Introduction
Alzheimer's disease (AD) is a neurodegenerative
disease of the central nervous system that is characterized by
progressive cognitive dysfunction, ultimately leading to dementia.
While the pathological hallmarks are mainly the formation of
extracellular senile plaques, intracellular neurofibrillary tangles
and neuronal loss, its pathogenesis has not been fully elucidated
(1). Previous studies have
demonstrated that energy dysmetabolism contributes significantly to
the pathogenesis of a variety of aging-associated diseases and
degenerative disease of the nervous system, including AD (2,3).
With aging, the imbalance of energy metabolism in the brain is
intensified, which promotes amyloid (A)β production. Aβ further
induces energy metabolism disorder in the brain (4,5).
When the energy metabolism of neurons in the brain is deregulated,
ATP synthesis is insufficient and leads to irreversible damage to
neuronal structure and function (6,7).
Since research has demonstrated that energy disorders of the brain
have an important impact on the development of AD, improving the
energy metabolism of nerve cells in the brain is expected to delay
the pathogenesis of AD (8–10).
Nicotinamide adenine dinucleotide (NAD+)
is an important coenzyme in the redox reaction, which can
participate in energy synthesis through glycolysis, the
tricarboxylic acid cycle and mitochondrial oxidative
phosphorylation (10). The
majority of NAD+ is synthesized by a salvage pathway
from the conversion of nicotinamide to a nicotinamide
mononucleotide, which is subsequently converted to NAD+
(11). Nicotinamide
phosphoribosyltransferase (NAMPT) is the rate-limiting enzyme in
the NAD+ rescue pathway and is involved in energy
metabolism by regulating NAD+ synthesis (12), maintaining the homeostasis of
energy metabolism in the brain and thereby increasing the
expression of NAD+ (the level of NAMPT has been proven
to regulate positively NAD+ levels) (13). However, few studies to date have
directly explored the potential therapeutic effect of targeting
NAMPT and NAD+ biosynthesis, despite it being known that
NAD+ is the starting point of the majority of key
metabolic pathways and therefore the key governor of cellular aging
and age-related processes (14,15).
How to regulate the expression of NAMPT to protect against
neurodegenerative diseases such as AD has become the focus of
recent research.
The present study used 6-month-old APPswe/PS1ΔE9
(APP/PS1) transgenic mice as early AD mouse models and observed the
mechanism of NAMPT-related pathways in the process of AD.
Materials and methods
Ethics statement
All procedures were performed in accordance with the
Guide for and Use of Medical Laboratory Animals (Ministry of Health
China, 1998) (16) and were
approved by the Shanghai University of Traditional Chinese Medicine
(Shanghai, China) Laboratory Animal Care and Use Committee (no.
PZSHUTCM19011807).
APP/PS1 transgenic mice and drug
treatment
In total, 18 male APP/PS1 transgenic mice (age: 24
weeks; weight: 26±3 g) and 6 male C57BL/6 mice (age: 24 weeks;
weight: 27±2 g) were used in the present study. The APP/PS1 mouse
strain is a double-transgenic hemizygote that expresses a chimeric
mouse/human amyloid precursor protein and mutant human
presenilin-1. These transgenic mice were used as they develop
behavioral and pathology features of AD similar to patients and
have been used in previous AD studies worldwide (17–19).
Separately, C57BL/6 mice were used as age-matched controls. The
APP/PS1 transgenic mice were purchased from the Model Animal
Research Center of Nanjing University, while C57BL/6 mice were
obtained from the Experimental Animal Center of Shanghai Academy.
The NAD+ group were intraperitoneally injected with
NAD+ (30 mg/kg, Sigma-Aldrich; Merck KGaA) at 20 weeks
of age and once every other day for 4 weeks. The FK866 (NAMPT
inhibitor) group were intraperitoneally injected with FK866 (1
mg/kg, Sigma-Aldrich; Merck KGaA) at 20 weeks of age and once every
other day for 4 weeks. The mice were housed in a controlled
specific-pathogen-free environment (22±3°C; 60% relative humidity;
12-h light/dark cycle) with ad libitum access to water and
food.
Morris water maze test
Spatial learning and memory of the mice were
assessed using a Morris water maze test (MWM) according to a
previous study (20) with minor
modifications. The water was made opaque with titanium dioxide and
water temperature was kept at 22±2°C. A platform was placed 1 cm
under the surface of the water. In the hidden platform experiment,
mice received 4 training trials per day for 5 consecutive days. On
the first trial of the first day, mice were placed on the platform
for 10 sec, after which they were placed in the water. The pool
area was conceptually divided into four quadrants of equal size.
Taking the quadrant of the platform as the first quadrant, the
second and fourth quadrants were taken as the starting point for 2
trials and the mice were placed facing the pool wall. If a mouse
failed to find the platform in 70 sec, it was gently guided to the
platform location and allowed to stay on it for 30 sec. The time to
find the platform was recorded as the escape latency. The
experiments were recorded with a camera connected a video recorder
and a computerized tracking system, with the automatic timer set to
70 sec.
In the probe trials, at 24 h after the last training
trial, the platform was removed and the mice were placed at the
second and fourth quadrants, and the time of the target quadrant
(first quadrant) and the number of times crossing the location that
previously contained the platform within 70 sec were recorded.
Thioflavin S staining
Mice were anesthetized with 5% chloral hydrate at a
dose of 400 mg/kg intraperitoneally and saline solution was used
for perfusion through the heart, which was followed by 4%
paraformaldehyde. The brains of the mice were then removed, fixed
in 4% paraformaldehyde for 24 h at room temperature and immersed in
30% sucrose until they sank. The brain tissue was serially
sectioned at a 30-µm thickness using a HM1950 freezing microtome.
The sections were permeabilized in xylene for 10 min, dehydrated
using anhydrous ethanol for 5 min, and then stained with 1%
thioflavin S (Sigma-Aldrich; Merck KGaA) at 37°C for 30 min. The
stained samples were differentiated in 70% alcohol solution for 5
min, mounted with glycerol gel, and observed using fluorescence
microscopy (magnification, 200×; Olympus BX51; Olympus
Corporation).
NAD+/NADH analysis
A NAD+/NADH quantification kit was used
to determine NAD+/NADH levels (cat. no. k337-100;
BioVision, Inc.), according to the manufacturer's instructions.
Hippocampus tissue (20 mg) was washed with precooled
phosphate-buffered saline (PBS), homogenized in 400 µl of the
NAD+/NADH extraction buffer and then centrifuged for 5 h
at 18,000 × g at 4°C. The resulting supernatant was labeled as
total NAD+ sample (NAD+t). Subsequently, 200
µl of the NAD+t sample was heated at 60°C for 30 min (to
consume all NAD+ from the sample, leaving only NADH to
be analyzed). Following cooling on ice, the sample was centrifuged
at 12,000 × g for 30 sec at 4°C and the resulting supernatant was
labeled as the NADH sample. NAD+/NADH extraction buffer
was added to bring the volume to 50 µl. Subsequently, 100 µl of
enzyme reaction mix and 10 µl of NADH developer were added to each
well, the mixture was incubated at room temperature for 2 h and the
absorbance was measured using a microplate reader (wavelength: 450
nm).
