Introduction
Angiogenesis, the formation of new blood vessels
sprouting from the pre-existing vasculature, is pivotal for the
growth of solid tumors and also plays a critical role in invasion
and metastasis. The role of vascular endothelial growth factor
(VEGF) in the regulation of angiogenesis has been under
investigation for over a decade (1). The VEGF family includes VEGF-A,
VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF) in
mammals (2,3), as well as two exogenous VEGF
subtypes, VEGF-E (virus genome-encoded VEGF) and VEGF-F (snake
venom-derived VEGF) (4,5). These VEGF molecules act through
binding to several high-affinity transmembrane endothelial cell
receptors, including Flt-1 (VEGFR1), KDR (VEGFR2) and Flt-4
(VEGFR3) with varying specificities. VEGF-A binds Flt-1 and KDR,
while PlGF-1 and vammin (one of VEGF-Fs) selectively bind Flt-1 and
KDR, respectively (2,3,6).
Binding of VEGF to these receptors leads to intracellular receptor
phosphorylation which initiates various intracellular downstream
receptor pathways, leading to endothelial cell proliferation and
blood vessel formation. Tumors establish their microvasculature in
part by secreting elevated amounts of VEGF. Increased levels of
VEGF mRNA are found in hypoxic areas of many solid tumors (7,8).
Tumor suppressors, p53 (9–12), p16 (13) and the von Hippel-Lindau gene
(14,15) have been shown to play a role in
VEGF downregulation.
In several types of cancer including bladder
(16), renal cell (17), head and neck (18), colorectal (19,20),
cervical (21), ovarian (22), lung (23) and breast cancer (24,25),
increased VEGF expression, either in the circulation or in tumor
tissue, has been found to be correlated with decreased disease-free
and shorter relapse-free intervals.
Four and a half LIM domains 1 (FHL1) belongs to a
family of LIM-only proteins, containing an N-terminal half LIM
domain, followed by four complete LIM domains. FHL1 regulates gene
transcription, cell proliferation, differentiation and apoptosis
(26). It has been reported that
FHL1 inhibited VEGF promoter activity induced by HIF-1α
overexpression (27,28) and that Smad4/deleted in pancreatic
carcinoma locus 4 (DPC4) restoration affected angiogenesis,
decreasing VEGF expression (29).
In a previous study, we showed that FHL physically interacted with
Smad4 as determined by co-immunoprecipitation (30). Furthermore, FHL1-Smad4 interaction
has been shown to affect TGF-β-responsive gene transcription
(30). Similarly, coexpression of
FHL2 with Smad4 synergistically repressed estrogen reporter
activity (31). These findings
suggest that the interaction of FHL1 with Smad4 is required for the
synergistic inhibition of VEGF signaling. The aim of this study was
to determine whether FHL1 and Smad4 inhibited VEGF signaling.
Materials and methods
Plasmids
The reporter construct VEGF promoter-containing
luciferase reporter (VEGF-Luc) and expression plasmids for FHL1
(32), Smad4 (33), FHL1 siRNA (32) and Smad4 siRNA (31) have been previously described.
Luciferase reporter assay
HEK293T embryonic kidney and HepG2 hepatoma cells
were routinely cultured in Dulbecco’s modified Eagle’s medium
(DMEM) (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine
serum (FBS) at 37°C in a humidified atmosphere of 5% CO2
in air. Cells were transfected using Lipofectamine™ 2000
(Invitrogen) with 0.2 μg of VEGF-Luc reporter plasmid, 0.1 μg of
β-galactosidase reporter, and 1 μg of expression vectors for FHL1
or Smad4, or small-interfering RNA (siRNA) vectors targeting FHL1
or Smad4. The respective empty vector was used to adjust the total
amount of DNA. Luciferase and β-galactosidase activities were
determined as previously described (34).
Quantitative real-time reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was prepared using TRIzol reagent
(Invitrogen) and reverse transcribed using SuperScript II Reverse
Transcriptase (Invitrogen). Real-time PCR was carried out with
VEGF- and GAPDH-specific primers. The sense primer for VEGF was
5′-TTCTGGGCT GTTCTCGCTTCG-3′ and the antisense primer was 5′-CCC
CTCTCCTCTTCCTTCTCT-3′. The sense primer for GAPDH was
5′-ACCACAGTCCATGCCATCAC-3′ and the antisense primer was
5′-TCCACCACCCTGTTGCTG TA-3′. The fold change in VEGF expression was
determined using the 2-ΔΔCt method, with GAPDH as an internal
control.
