Introduction
Doxorubicin (DOX) is widely used in the treatment of
numerous types of human solid and hematological malignancies,
including acute leukemia, lymphoma, Kaposi’s sarcoma and bone
tumors, as well as stomach, breast and ovarian cancer (1). However, the clinical use of DOX is
limited by severe side effects, including cardiotoxicity, which
leads to heart failure (2–4). The cause of DOX-induced
cardiotoxicity is multifactorial; however, cardiac inflammation and
the generation of oxidative stress are known to participate in this
clinical event. DOX has been shown to induce a significant increase
in the levels of inflammatory markers, including interleukin
(IL)-6, tumor necrosis factor-α (TNF-α) (5–8) and
cyclooxygenase-2 (COX-2) (9). In a
murine model of DOX-induced heart failure, an inhibitor of COX-2
was able to improve left ventricular function and mortality
(10), suggesting the involvement
of COX-2 in DOX-induced cardiotoxicity.
Nuclear factor-κB (NF-κB) may also be a key
contributor to DOX-induced cardiotoxicity. NF-κB is a positive
regulator of COX-2 expression in response to various cytokines and
growth factors (11,12). The NF-κB family is composed of five
proteins, Rel A (p65), Rel B, c-Rel, NF-κB1 (p50) and NF-κB2 (p52),
each of which may form homo- or heterodimers. NF-κB is a dimeric
transcription factor that regulates genes associated with stress
responses, including inflammation, oxidative stress and apoptosis.
DOX has been shown to induce NF-κB (13–15).
We have recently demonstrated that the inhibition of NF-κB
attenuates the cytotoxicity and levels of IL-6 and IL-8, as well as
the overexpression of COX-2 in chemical hypoxia-treated HaCaT cells
(16,17). These findings indicate the
modulatory effect of NF-κB on inflammatory factors. However, it is
unclear whether there is an association between NF-κB and
inflammatory factors in DOX-induced cardiotoxicity.
The role of p38 mitogen-activated protein kinase
(MAPK) in DOX-induced cardiotoxicity has been examined in several
studies (18–21). p38 MAPK is a subfamily of the MAPK
superfamily. This subfamily is composed of four isoforms, p38α,
p38β, p38γ and p38δ (22,23), and is important in the inflammatory
stress response and cell differentiation (24,25).
Kang et al(18) have shown
that the activation of p38 MAPK is implicated in DOX-induced
apoptosis. DOX is able to activate p38α and p38β, which contribute
to DOX-induced cardiomyocyte apoptosis by degradation of the
transcriptional co-factor p300 (21). Our recent study has indicated that
the activation of p38 MAPK is capable of enhancing the generation
of reactive oxygen species (ROS) (26) and mediating chemical
hypoxia-induced inflammation (data not shown), strongly indicating
that p38 MAPK activation may contribute to the DOX-induced
inflammatory response. The present study aimed to investigate the
molecular mechanisms underlying DOX-induced inflammation in order
to clarify the association between p38 MAPK and NF-κB and the roles
of these two pathways in the induction of inflammatory factors,
including IL-1β, IL-6 and TNF-α by DOX. The findings of the present
study demonstrated that the p38 MAPK/NF-κB pathway is critical in
the induction of the inflammatory response in DOX-treated H9c2
cardiac cells.
Materials and methods
Materials
DOX, SB203580 and pyrrolidine dithiocarbamate (PDTC)
were purchased from Sigma-Aldrich (St. Louis, MO, USA). The Cell
Counter kit-8 (CCK-8) was purchased from Dojindo Laboratories
(Kumamoto, Japan). DMEM-F12 medium and fetal bovine serum (FBS)
were purchased from Gibco-BRL (Carlsbad, CA, USA).
Cell culture and treatment
H9c2 embryonic rat cardiac cells (Sun Yat-sen
University Experimental Animal Center, Guangzhou, China) were
cultured in DMEM-F12 medium supplemented with 10% FBS at 37°C in an
atmosphere of 5% CO2. To examine the effects of PDTC and
SB203580 on DOX-induced injury, H9c2 cells were pretreated with
PDTC (a selective inhibitor of NF-κB) for 30 min or SB203580 for 60
min prior to treatment with DOX.