ATP content detection
Cortical or hippocampus tissue (10 mg) was washed
with precooled PBS, homogenized in 100 µl of lysis buffer (cat. no.
P0013B: Beyotime Institute of Biotechnology) and centrifuged at
12,000 × g at 4°C for 5 min. The resulting supernatant was
transferred to a new Eppendorf tube and then the reaction solution
(ATP Assay Buffer, ATP probe, ATP Converter and Develop Mix;
Sigma-Aldrich; Merck KGaA) was added to bring the volume to 50 µl.
The reaction solution was shaken in a shaker for 30 min at room
temperature. To correct for the background of the samples, a well
per sample was used as the sample background well. The absorbance
was measured at a wavelength of 570 nm using a microplate
reader.
Immunohistochemical staining
Following a behavioral test, three mice were
randomly selected in each group. Following anesthesia, the brains
were perfused with 4% paraformaldehyde for 4 h and then transferred
to a 30% sucrose gradient at room temperature. After the brains had
sunk to the bottom, continuous coronal sectioning was performed (30
µm thickness) using a HM1950 freezing microtome. The sections were
permeabilized in xylene for 10 min and dehydrated using anhydrous
ethanol for 5 min. The sections were washed with 0.01 M of PBS and
blocked with 5% BSA (Sigma-Aldrich; Merck KGaA) for 1 h at room
temperature. Then the sections were incubated with polyclonal
anti-NAMPT antibody (1:200; cat. no. ab45890; Abcam) overnight at
4°C. Following rinsing with PBS, sections were incubated with
horseradish peroxidase (HRP)-conjugated anti-rabbit IgG antibody
(1:500; cat. no. SA00001-2; ProteinTech Group, Inc.) for 1 h at
37°C, washed with PBS, and streptavidin-biotin complex (Beyotime
Institute of Biotechnology) was added. Then the sections were
developed with 3,3′-diaminobenzidine for 10 min at room temperature
and dehydrated with gradient alcohol, cleared with xylene, sealed
with neutral gum and observed with using a light microscope
(magnification, ×200; Olympus BX51; Olympus Corporation).
Western blot analysis
The isolated hippocampus tissue was collected,
homogenized in lysis buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology), lysed on ice for 30 min and centrifuged at 12,000 ×
g for 20 min at 4°C. Total protein concentration was determined
using a Micro BCA Protein Assay kit (Thermo Fisher Scientific,
Inc.). Then, 30 µg of protein sample was heated at 95°C for 5 min
and separated by 10% SDS-polyacrylamide gel electrophoresis, after
which it was transferred onto a PVDF membrane. The membrane was
blocked with 3% BSA at 37°C for 1 h. Following blocking, the
membranes were incubated with primary antibodies (anti-NAMPT 1:500,
cat. no. ab45890, Abcam; anti-SIRT1, 1:1,000, cat. no. ab110304,
Abcam; and GAPDH, 1:1,000; cat. no. 5174; Cell Signaling
Technology, Inc.) overnight at 4°C. The membrane was washed with
TBST three times for 10 min and then treated with HRP-labeled goat
anti-rabbit secondary antibody (1:2,000; cat. no. SA00001-2;
ProteinTech Group, Inc.) and HRP-labeled goat anti-mouse secondary
antibody (1:2,000; cat. no. SA00001-1; ProteinTech Group, Inc.) for
1 h at 37°C. Blots were washed three times for 10 min in TBST.
After incubation with enhanced chemiluminescence chromogenic
solution (cat. no. P0018s; Beyotime Institute of Biotechnology) for
1 min at room temperature, quantification of the proteins of
interest was determined relative to GAPDH using ImageJ software
(version 1.4; National Institutes of Health).
Statistical analysis
Statistical analysis was performed using GraphPad
software 6.0 (GraphPad Software, Inc.). Measurement data are
presented as the mean ± standard error of the mean. Two-way ANOVA
followed by Bonferroni post hoc test was used to analyze group
differences in the escape latency. A one-way ANOVA followed by LSD
multiple-range test was used in the probe trial. For the
non-normally distributed data or for data with heterogeneous
variance, a Kruskal-Wallis test was used. P<0.05 was considered
to indicate a statistically significant difference.
Results
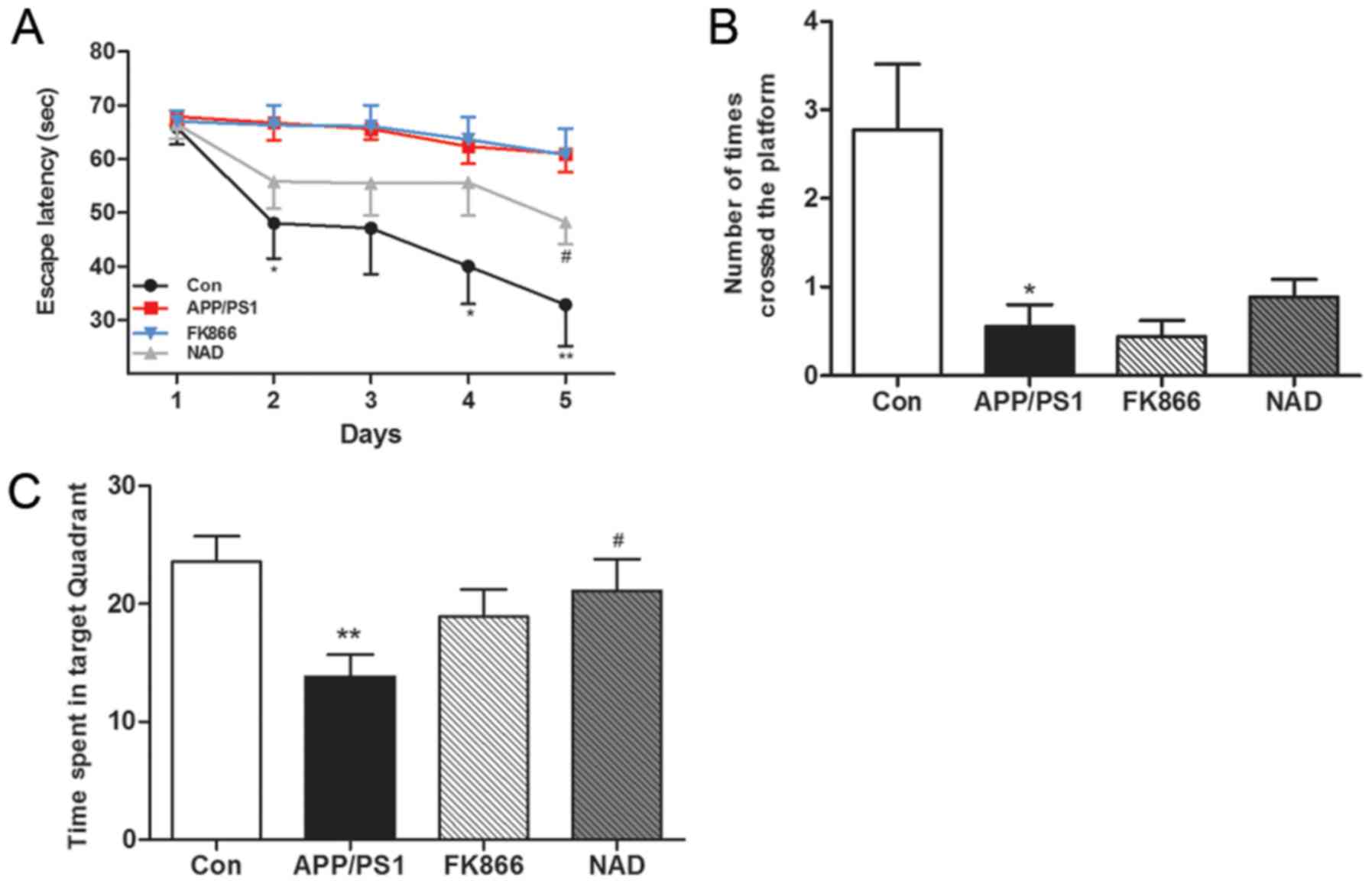
APP/PS1 mice exhibit impaired
performance and effects of NAD+ and FK866 treatment in
the MWM tasks
Memory impairment in APP/PS1 transgenic mice was
determined using the MWM test in terms of the escape latency. The
results demonstrated that, with the increase in the number of
training days, the escape latency gradually shortened. On the fifth
day, the escape latency of the APP/PS1 group (AD) mice was
significantly longer compared with the blank control group
(P<0.01), suggesting that the learning and memory abilities of
AD mice were significantly impaired. Furthermore, the escape
latency of NAD+ mice was significantly shorter than that
of APP/PS1 mice after intraperitoneal injection of NAD+
(P<0.01); while, after FK866 intervention, there was no
significant change in escape latency in the FK866 mice compared
with the APP/PS1 mice (Fig.