Enzyme-linked immunosorbent assay
(ELISA)
Cells were transiently transfected with expression
vectors for FHL1 or Smad4, or siRNA vectors targeting FHL1 or
Smad4. At 48 h post-transfection, media were harvested for VEGF
secretion assay. Human VEGF-A protein concentrations were assessed
by ELISA analysis according to the manufacturer’s protocol (R&D
Systems, Minneapolis, MN, USA). The values obtained were normalized
to the total protein concentration in the total cell extracts
prepared from each dish.
Statistical analysis
Statistical analysis was performed by using SPSS
version 11.0 software. Statistical significance in the luciferase
activity assays between two groups of data was determined using the
unpaired t-test. Data were presented as the means ± SD. P<0.05
was considered to indicate a statistically significant
difference.
Results
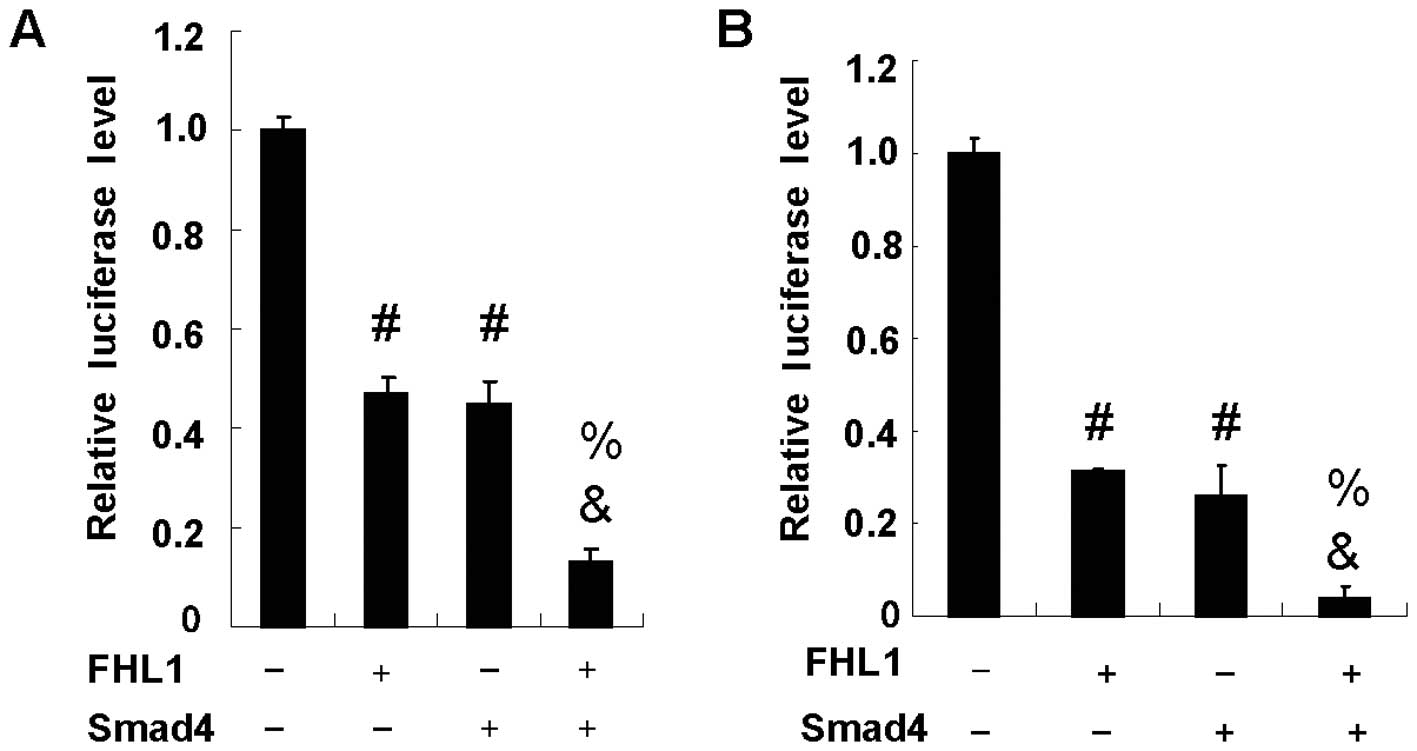
FHL1 and Smad4 synergistically inhibit
VEGF promoter activity
FHL1 has been shown to repress VEGF promoter
activity in hepatocellular carcinoma cells (27,28).