Cell viability assay
After the H9c2 cells were cultured in 96-well plates
and administered with the indicated treatments, 10 μl CCK-8
solution was added to each well at a 1/10 dilution, followed by a
2-h incubation. The absorbance was measured at 450 nm with a
microplate reader (Multiskan MK3 Microplate reader; Thermo Fisher
Scientific Inc., Waltham, MA, USA). The mean optical density (OD)
of five wells in the indicated groups was used to calculate the
percentage of cell viability according to the following formula:
Cell viability (%) = OD treatment group/OD control group × 100. All
the experiments were performed in triplicate.
Measurement of inflammatory cytokine
levels using ELISA
The H9c2 cells were plated in 96-well plates.
Following the administration of the indicated treatments, the
relative content of each secreted inflammatory cytokine (IL-1β,
IL-6 and TNF-α) in the supernatant was measured using the Cytokine
ELISA kit (Boster BioTech, Wuhan, China) according to the
manufacturer’s instructions. The plates were read at a wavelength
of 450 nm using a microplate reader (Multiskan MK3 Microplate
reader; Thermo Fisher Scientific Inc.). The relative content of
inflammatory cytokines in the culture medium was corrected by cell
viability. All the experiments were performed in triplicate.
Western blot assay
Following the administration of the indicated
treatments, the H9c2 cells were harvested and lysed, and the
homogenate was centrifuged. After the total protein in the