1A).
In the space exploration experiment on the sixth
day, after removing the platform it was found that APP/PS1 mice
significantly reduced the target quadrant residence time
(P<0.01) compared with control group mice, indicating that the
spatial memory ability of AD mice was impaired. Following
NAD+ treatment, the NAD+ mice spent more time
in the target quadrant compared with the APP/PS1 mice (P<0.01);
however, there was no significant change in the target quadrant
residence time of the mice in the APP/PS1 group compared with the
FK866 group (Fig. 1B). In
comparison with the control group, the number of platform crossings
was significantly reduced in the APP/PS1 group (P<0.05) and the
NAD+ mice exhibited an increased number of platform
crossings compared with the APP/PS1 mice, but these findings were
not statistically significant (P>0.05). Overall, there was no
significant change between the APP/PS1 group and FK866 group
(Fig. 1C).
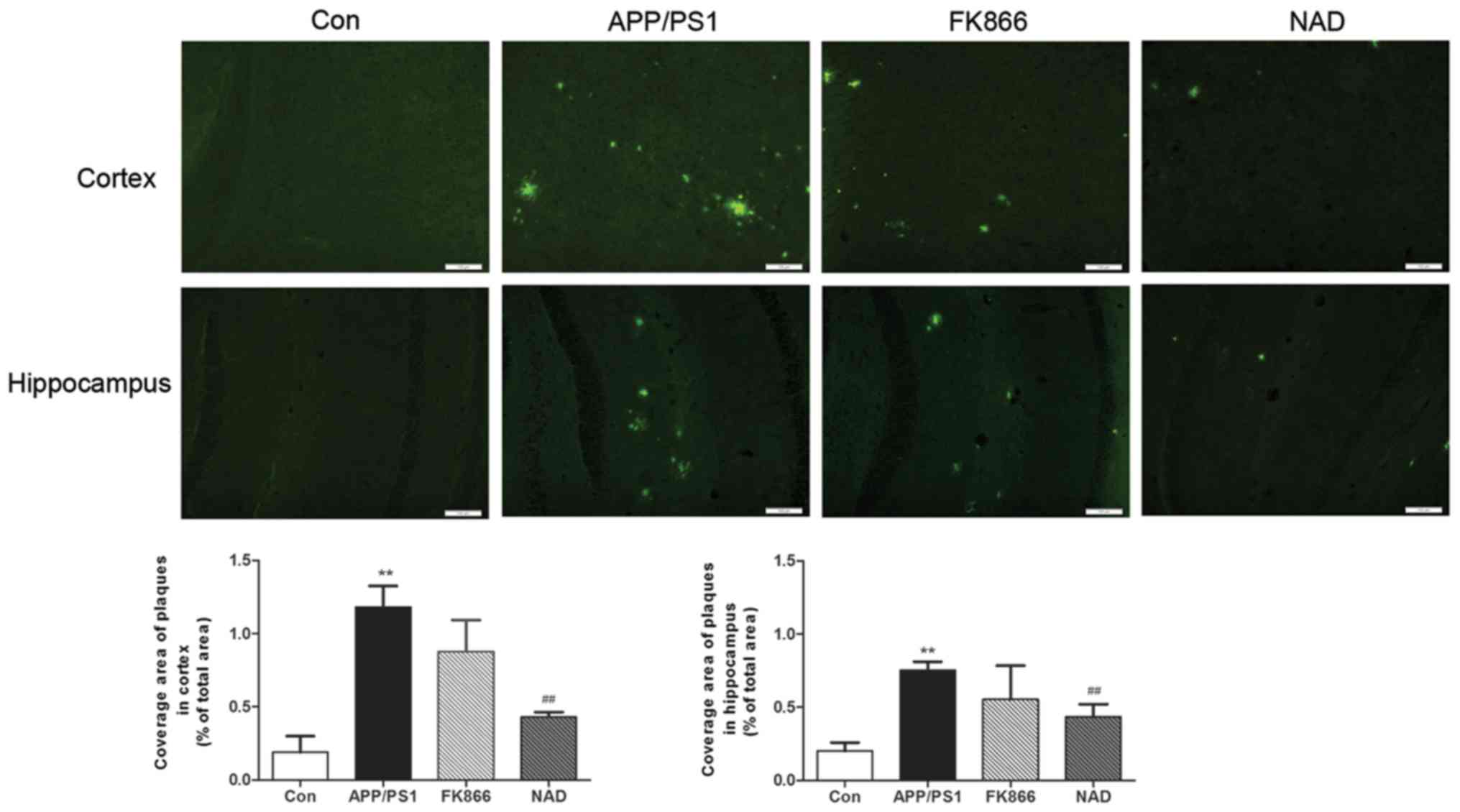
NAD+ treatment reduces
amyloid plaques of cortex and hippocampus in APP/PS1 transgenic
mice
Staining with Thioflavin S for senile plaques in the
cortex and hippocampus was conducted. As expected, the AD mice
presented numerous plaques at 6 months of age as compared with the
control mice in cortex and hippocampus; however, following
NAD+ treatment, scattered green fluorescent plaques were
observed in the cortex and hippocampus of every group and, after
FK866 injection, multiple green fluorescent plaques were also seen
in the cortex and hippocampus in AD mice (Fig. 2).