Similar to FHL1, Smad4 has also been demonstrated to inhibit VEGF
expression in pancreatic carcinoma cells. To determine whether FHL1
and Smad4 have a synergistic effect on VEGF promoter activity,
hepatocellular carcinoma HepG2 cells were cotransfected with the
VEGF-Luc reporter, Smad4 and FHL1. As previously reported (27,28),
FHL1 overexpression significantly reduced VEGF-Luc reporter
activity in HepG2 cells. Similar to FHL1, Smad4 overexpression also
inhibited the reporter activity. Notably, the coexpression of FHL1
with Smad4 synergistically inhibited the reporter activity due to
the fact that coexpression of FHL1 with Smad4 achieved levels of
inhibition greater than the sum of each individual gene expression
(Fig. 1A). Similar results were
observed in HEK293T cells (Fig.
1B).
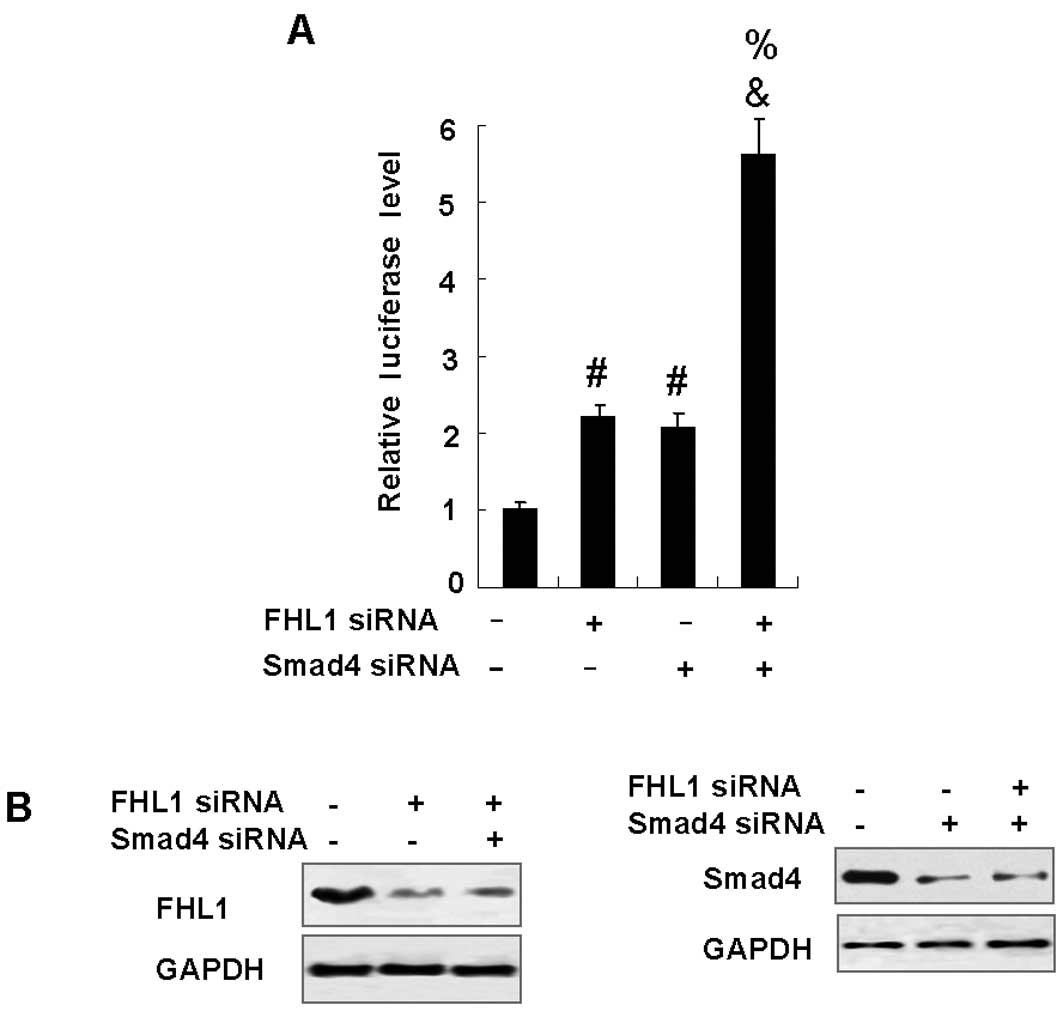
Knockdown of FHL1 and Smad4
synergistically enhances VEGF promoter activity
To examine the role of endogenous FHL1 and Smad4 in
the repression of VEGF promoter activity, HepG2 cells were
transfected with FHL1 siRNA and Smad4 siRNA. As shown in Fig. 2B, FHL1 siRNA effectively inhibited
the expression of FHL1 protein at 48 h following transfection,
whereas control siRNA had no effect. Similar results were observed
when Smad4 siRNA was used. As expected, reduction of FHL1 or Smad4
enhanced the VEGF-Luc reporter activity. Similar to the results
that overexpression of FHL1 and Smad4 synergistically inhibited
VEGF promoter activity, knockdown of endogenous FHL1 and Smad4
synergistically enhanced VEGF promoter activity (Fig. 2A).
