insulin glargine
was changed to oral metformin (250 mg orally three times/day) and
acarbose (50 mg orally three times/day). The HbA1c level was 7.0%
at 9 months following discharge. The comprehensive clinical
manifestations and treatment outcomes were largely in line with the
characteristics of MODY4.
In terms of the father and uncle of the proband (II2
and II1), their age at onset was similar (35–40 years). Following
oral 75 g glucose stimulation, the secretion of insulin disappeared
in phase 1. According to conventional treatment experience, such
patients with poor islet function should be treated with insulin or
secretagogues. However, the HbA1c levels of the father and uncle
decreased to 7.8 and 8.1% following regular oral metformin
administration for 3 months. Furthermore, genetic analysis revealed
that both were likely to have MODY4. Therefore, consistent with
previous literature on treatment experience of patients with MODY4,
the dose of metformin was halved. The HbA1c levels were 6.8 and
7.1% at 6 months, and metformin was discontinued. At 9 months, the
HbA1c levels were 9.9 and 7.0%. The age at onset, islet function
and treatment outcomes were consistent with the characteristics of
MODY4.
The findings of the present study confirmed the
presence of MODY4 in families caused by PDX1 gene mutation in the
Chinese population, and suggested that attention should be paid to
screening for MODY among diabetic patients with a strong family
genetic background and onset in adolescence. Such patients have
early manifestations of insufficient insulin secretion and may be
clinically misdiagnosed with type I or atypical type II diabetes.
The traceability of their family history and family members'
clinical characteristics is important, and precision medicine is
crucial for accurate diagnosis, subsequent treatment and disease
outcome. There is no uniform clinical diagnostic standard for MODY,
and only genetic testing can confirm the diagnosis. With the
continuous advancement of genetic testing technology, it may become
easier to accurately diagnose MODY and to identify specific MODY
subtypes. Furthermore, it is crucial for clinicians to design
individualized treatment plans and provide genetic counseling for
the relatives of such patients.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of materials and data
Any datasets generated and/or analyzed in the
present study are available from the corresponding author on
reasonable request.
Authors' contributions
HY performed experiments and was involved in writing
the manuscript; JL performed experiments and was involved in
writing the manuscript; XL and FM were involved in data analysis;
YY was involved in processing data and writing the manuscript. All
the authors have read and approved the final version of this
manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Shanxi Provincial People's Hospital.
Patient consent for publication
Informed written consent was obtained from the
patient for the publication of this case report and any
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
MODY
|
maturity-onset diabetes of the
young
|
|
WES
|
whole-exome sequencing
|
|
IDDM
|
insulin-dependent diabetes
mellitus
|
|
MODY4
|
maturity-onset diabetes of the young
type 4
|
References
|
1
|
Anık A, Çatlı G, Abacı A and Böber E:
Maturity-onset diabetes of the young (MODY): An update. J Pediatr
Endocrinol Metab. 28:251–263. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim SH: Maturity-onset diabetes of the
young: What do clinicians need to know? Diabetes Metab J.
39:468–477. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kleinberger JW and Pollin TI: Undiagnosed
MODY: Time for action. Curr Diab Rep. 15:1102015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Anık A, Çatlı G, Abacı A, Sarı E,
Yeşilkaya E, Korkmaz HA, Demir K, Altıncık A, Tuhan HÜ, Kızıldağ S,
et al: Molecular diagnosis of maturity-onset diabetes of the young
(MODY) in Turkish children by using targeted next-generation
sequencing. J Pediatr Endocrinol Metab. 28:1265–1271. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kwak SH, Jung CH, Ahn CH, Park J, Chae J,
Jung HS, Cho YM, Lee DH, Kim JI and Park KS: Clinical whole exome
sequencing in early onset diabetes patients. Diabetes Res Clin
Pract. 122:71–77. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Johansson S, Irgens H, Chudasama KK,
Molnes J, Aerts J, Roque FS, Jonassen I, Levy S, Lima K, Knappskog
PM, et al: Exome sequencing and genetic testing for MODY. PLoS One.
7:e380502012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bonnefond A, Philippe J, Durand E, Muller
J, Saeed S, Arslan M, Martínez R, De Graeve F, Dhennin V,
Rabearivelo I, et al: Highly sensitive diagnosis of 43 monogenic
forms of diabetes or obesity through one-step PCR-based enrichment
in combination with next-generation sequencing. Diabetes Care.