NAD+ treatment improves ATP
content of cortex and hippocampus in APP/PS1 transgenic mice
ATP content in the cortex and hippocampus was
measured in each group of mice. The results demonstrated that,
compared with the control group, the ATP content in the cortex in
the APP/PS1 group was significantly decreased (P<0.05), while,
in comparison with the APP/PS1 model group, the ATP content in the
cortex in the NAD+ group was significantly increased
(P<0.05). Following FK866 intervention, the ATP content of the
cortex decreased compared with APP/PS1 mice, but without
statistical significance (P>0.05). Additionally, in comparison
with the control group, the ATP content in the hippocampus in the
APP/PS1 group was significantly decreased (P<0.05), while,
compared with the APP/PS1 model group, the ATP content in the
hippocampus in the NAD+ group was significantly
increased (P<0.05). Following FK866 intervention, there was no
significant change in the NAD+ group compared with
APP/PS1 model group (Fig. 3).
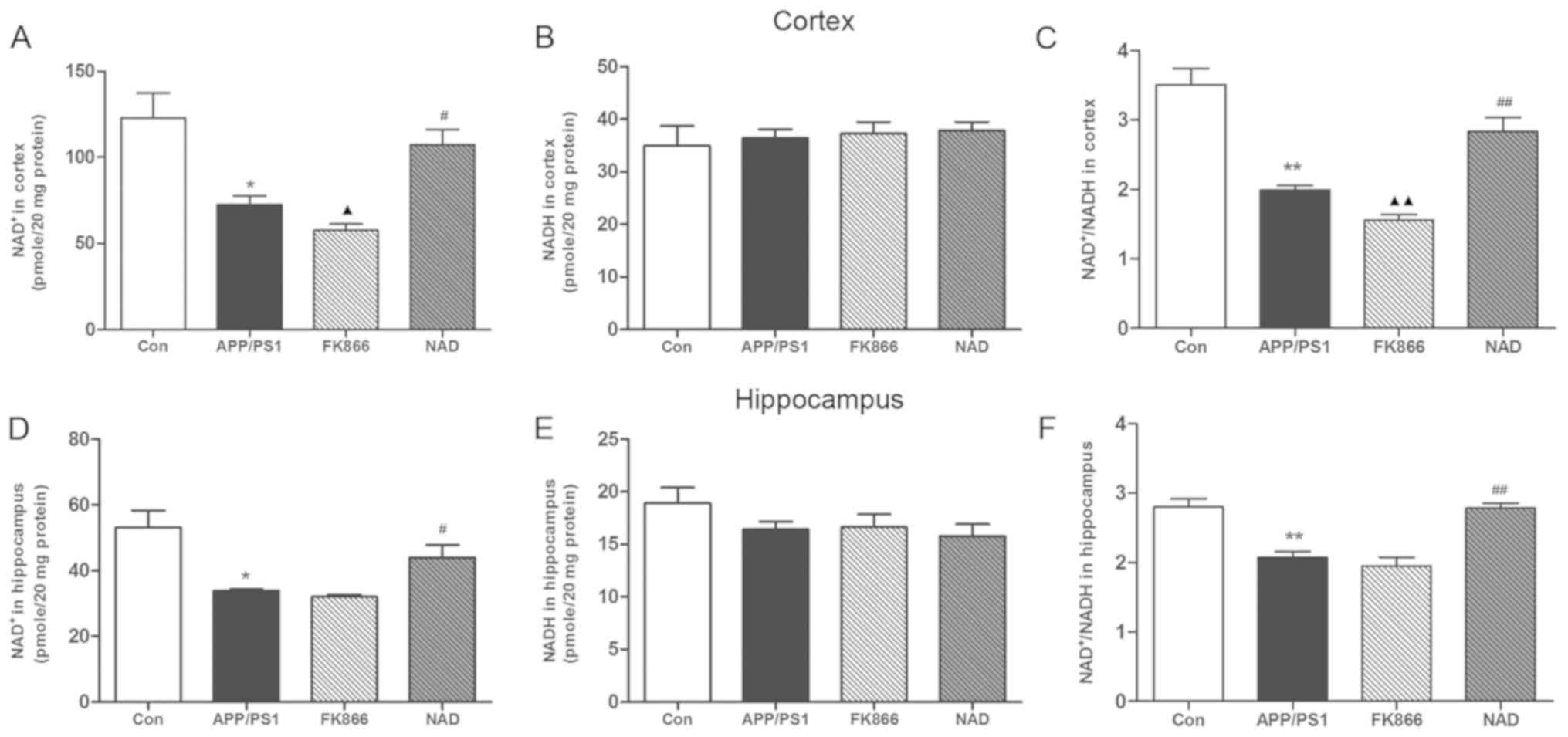
NAD+ treatment increases
NAD+ level and NAD+/NADH ratio in APP/PS1
transgenic mice
NAD+ and NADH are essential coenzymes for
cell energy metabolism. Under normal physiological conditions, the
NAD+/NADH ratio reflects the redox state of cells. When
the energy metabolism of the cells is deregulated, the levels of
NAD+ and NADH will change, which is reflected as an
imbalance in the NAD+/NADH ratio (21). The present study detected
NAD+ and NADH levels in the cortex and hippocampus. The
NAD+ content in the cortex in the APP/PS1 group was
significantly decreased compared with the control group (P<0.05)
and the NAD+/NADH ratio was similarly decreased
(P<0.01), although the NADH level was not significantly changed.
Compared with the APP/PS1 model group, the NAD+ content
of the cortex was significantly increased in the NAD+
group (P<0.05) and the NAD+/NADH ratio was
significantly increased (P<0.01), while the NADH level was not
significantly changed. Separately, in comparison with APP/PS1 mice,
NAD+ content in the cortex decreased significantly in
the FK866 group (P<0.05) and the NAD+/NADH ratio
decreased (P<0.01), although the NADH level did not change
significantly between the two groups.
Compared with the control group, the NAD+
content in the hippocampus in the APP/PS1 group was significantly
decreased (P<0.05), while the NADH level was not significantly
changed and the NAD+/NADH ratio was decreased
(P<0.01). Furthermore, compared with the APP/PS1 model group,
the NAD+ content in the hippocampus in the
NAD+ group was significantly increased (P<0.05) and
the NAD+/NADH ratio was significantly increased
(P<0.01), while the NADH level was not significantly different.
Regarding the NAD+ content, NADH level and
NAD+/NADH ratio, there was no significant change in the
hippocampus in the FK866 group compared with the other mice
(Fig. 4).