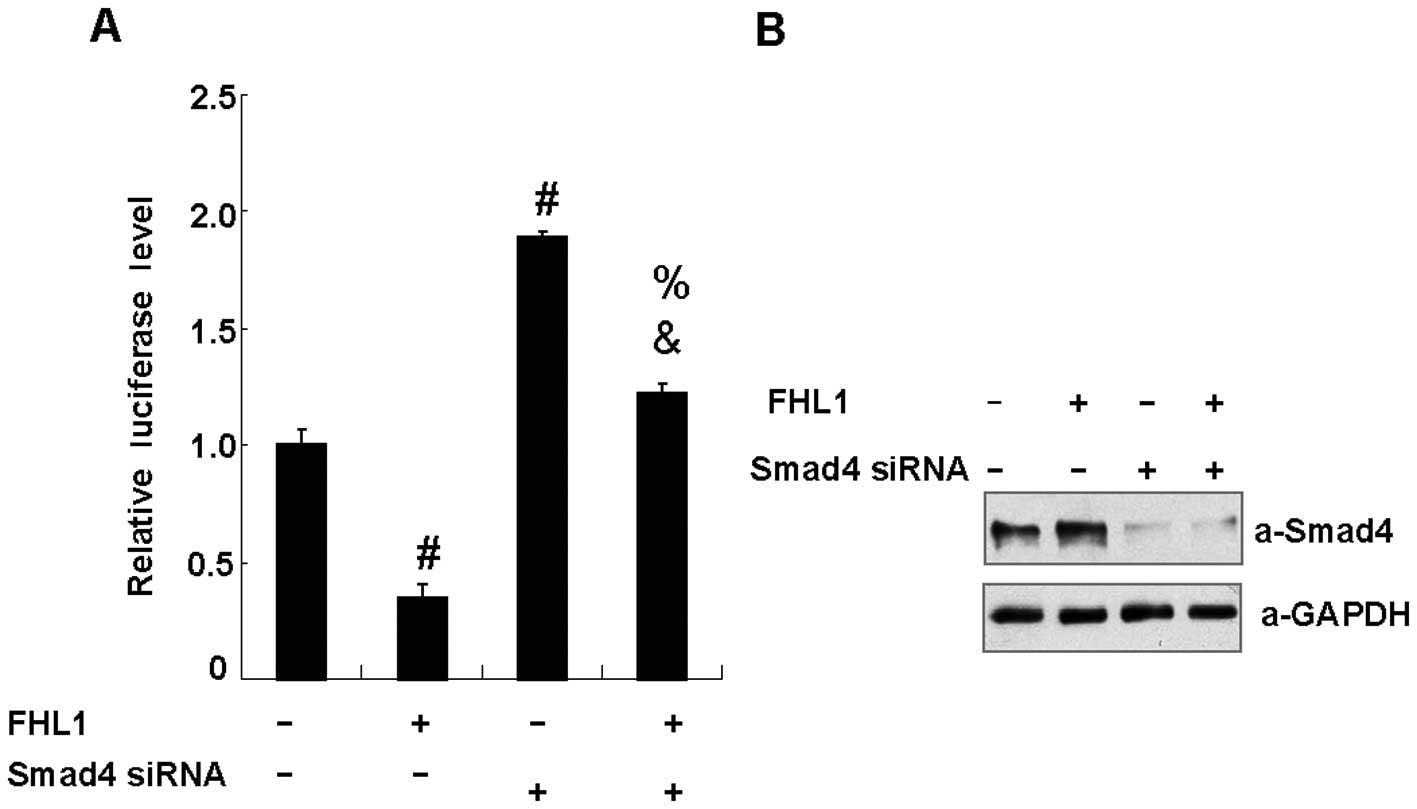
Abrogation of FHL1 inhibition of VEGF
promoter activity by reducing endogenous Smad4
To examine the role of endogenous Smad4 in FHL1
repression of VEGF promoter activity, HepG2 cells were transfected
with FHL1 or Smad4 siRNA. As shown in Fig. 3B, Smad4 siRNA effectively inhibited
the expression of Smad4 protein 48 h after transfection, whereas
control siRNA had no effect. Reduction of Smad4 enhanced the
VEGF-Luc reporter activity. Notably, reduction of Smad4 eliminated
FHL1 inhibition of the reporter activity, suggesting that FHL1
represses VEGF promoter activity in a Smad4-dependent manner
(Fig. 3A).