supernatant was quantified using the BCA protein assay kit (Thermo
Fisher Scientific Inc., Rockford, IL, USA), the protein (30 μg from
each sample) was fractionated by 12% SDS-PAGE and then transferred
onto a polyvinylidene difluoride (PVDF) membrane. The membrane was
blocked with 5% free-fat milk in TBS-T for 1 h at room temperature,
and then incubated with monoclona rabbit primary antibodies
specific to p38 MAPK (#2371; Cell Signaling Technology Inc.,
Beverly, MA, USA) and phosphorylated (p)-p38 MAPK (#4631; Cell
Signaling Technology Inc.) (1:4,000), NF-κB p65 (#4764; Cell
Signaling Technology Inc.) and p-NF-κB p65 (#3033; Cell Signaling
Technology Inc.) (1:2,000) or GAPDH with gentle agitation at 4°C
overnight and subsequent incubation with horseradish peroxidase
(HRP)-conjugated secondary antibodies (1:5,000 dilution) for 1.5 h
at room temperature. Following three washes with TBS-T, the
membranes were developed using enhanced chemiluminescence and
exposed to X-ray films. To quantify protein expression, the X-ray
films were scanned and analyzed with ImageJ 1.41o software
(National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All data are presented as the mean ± standard error
(SE). Differences between groups were analyzed by one-way analysis
of variance (ANOVA) using SPSS 13.0 (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
result.
Results
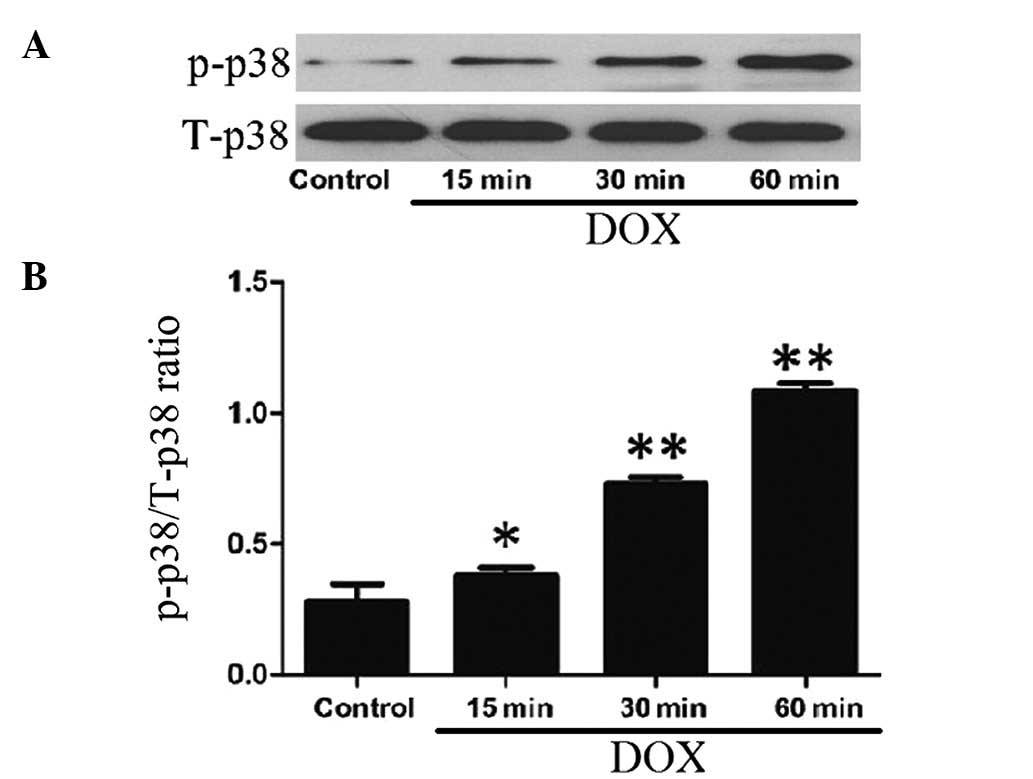
DOX induces the activation of p38 MAPK in
H9c2 cells
After the H9c2 cells were treated with 5 μmol/l DOX