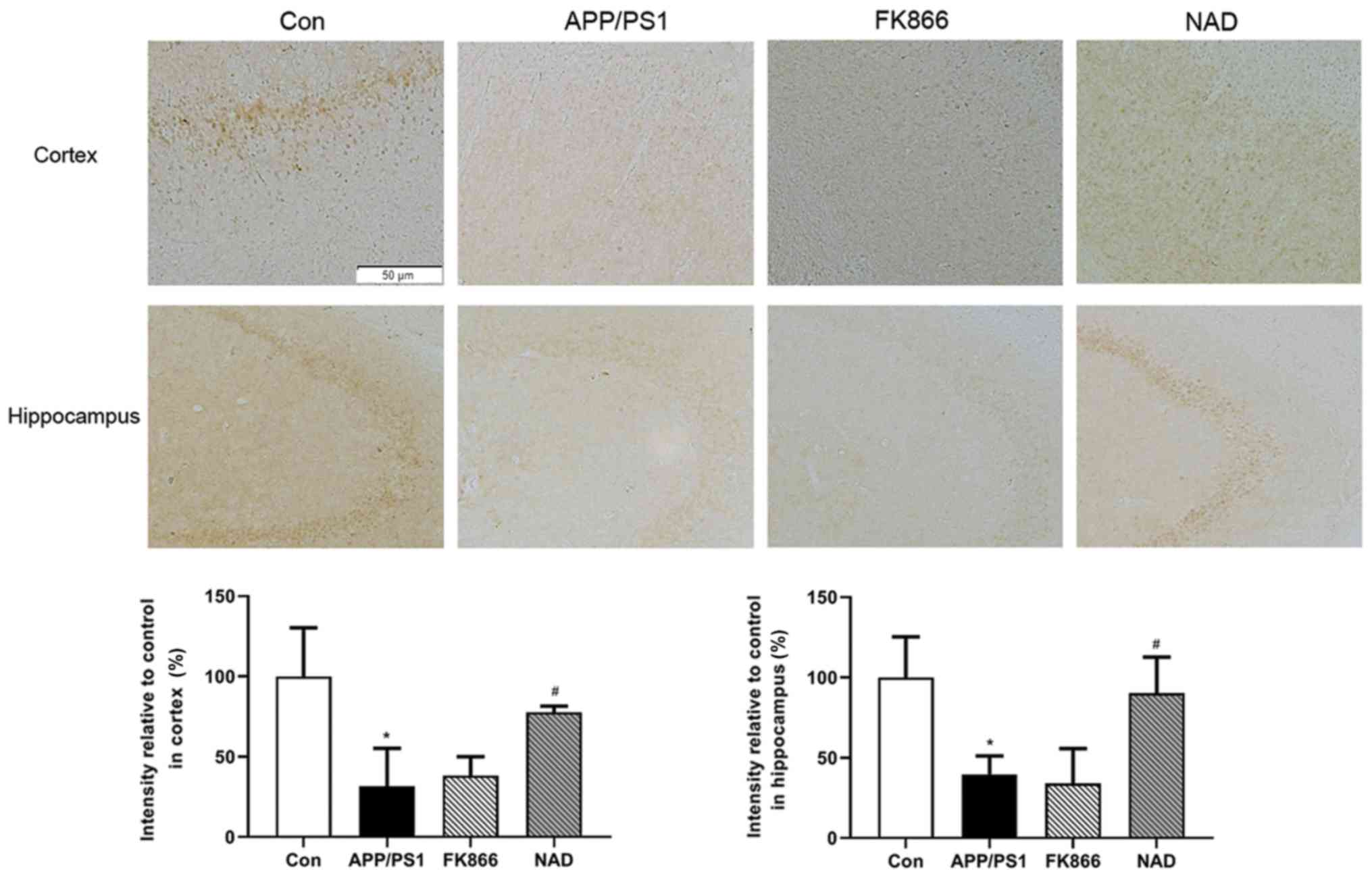
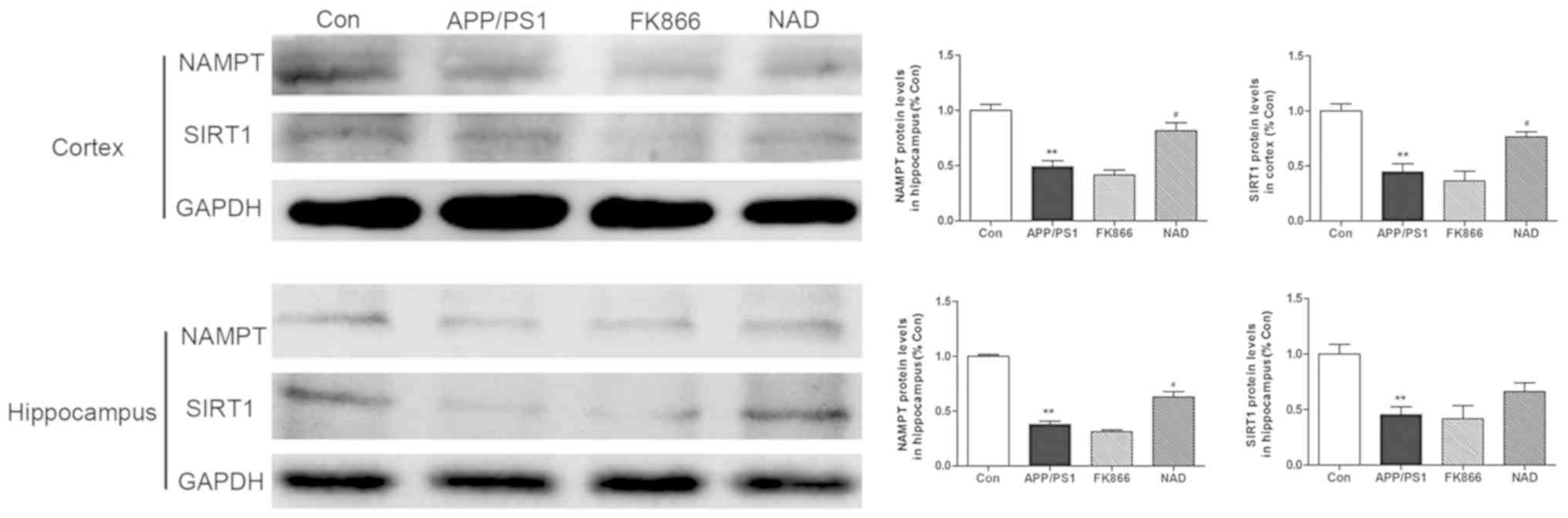
Expression of NAMPT protein decreases
in APP/PS1 transgenic mice and NAD+ treatment improves
the expression
In order to confirm the expression of NAMPT proteins
in the cortex and hippocampus in APP/PS1 transgenic mice,
immunohistochemical staining was performed. It was observed that
the brown positive expression of NAMPT protein in the cortex and
hippocampus in APP/PS1 group was decreased compared with the
control group (P<0.05). Following intraperitoneal injection of
NAD+, the positive expression of NAMPT protein increased
compared with the APP/PS1 group (P<0.05), while there was no
significant change in NAMPT levels in the cortex and hippocampus in
FK866 mice compared with APP/PS1 mice (P>0.05; Fig. 5).