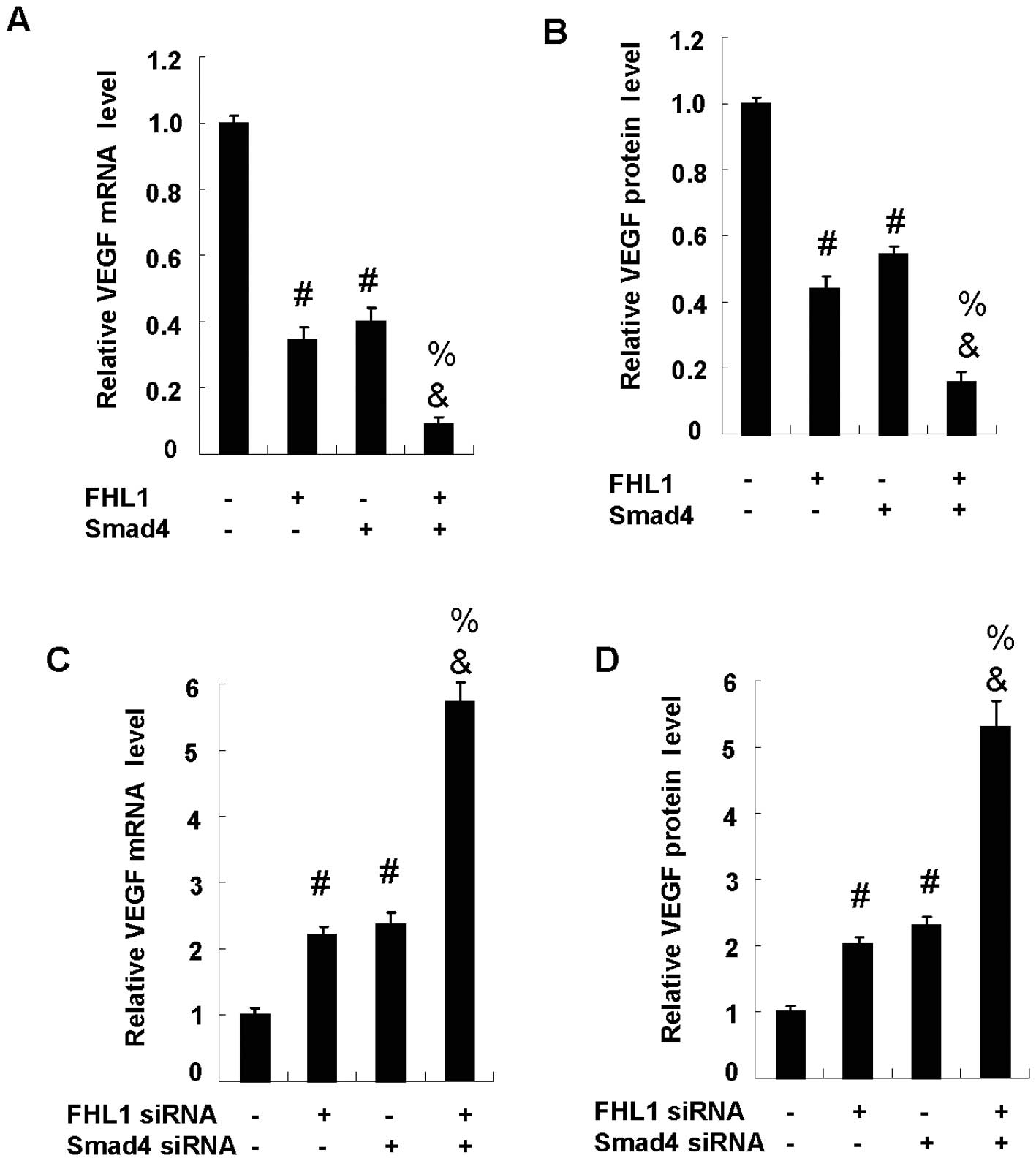
FHL1 and Smad4 synergistically inhibit
VEGF expression
To determine the effect of FHL1 and Smad4 on VEGF
expression, HepG2 cells were cotransfected with FHL1 and Smad4, or
with FHL1 siRNA and Smad4 siRNA. Overexpression of FHL1
significantly reduced VEGF mRNA expression. As with FHL1, Smad4
overexpression also inhibited the mRNA expression. Notably, the
coexpression of FHL1 with Smad4 synergistically inhibited the mRNA
expression due to the fact that coexpression of FHL1 with Smad4
achieved levels of inhibition greater than the sum of each
individual gene expression (Fig.