for 15, 30 and 60 min, the expression levels of p-p38 MAPK
increased in a time-dependent manner, indicating the activation of
p38 MAPK by DOX treatment (Fig.
1). Alone, DOX at 5 μmol/l did not alter the expression of
total p38 MAPK.
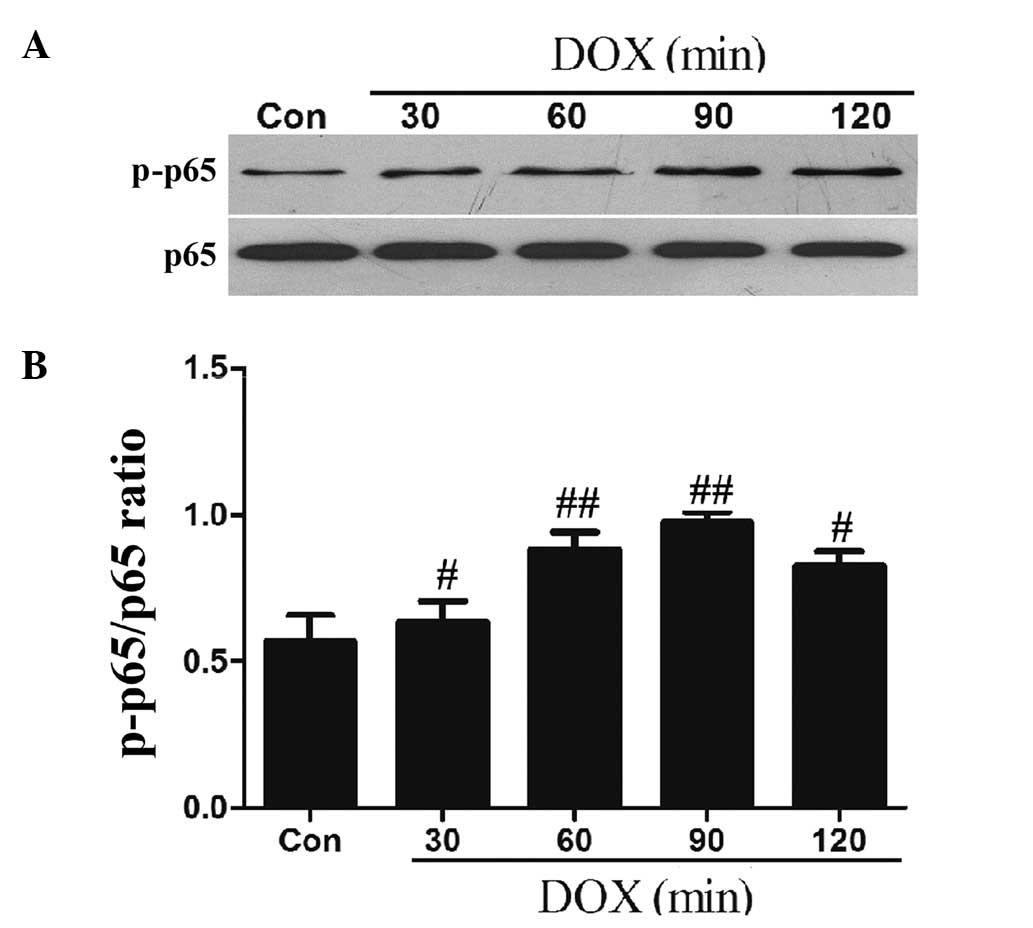
DOX upregulates the phosphorylation of
NF-κB p65 in H9c2 cells
NF-κB is important in regulating genes that
contribute to the onset of oxidative stress and the inflammatory
response. Therefore, we observed the effect of DOX on the
phosphorylation of the NF-κB p65 subunit (an essential step of
NF-κB activation). The results of the western blot analysis
demonstrated that after the H9c2 cells were exposed to 5 μmol/l DOX
for 60 min, the expression levels of p-NF-κB p65 significantly
increased, reaching peak levels at 90 min, with the higher levels
being sustained until 120 min (Fig.
2).
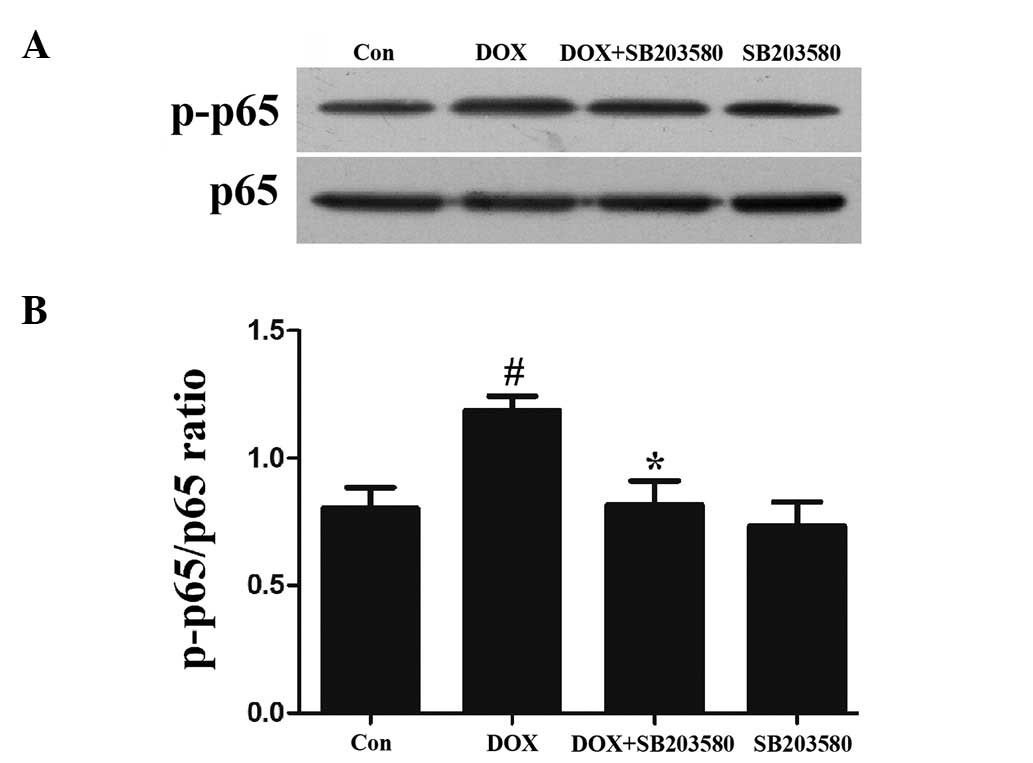
p38 MAPK participates in the activation
of NF-κB p65 by DOX in H9c2 cells
To examine the effect of the activation of p38 MAPK
on the increased phosphorylation of NF-κB p65 by DOX, H9c2 cells
were pretreated with 3 μmol/l SB203580, a specific inhibitor of p38
MAPK, for 60 min prior to exposure to 5 μmol/l DOX. As shown in
Figs. 3A and 2B, the exposure of cells to 5 μmol/l DOX
for 90 min markedly enhanced the expression levels of p-NF-κB p65,
which were attenuated by treatment with SB203580, suggesting the
involvement of p38 MAPK in the DOX-induced activation of NF-κB p65.