Protein extracts were also prepared and analyzed by
western blotting to determine protein levels. The results
demonstrated that the protein expression of NAMPT and SIRT1 in the
cortex in the APP/PS1 group was significantly decreased when
compared with the control group (P<0.01). Intraperitoneally
injecting NAD+ increased the expression of NAMPT in the
cortex in the NAD+ mice (P<0.05). There was no
significant change in NAMPT protein levels in the FK866 mice
compared with in APP/PS1 mice (P>0.05). Consistent with the
changes in the cortex, the results demonstrated that the NAMPT and
SIRT1 level in the hippocampus in the APP/PS1 group was decreased
compared with the control group (P<0.01). NAMPT expression
levels in the hippocampus in the NAD+ group were also
significantly increased after intraperitoneal injection of
NAD+ (P<0.05); however, injecting NAD+ did
not increase SIRT1 expression levels. Finally, there was no
significant change in NAMPT and SIRT1 protein expression levels in
the hippocampus in FK866 mice, compared with in APP/PS1 mice
(P>0.05; Fig. 6).
Discussion
AD is a degenerative disease of the nervous system.
Studies have demonstrated that energy metabolism abnormalities are
closely related to the development of early AD (22–25).
The energy metabolism of failing neurons often precedes, or occurs
simultaneously with, cognitive impairment (26). The mechanism may be that the energy
metabolism disorder of the brain can upregulate amyloid precursor
protein amyloidal digestion pathway to promote the production of Aβ
and Aβ deposition, further causing mitochondrial dysfunction and
leading to abnormal energy metabolism (27,28).
In addition, abnormal energy metabolism of the brain has also been
associated with hyperphosphorylation of the protein τ, which can
affect synaptic function and aggravate cognitive impairment by
activating the p38/mitogen-activated protein kinase-related pathway
(29).
NAD+ is required for mitochondrial
respiration and ATP synthesis and maintenance of NAD+
level is important for the prevention of mitochondrial dysfunction
(30). NAD+ level
declines with age in many cell and tissue types, with altered
NAD+ metabolism and concurrent alterations in the
mitochondrial function being inherent in AD (31). NAD+ can be synthesized
via different precursors; the main two synthetic pathways are
tryptophan synthetic pathways de novo and salvage pathways
(32,33). The de novo synthesis pathway
of tryptophan is mainly carried out in the liver and kidneys, which
only synthesizes a very small amount of NAD+. The
salvage pathway, using nicotinic acid (NA) and nicotinamide (NAM)
as raw materials, is the main source of NAD+ synthesis.
NA and NAM produce nicotinamide mononucleotide (NMN) through
catalyzation by nicotinic acid phosphoribosyltransferase. NAMPT,
originally identified as pre-B-cell colony enhancing factor, is the
rate-limiting enzyme that catalyzes the first step in the
biosynthesis of NAD+ from nicotinamide (33,34).
Inhibition of NAMPT results in significant depletion of
NAD+ which can modulate the activity of the TCA cycle
and most of sirtuins (35).
Previous studies have found that NAD+
precursors such as NMN and NAM can improve the learning and memory
abilities and mitochondrial function of AD model mice, suggesting
that NAD+ upregulation can reverse the damage of brain
energy metabolism (36,37). Other studies have suggested that
NAD+ mediates a number of major biological processes,
including calcium homeostasis, mitochondrial functions and energy
metabolism, aging, and cell death in various tissues including the
brain (38–40). NAD+ acts as a
neuroprotective agent via several mechanisms, including the
prevention of ATP depletion (41)
NAMPT, a cytokine secreted by fat cells, is expressed in various
tissues including the liver, kidneys, skeletal muscle and brain
tissue and is involved in the oxidation of cells due to the
biosynthesis of nicotinamide adenine dinucleotide (42). NAMPT affects energy metabolism,
protein modification, DNA repair and other important processes by
participating in the synthesis of NAD+. As a
rate-limiting enzyme of the NAD+ salvage pathway, NAMPT
can affect cell metabolism and NAD+ synthesis,
interfering with NAD+ dependent-related protein
expression (38,42). Recently, studies have demonstrated
that NAMPT is associated with central nervous system diseases, in
that it can protect neurons and delay the occurrence of axonal
degeneration (43,44). With increasing age, the expression
of NAMPT in the brain of AD patients is decreased, causing the
level of NAD+ and the antioxidant enzyme glutathione to
decrease, aggravating neuronal degeneration (45,46).
Previous studies have demonstrated that NAMPT is mainly expressed
in neurons in the mouse brain (47–49).
With aging, the essential cofactor NAD+ decreases; the
age-associated decrease in NAD+ levels is due to a
decline in protein level NAMPT (50) and AD is an age-related
neurodegenerative disease (51).
The present study has demonstrated that cortex and
hippocampal NAD+ levels and NAMPT expression declined in
early stage AD model mice. Based on previous studies (52,53),
it is hypothesized that NAD+ administration could combat
NAMPT decrease in the early stage of AD. The present study
demonstrated that the decrease of NAMPT lead to NAD+
decrease, while NADH levels were not affected, leading to a
decrease in the NAD/NADH ratio. Maintenance of NAD+/NADH
ratio is critical for mitochondrial function, such as ATP synthesis
in TCA cycle and oxidative phosphorylation in mitochondria
(54), and it was identified that
ATP decreased in the cortex and hippocampus. As NAD+ is
able to travel freely inside the cell, decreased cellular
NAD+ levels may result in reduced activity of the
enzymes that use NAD+ as a substrate, including SIRT1
(55). In various animal models,
age-related metabolic decline is positively correlated with the
decline of NAD+, and the change of NAD+/NADH
ratio can regulate sirtuin enzymes, including SIRT1. SIRT1 is
expressed in both brain and peripheral tissues, and may shuttle to
the cytoplasm during neuronal differentiation and neurite outgrowth
and apoptosis. SIRT1 has an essential role in regulating cellular
homeostasis by influencing neuron survival and mitochondrial
biogenesis (56,57). The present study demonstrated that
the intraperitoneal injection NAD+ levels in APP/PS1
mice was also associated with increased level of SIRT1. The
NAD+ level in APP/PS1 mice after treatment with FK866
was measured and, as expected, FK866 treated APP/PS1 mice contained
significantly less total NAD+ in the cortex and a lower
NAD+/NADH ratio compared with APP/PS1 mice without any
treatment. In the hippocampus, there was no significance difference
between the FK866 and APP/PS1 groups in the level of
NAD+ and the NAD+/NADH ratio, which may be
related to the pathological development of early AD mice model.
However, it is considered that these results indicated that NAMPT
inhibition caused NAD+ deficiency, which demonstrated
that improvement of NAMPT can increase the generation NAD.
The present study demonstrated that NAD+
depletion with decreased NAMPT is reversible and that
NAD+ replenishment can reverse the impaired brain-energy
metabolism that are implicated in cognitive decline and mitigates
neurodegeneration. Given that NAMPT plays an important role in NAD
synthesis, combining NAD+ or a drug increasing NAMPT may
constitute a therapy for AD in the future.
Acknowledgements
Not applicable.