4A). Similar results were observed in the VEGF protein
expression (Fig. 4B). As expected,
the reduction of Smad4 or FHL1 enhanced VEGF expression at both
mRNA (Fig. 4C) and protein levels
(Fig. 4D). Of note, the reduction
of FHL1 and Smad4 synergistically enhanced the mRNA and protein
expression.
Discussion
In this study, we have demonstrated for the first
time that FHL1 and Smad4 synergistically inhibit VEGF signaling,
which has been validated by a number of in vitro
experiments, including luciferase reporter, real-time reverse
transcription-PCR and ELISA assays.
FHL1 was reported to play important roles in
skeletal and cardiac muscle growth. FHL1 is also involved in
several types of human cancer, such as breast, liver, kidney,
prostate, bladder and gastric cancer (32,35).
Recently, FHL1 proteins have been shown to regulate the activity of
transcription factors including Smad4 and HIF1 (27). FHL1 has been reported to interact
with HIF1α, and inhibit HIF1α-induced VEGF promoter activity and
VEGF expression by blockade of HIF1α-HIF1β heterodimerization
(28) and HIF-1α binding to the
general co-activators p300/CBP (27).
Smad4, initially identified as a candidate tumor
suppressor gene, is functionally inactivated at high frequency in
pancreatic carcinomas and metastatic colon carcinoma (29,36).
Smad4 belongs to the evolutionarily conserved family of Smad
proteins that are crucial for transmitting transforming growth
factor-β (TGF-β) superfamily signals from the cell surface to the
nucleus. These proteins regulate proliferation, differentiation and
cell death. Smad pathway impairment may contribute to
carcinogenesis by the promotion of cell proliferation, and Smad4
inactivation in tumor patients has been closely associated with
poor prognosis (37). The in
vitro and in vivo experiments showed that Smad4
decreased the expression of VEGF and increased the levels of the
angiogenesis inhibitor thrombospondin-1, causing human pancreatic
carcinoma cells to switch from potently angiogenic to
antiangiogenic (29). In a
previous study, we showed that FHL1 physically and functionally
interacts with Smad2, Smad3 and Smad4, and suppresses human
hepatoma cell growth (30). In
this study, FHL1 and Smad4 were shown to synergistically inhibit
VEGF promoter reporter activity and VEGF mRNA expression and
secretion levels, suggesting that FHL1-Smad4 interaction
contributes to VEGF signaling-related diseases. The reduction of
Smad4 eliminated the FHL1 and Smad4-mediated synergistic inhibition
of the VEGF-Luc reporter activity, indicating that the inhibition
of FHL1 on VEGF promoter activity occurred in a Smad4-dependent
manner. However, the mechanism by which FHL1 and Smad4
synergistically control VEGF promoter activity and expression has
yet to be fully elucidated. Considering that VEGF promoter harbors
several potential Smad-binding sequences (29) and that FHL1 interacts with Smad4
physically and functionally, it could be concluded that FHL1 acts
as a co-repressor for Smad4 in the regulation of VEGF
expression.
Besides recruiting co-factors, Smad regulate
transcriptional responses through physical and functional
interaction with different transcription factors. Papageorgis et
al(36) reported that there
was physical interaction between Smad4 and HIF1α, which are
important transcriptional factors regulating VEGF expression,
providing a molecular basis for the differential regulation of
target genes. Furthermore, Sánchez-Elsner et al(38) identified that HIF1 and Smad-binding
motifs were within fragment -1006/-954 of the human VEGF promoter.