37:460–467. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gragnoli C, Stanojevic V, Gorini A, Von
Preussenthal GM, Thomas MK and Habener JF: IPF-1/MODY4 gene
missense mutation in an Italian family with type 2 and gestational
diabetes. Metabolism. 54:983–988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Weng J, Macfarlane WM, Lehto M, Gu HF,
Shepherd LM, Ivarsson SA, Wibell L, Smith T and Groop LC:
Functional consequences of mutations in the MODY4 gene (IPF1) and
coexistence with MODY3 mutations. Diabetologia. 44:249–258. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stoffers DA, Ferrer J, Clarke WL and
Habener JF: Early-onset type-II diabetes mellitus (MODY4) linked to
IPF1. Nat Genet. 17:138–139. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ağladıoğlu SY, Aycan Z, Çetinkaya S, Baş
VN, Önder A, Peltek Kendirci HN, Doğan H and Ceylaner S: Maturity
onset diabetes of youth (MODY) in Turkish children: Sequence
analysis of 11 causative genes by next generation sequencing. J
Pediatr Endocrinol Metab. 29:487–496. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Szopa M, Ludwig-Gałęzowska A, Radkowski P,
Skupień J, Zapała B, Płatek T, Klupa T, Kieć-Wilk B, Borowiec M,
Młynarski W, et al: Genetic testing for monogenic diabetes using
targeted next-generation sequencing in patients with maturity-onset
diabetes of the young. Pol Arch Med Wewn. 125:845–851.
2015.PubMed/NCBI
|
|
13
|
Andrews S: FastQC: A quality control tool
for high throughput sequence data. BibSonomy; 2010
|
|
14
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Faust GG and Hall IM: SAMBLASTER: Fast
duplicate marking and structural variant read extraction.
Bioinformatics. 30:2503–2505. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R; 1000 Genome
Project Data Processing Subgroup, : The sequence alignment/map
format and SAMtools. Bioinformatics. 25:2078–2079. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garrison E and Marth G: Haplotype-based
variant detection from short-read sequencing. arXiv preprint
arXiv:1207.3907. 2012.
|
|
19
|
Rimmer A, Phan H, Mathieson I, Iqbal Z,
Twigg SRF; WGS500 Consortium, ; Wilkie AOM, McVean G and Lunter G:
Integrating mapping-, assembly-and haplotype-based approaches for
calling variants in clinical sequencing applications. Nat Genet.
46:9122014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koboldt DC, Zhang Q, Larson DE, Shen D,
McLellan MD, Lin L, Miller CA, Mardis ER, Ding L and Wilson RK:
VarScan 2: Somatic mutation and copy number alteration discovery in
cancer by exome sequencing. Genome Res. 22:568–576. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chapla A, Mruthyunjaya MD, Asha HS,
Varghese D, Varshney M, Vasan SK, Venkatesan P, Nair V, Mathai S,
Paul TV and Thomas N: Maturity onset diabetes of the young in
India-a distinctive mutation pattern identified through targeted
next-generation sequencing. Clin Endocrinol (Oxf). 82:533–542.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dusatkova P, Fang M, Pruhova S, Gjesing
AP, Cinek O, Hansen T, Pedersen OB, Xu X and Lebl J: Lessons from
whole-exome sequencing in MODYX families. Diabetes Res Clin Pract.
104:e72–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang X, Sterr M, Burtscher I, Chen S,
Hieronimus A, Machicao F, Staiger H, Häring HU, Lederer G and
Meitinger T: Genome-wide analysis of PDX1 target genes in human
pancreatic progenitors. Mol Metab. 9:57–68. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fajans SS, Bell GI, Paz VP, Below JE, Cox
NJ, Martin C, Thomas IH and Chen M: Obesity and hyperinsulinemia in
a family with pancreatic agenesis and MODY caused by the IPF1
mutation Pro63fsX60. Transl Res. 156:7–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Winter WE and Silverstein JH: Molecular
and genetic bases for maturity onset diabetes of youth. Curr Opin
Pediatr. 12:388–393. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guazzarotti L, Bartolotta E and Chiarelli
F: Maturity-onset diabetes of the young (MODY): A new challenge for
pediatric diabetologists. J Pediatr Endocrinol Metab. 12:487–497.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Froguel P and Velho G: Molecular genetics
of maturity-onset diabetes of the young. Trends Endocrinol Metab.
10:142–146. 1999. View Article : Google Scholar : PubMed/NCBI
|