Funding
Funding was provided by the National Natural Science
Foundation of China (grant no. 81503626) and the Shanghai Health
Bureau Youth Fund (grant no. 201540254).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
SX was the principal investigator and designed the
study. YH performed statistical analysis, conducted the study and
wrote the manuscript. XH performed experiments. DS performed
statistical analysis and data collection. CC contributed to
experimental design and manuscript preparation. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures were performed in accordance with the
Guide for and Use of Medical Laboratory Animals (Ministry of Health
of China, 1998) and approved by the Shanghai University of
Traditional Chinese Medicine Laboratory Animal Care and Use
Committee (no. PZSHUTCM19011807).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hardy J and Selkoe DJ: The amyloid
hypothesis of Alzheimer's disease: Progress and problems on the
road to therapeutics. Science. 297:353–356. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yin F, Boveris A and Cadenas E:
Mitochondrial energy metabolism and redox signaling in brain aging
and neurodegeneration. Antioxid Redox Signal. 20:353–371. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yin F, Sancheti H, Patil I and Cadenas E:
Energy metabolism and inflammation in brain aging and Alzheimer's
disease. Free Radic Biol Med. 100:108–122. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cabezas-Opazo FA, Vergara-Pulgar K, Pérez
MJ, Jara C, Osorio-Fuentealba C and Quintanilla RA: Mitochondrial
dysfunction contributes to the pathogenesis of Alzheimer's disease.
Oxid Med Cell Longev. 2015:5096542015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Han XJ, Hu YY, Yang ZJ, Jiang LP, Shi SL,
Li YR, Guo MY, Wu HL and Wan YY: Amyloid β-42 induces neuronal
apoptosis by targeting mitochondria. Mol Med Rep. 16:4521–4528.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi
L, Büeler H, Bhat NR, Williams RW, Park EA and Liao FF: Metabolic
stress modulates Alzheimer's β-secretase gene transcription via
SIRT1-PPARү-PGC-1 in neurons. Cell Metab. 17:685–694. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu MF, Yin JH, Hwang CS, Tang CM and Yang
DI: NAD attenuates oxidative DNA damages induced by amyloid
beta-peptide in primary rat cortical neurons. Free Radic Res.
48:794–805. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seixas da Silva GS, Melo HM, Lourenco MV,
Lyra E Silva NM, de Carvalho MB, Alves-Leon SV, de Souza JM, Klein
WL, da-Silva WS, Ferreira ST and De Felice FG: Amyloid-β oligomers
transiently inhibit AMP-activated kinase and cause metabolic
defects in hippocampal neurons. J Biol Chem. 292:7395–7406. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rijpma A, van der Graaf M, Meulenbroek O,
Olde Rikkert MGM and Heerschap A: Altered brain high-energy
phosphate metabolism in mild Alzheimer's disease: A 3-dimensional
31P MR spectroscopic imaging study. Neuroimage Clin.
18:254–261. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rajmohan R and Reddy PH: Amyloid-beta and
phosphorylated tau accumulations cause abnormalities at synapses of
Alzheimer's disease Neurons. J Alzheimers Dis. 57:975–999. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elhassan YS, Philp AA and Lavery GG:
Targeting NAD+ in metabolic disease: New insights into an old
molecule. J Endocr Soc. 1:816–835. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rongvaux A, Shea RJ, Mulks MH, Gigot D,
Urbain J, Leo O and Andris F: Pre-B-cell colony-enhancing factor,
whose expression is up-regulated in activated lymphocytes, is a
nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved
in NAD biosynthesis. Eur J Immunol. 32:3225–3234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garten A, Schuster S, Penke M, Gorski T,
de Giorgis T and Kiess W: Physiological and pathophysiological
roles of NAMPT and NAD metabolism. Nat Rev Endocrinol. 11:535–546.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ruggieri S, Orsomando G, Sorci L and
Raffaelli N: Regulation of NAD biosynthetic enzymes modulates
NAD-sensing processes to shape mammalian cell physiology under
varying biological cues. Biochim Biophys Acta. 1854:1138–1149.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Verdin E: NAD+ in aging,
metabolism, and neurodegeneration. Science. 350:1208–1213. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Zhang Y, Li X, Zhang M and Lv K:
Microarray analysis of circular RNA expression patterns in
polarized macrophages. Int J Mol Med. 39:373–379. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Trinchese F, Liu S, Battaglia F, Walter S,
Mathews PM and Arancio O: Progressive age-related development of
alzheimer-like pathology in app/ps1 mice. Ann Neurol. 55:801–814.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Janus C, Flores AY, Xu G and Borchelt DR:
Behavioral abnormalities in app swe/ps1de9 mouse model of ad-like
pathology: Comparative analysis across multiple behavioral domains.
Neurobiol Aging. 36:2519–2532. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Balducci C, Mancini S, Minniti S, La
Vitola P, Zotti M, Sancini G, Mauri M, Cagnotto A, Colombo L,
Fiordaliso F, et al: Multifunctional liposomes reduce brain
β-amyloid burden and ameliorate memory impairment in Alzheimer's
disease mouse models. J Neurosci. 34:14022–14031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Caccamo A, Branca C, Talboom JS, Shaw DM,
Turner D, Ma L, Messina A, Huang Z, Wu J and Oddo S: Reducing
ribosomal protein S6 Kinase 1 expression improves spatial memory
and synaptic plasticity in a mouse model of Alzheimer's disease. J
Neurosci. 35:14042–14056. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SY, Cohen BM, Chen X, Lukas SE, Shinn
AK, Yuksel AC, Li T, Du F and Öngür D: Redox Dysregulation in
schizophrenia revealed by in vivo NAD+/NADHH measurement. Schizophr
Bull. 43:197–204. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Blass JP, Sheu RK and Gibson GE: Inherent
abnormalities in energy metabolism in Alzheimer disease.
Interaction with cerebrovascular compromise. Ann N Y Acad Sci.
903:204–221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lewczuk P, Riederer P, O'Bryant SE,
Verbeek MM, Dubois B, Visser PJ, Jellinger KA, Engelborghs S,
Ramirez A, Parnetti L, et al: Cerebrospinal fluid and blood
biomarkers for neurodegenerative dementias: An update of the
consensus of the task force on biological markers in psychiatry of
the world federation of societies of biological psychiatry. World J
Biol Psychiatry. 19:244–328. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pérez MJ, Ponce DP, Aranguiz A, Behrens MI
and Quintanilla RA: Mitochondrial permeability transition pore
contributes to mitochondrial dysfunction in fibroblasts of patients
with sporadic Alzheimer's disease. Redox Biol. 19:290–300. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Swerdlow RH, Koppel S, Weidling I, Hayley
C, Ji Y and Wilkins HM: Mitochondria, Cybrids, Aging, and
Alzheimer's disease. Prog Mol Biol Transl Sci. 146:259–302. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adiele RC and Adiele CA: Mitochondrial
regulatory pathways in the pathogenesis of Alzheimer's disease. J
Alzheimers Dis. 53:1257–1270. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Velliquette RA, O'Connor T and Vassar R:
Energy inhibition elevates beta-secretase levels and activity and
is potentially amyloidogenic in APP transgenic mice: Possible early
events in Alzheimer's disease pathogenesis. J Neurosci.