Based on these findings, it is suggested that Smad4, HIF1 and their
co-repressor FHL1 may form a complex on the promoter of VEGF and
function as transcriptional co-modulators in the regulation of VEGF
promoter activity and expression, and that FHL1 and Smad4 may
negatively regulate HIF1α-induced VEGF expression
synergistically.
Therefore, in this study, the cooperative
transcriptional regulation of VEGF signaling by FHL1 and Smad4 was
demonstrated, which might provide a novel therapeutic intervention
target for VEGF signaling-related diseases.
Acknowledgements
This study was supported by the National Natural
Science Foundation (81071954).
References
|
1
|
Ferrara N: VEGF and the quest for tumour
angiogenesis factors. Nat Rev Cancer. 2:795–803. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrara N, Gerber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meyer M, Clauss M, Lepple-Wienhues A, et
al: A novel vascular endothelial growth factor encoded by Orf
virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2
(KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J.
18:363–374. 1999. View Article : Google Scholar
|
|
5
|
Yamazaki Y and Morita T: Molecular and
functional diversity of vascular endothelial growth factors. Mol
Divers. 10:515–527. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamazaki Y, Takani K, Atoda H and Morita
T: Snake venom vascular endothelial growth factors (VEGFs) exhibit
potent activity through their specific recognition of KDR (VEGF
receptor 2). J Biol Chem. 278:51985–51988. 2003. View Article : Google Scholar
|
|
7
|
Plate KH, Breier G, Weich HA and Risau W:
Vascular endothelial growth factor is a potential tumour
angiogenesis factor in human gliomas in vivo. Nature. 359:845–848.
1992. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shweiki D, Itin A, Soffer D and Keshet E:
Vascular endothelial growth factor induced by hypoxia may mediate
hypoxia-initiated angiogenesis. Nature. 359:843–845. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dameron KM, Volpert OV, Tainsky MA and
Bouck N: Control of angiogenesis in fibroblasts by p53 regulation
of thrombospondin-1. Science. 265:1582–1584. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kieser A, Weich HA, Brandner G, Marme D
and Kolch W: Mutant p53 potentiates protein kinase C induction of
vascular endothelial growth factor expression. Oncogene. 9:963–969.
1994.PubMed/NCBI
|
|
11
|
Mukhopadhyay D, Tsiokas L and Sukhatme VP:
Wild-type p53 and v-Src exert opposing influences on human vascular
endothelial growth factor gene expression. Cancer Res.
55:6161–6165. 1995.PubMed/NCBI
|
|
12
|
Volpert OV, Dameron KM and Bouck N:
Sequential development of an angiogenic phenotype by human
fibroblasts progressing to tumorigenicity. Oncogene. 14:1495–1502.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Harada H, Nakagawa K, Iwata S, et al:
Restoration of wild-type p16 down-regulates vascular endothelial
growth factor expression and inhibits angiogenesis in human
gliomas. Cancer Res. 59:3783–3789. 1999.PubMed/NCBI
|
|
14
|
Siemeister G, Weindel K, Mohrs K, Barleon
B, Martiny-Baron G and Marme D: Reversion of deregulated expression
of vascular endothelial growth factor in human renal carcinoma
cells by von Hippel-Lindau tumor suppressor protein. Cancer Res.
56:2299–2301. 1996.
|
|
15
|
Mukhopadhyay D, Knebelmann B, Cohen HT,
Ananth S and Sukhatme VP: The von Hippel-Lindau tumor suppressor
gene product interacts with Sp1 to repress vascular endothelial
growth factor promoter activity. Mol Cell Biol. 17:5629–5639.
1997.PubMed/NCBI
|
|
16
|
Inoue K, Slaton JW, Karashima T, et al:
The prognostic value of angiogenesis factor expression for
predicting recurrence and metastasis of bladder cancer after
neoadjuvant chemotherapy and radical cystectomy. Clin Cancer Res.
6:4866–4873. 2000.
|
|
17
|
Jacobsen J, Rasmuson T, Grankvist K and
Ljungberg B: Vascular endothelial growth factor as prognostic
factor in renal cell carcinoma. J Urol. 163:343–347. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eisma RJ, Spiro JD and Kreutzer DL:
Vascular endothelial growth factor expression in head and neck
squamous cell carcinoma. Am J Surg. 174:513–517. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wong MP, Cheung N, Yuen ST, Leung SY and
Chung LP: Vascular endothelial growth factor is up-regulated in the
early pre-malignant stage of colorectal tumour progression. Int J
Cancer. 81:845–850. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Amaya H, Tanigawa N, Lu C, et al:
Association of vascular endothelial growth factor expression with
tumor angiogenesis, survival and thymidine
phosphorylase/platelet-derived endothelial cell growth factor
expression in human colorectal cancer. Cancer Lett. 119:227–235.
1997. View Article : Google Scholar
|
|
21
|
Guidi AJ, Abu-Jawdeh G, Berse B, et al:
Vascular permeability factor (vascular endothelial growth factor)
expression and angiogenesis in cervical neoplasia. J Natl Cancer
Inst. 87:1237–1245. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boocock CA, Charnock-Jones DS, Sharkey AM,
et al: Expression of vascular endothelial growth factor and its
receptors flt and KDR in ovarian carcinoma. J Natl Cancer Inst.
87:506–516. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
O’Byrne KJ, Koukourakis MI, Giatromanolaki
A, et al: Vascular endothelial growth factor, platelet-derived
endothelial cell growth factor and angiogenesis in non-small-cell
lung cancer. Br J Cancer. 82:1427–1432. 2000.
|
|
24
|
Brown LF, Berse B, Jackman RW, et al:
Expression of vascular permeability factor (vascular endothelial
growth factor) and its receptors in breast cancer. Hum Pathol.
26:86–91. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yoshiji H, Gomez DE, Shibuya M and
Thorgeirsson UP: Expression of vascular endothelial growth factor,
its receptor, and other angiogenic factors in human breast cancer.
Cancer Res. 56:2013–2016. 1996.PubMed/NCBI
|
|
26
|
Kadrmas JL and Beckerle MC: The LIM
domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell
Biol. 5:920–931. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hubbi ME, Gilkes DM, Baek JH and Semenza
GL: Four-and-a-half LIM domain proteins inhibit transactivation by
hypoxia-inducible factor 1. J Biol Chem. 287:6139–6149. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin J, Qin X, Zhu Z, Mu J, et al: FHL
family members suppress vascular endothelial growth factor
expression through blockade of dimerization of HIF1α and HIF1β.
IUBMB Life. 64:921–930. 2012.PubMed/NCBI
|
|
29
|
Schwarte-Waldhoff I, Volpert OV, Bouck NP,
et al: Smad4/DPC4-mediated tumor suppression through suppression of
angiogenesis. Proc Natl Acad Sci USA. 97:9624–9629. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding L, Wang Z, Yan J, et al: Human
four-and-a-half LIM family members suppress tumor cell growth
through a TGF-beta-like signaling pathway. J Clin Invest.
119:349–361. 2009.PubMed/NCBI
|
|
31
|
Xiong Z, Ding L, Sun J, et al: Synergistic
repression of estrogen receptor transcriptional activity by FHL2
and Smad4 in breast cancer cells. IUBMB Life. 62:669–676. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin J, Ding L, Jin R, et al: Four and a
half LIM domains 1 (FHL1) and receptor interacting protein of
140kDa (RIP140) interact and cooperate in estrogen signaling. Int J
Biochem Cell Biol. 41:1613–1618. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Sun Y, Ding L, Zhang H, et al:
Potentiation of Smad-mediated transcriptional activation by the
RNA-binding protein RBPMS. Nucleic Acids Res. 34:6314–6326. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ding L, Yan J, Zhu J, et al:
Ligand-independent activation of estrogen receptor alpha by XBP-1.
Nucleic Acids Res. 31:5266–5274. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsumoto M, Kawakami K, Enokida H, et al:
CpG hypermethylation of human four-and-a-half LIM domains 1
contributes to migration and invasion activity of human bladder
cancer. Int J Mol Med. 26:241–247. 2010.
|
|
36
|
Papageorgis P, Cheng K, Ozturk S, et al:
Smad4 inactivation promotes malignancy and drug resistance of colon
cancer. Cancer Res. 71:998–1008. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yatsuoka T, Sunamura M, Furukawa T, et al:
Association of poor prognosis with loss of 12q, 17p, and 18q, and
concordant loss of 6q/17p and 12q/18q in human pancreatic ductal
adenocarcinoma. Am J Gastroenterol. 95:2080–2085. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sánchez-Elsner T, Botella LM, Velasco B,
et al: Synergistic cooperation between hypoxia and transforming
growth factor-beta pathways on human vascular endothelial growth
factor gene expression. J Biol Chem. 276:38527–38535.
2001.PubMed/NCBI
|