25:10874–10883. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu Z, Zhu Y, Cao X, Sun S and Zhao B:
Mitochondrial toxic effects of Aβ through mitofusins in the early
pathogenesis of Alzheimer's disease. Mol Neurobiol. 50:986–996.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lauretti E, Li JG, Di Meco A and Pratico
D: Glucose deficit triggers tau pathology and synaptic dysfunction
in a tauopathy mouse model. Transl Psychiatry. 7:e10202017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fang EF: Mitophagy and NAD+
inhibit Alzheimer disease. Autophagy. 15:1112–1114. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ghosh D, Levault KR and Brewer GJ:
Relative importance of redox buffers GSH and NAD+(P)H in
age-related neurodegeneration and Alzheimer disease-like mouse
neurons. Aging Cell. 13:631–640. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang Y and Sauve AA: NAD(+) metabolism:
Bioenergetics, signaling and manipulation for therapy. Biochim
Biophys Acta. 1864:1787–1800. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pehar M, Harlan BA, Killoy KM and Vargas
MR: Nicotinamide adenine dinucleotide metabolism and
Neurodegeneration. Antioxid Redox Signal. 18:1652–1668. 2018.
View Article : Google Scholar
|
|
34
|
Stein LR and Imai S: The dynamic
regulation of NAD metabolism in mitochondria. Trends Endocrinol
Metab. 23:420–428. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Long AN, Owens K, Schlappal AE, Kristian
T, Fishman PS and Schuh RA: Effect of nicotinamide mononucleotide
on brain mitochondrial respiratory deficits in an Alzheimer's
disease-relevant murine model. BMC Neurol. 15:192015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yao Z, Yang W, Gao Z and Jia P:
Nicotinamide mononucleotide inhibits JNK activation to reverse
Alzheimer disease. Neurosci Lett. 647:133–140. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang X, Zhang Q, Bao R, Zhang N, Wang Y,
Polo-Parada L, Tarim A, Alemifar A, Han X, Wilkins HM, et al:
Deletion of NAMPT in projection neurons of adult mice leads to
motor dysfunction, Neurodegeneration, and death. Cell Rep.
20:2184–2200. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Katsyuba E and Auwerx J: Modulating
NAD+ metabolism, from bench to bedside. EMBO J.
36:2670–2683. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nikiforov A, Kulikova V and Ziegler M: The
human NAD metabolome: Functions, metabolism and
compartmentalization. Crit Rev Biochem Mol Biol. 50:284–297. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ryu KW, Nandu T, Kim J, Challa S,
DeBerardinis RJ and Kraus WL: Metabolic regulation of transcription
through compartmentalized NAD+ biosynthesis. Science.
360(pii): eaan57802018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhu XH, Lu M, Lee BY, Ugurbil K and Chen
W: In vivo NAD assay reveals the intracellular NAD contents and
redox state in healthy human brain and their age dependences. Proc
Natl Acad Sci USA. 112:2876–2881. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Revollo JR, Grimm AA and Imai S: The
regulation of nicotinamide adenine dinucleotide biosynthesis by
Nampt/PBEF/visfatin in mammals. Curr Opin Gastroenterol.
23:164–170. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lin JB, Kubota S, Ban N, Yoshida M,
Santeford A, Sene A, Nakamura R, Zapata N, Kubota M, Tsubota K, et
al: NAMPT-Mediated NAD(+) Biosynthesis is essential for vision in
mice. Cell Rep. 17:69–85. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yamamoto T, Byun J, Zhai P, Ikeda Y, Oka S
and Sadoshima J: Nicotinamide mononucleotide, an intermediate of
NAD+ synthesis, protects the heart from ischemia and reperfusion.
PLoS One. 9:e989722014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
LoCoco PM, Risinger AL, Smith HR, Chavera
TS, Berg KA and Clarke WP: Pharmacological augmentation of
nicotinamide phosphoribosyltransferase (NAMPT) protects against
paclitaxel-induced peripheral neuropathy. Elife. 6:e296262017.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ghosh D, LeVault KR, Barnett AJ and Brewer
GJ: A reversible early oxidized redox state that precedes
macromolecular ROS damage in aging nontransgenic and 3×Tg-AD mouse
neurons. J Neurosci. 32:5821–5832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Sasaki Y, Araki T and Milbrandt J:
Stimulation of nicotinamide adenine dinucleotide biosynthetic
pathways delays axonal degeneration after axotomy. J Neurosci.
26:8484–8491. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen J, Sysol JR, Singla S, Zhao S,
Yamamura A, Valdez-Jasso D, Abbasi T, Shioura KM, Sahni S, Reddy V,
et al: Nicotinamide Phosphoribosyltransferase promotes pulmonary
vascular remodeling and is a therapeutic target in pulmonary
arterial hypertension. Circulation. 135:1532–1546. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang SN and Miao CY: Targeting NAMPT as a
therapeutic strategy against stroke. Stroke Vasc Neurol. 4:83–89.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Imai S and Yoshino J: The importance of
NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and aging.
Diabetes Obes Metab. 15 (Suppl 3):S26–S33. 2013. View Article : Google Scholar
|
|
51
|
Stein LR and Imai S: Specific ablation of
Nampt in adult neural stem cells recapitulates their functional
defects during aging. EMBO J. 33:1321–1340. 2014.PubMed/NCBI
|
|
52
|
Kiss G, Konrad C, Pour-Ghaz I, Mansour JJ,
Németh B, Starkov AA, Adam-Vizi V and Chinopoulos C: Mitochondrial
diaphorases as NAD+ donors to segments of the cirtric acid cycle
that support substrate-level phosphorylation yielding ATP during
respiratory inhibition. FASEB J. 28:1682–1697. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hou Y, Lautrup S, Cordonnier S, Wang Y,
Croteau DL, Zavala E, Zhang Y, Moritoh K, O'Connell JF, Baptiste
BA, et al: NAD+ supplementation normalizes key Alzheimer's features
and DNA damage responses in a new AD mouse model with introduced
DNA repair deficiency. Proc Natl Acad Sci USA. 115:1876–1885. 2018.
View Article : Google Scholar
|
|
54
|
Wang X, Li H and Ding S: The effects of
NAD+ on apoptotic neuronal death and mitochondrial biogenesis and
function after glutamate Excitotoxicity. Int J Mol. 15:20449–20468.
2014. View Article : Google Scholar
|
|
55
|
Guan Y, Wang SR, Huang XZ, Xie QH, Xu YY,
Shang D and Han CM: Nicotinamide Mononucleotide, an NAD+ Precursor,
Rescues Age-associated susceptibility to AKI in a sirtuin
1-dependent manner. J Am Soc Nephrol. 28:2337–2352. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Du LL, Xie JZ, Cheng XS, Li XH, Kong FL,
Jiang X, Ma ZW, Wang JZ, Chen C and Zhou XW: Activation of sirtuin
1 attenuates cerebral ventricular streptozotocin-induced tau
hyperphosphorylation and cognitive injuries in rat hippocampi. Age
(Dordr). 36:613–623. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bonfili L, Cecarini V, Cuccioloni M,
Angeletti M, Berardi S, Scarpona S, Rossi G and Eleuteri AM: SLAB51
probiotic formulation activates SIRT1 pathway promoting antioxidant
and neuroprotective effects in an AD mouse model. Mol Neurobiol.
55:7987–8000. 2018. View Article : Google Scholar : PubMed/NCBI
|