SB203580 at 3 μmol/l alone did not change the basal expression
level of p-NF-κB p65 in H9c2 cells (Fig. 3A and B).
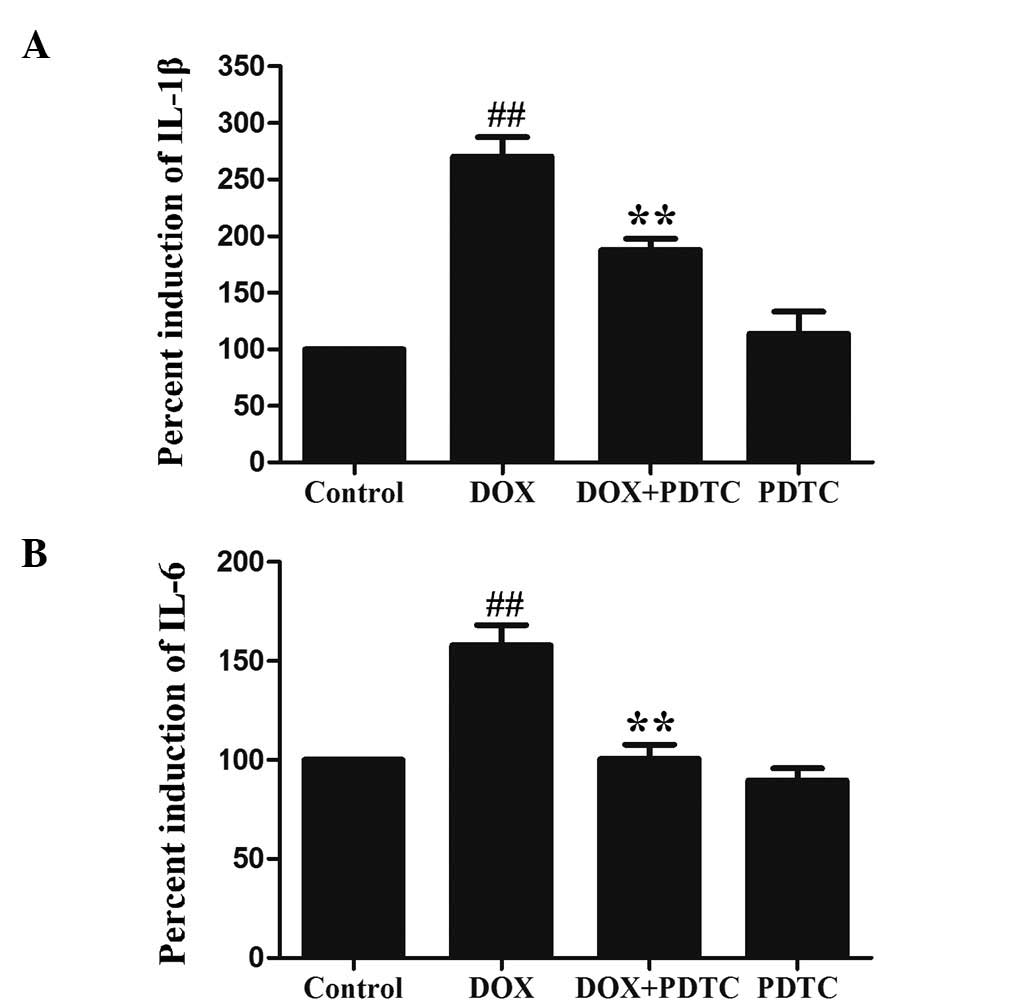
Activation of NF-κB p65 contributes to
DOX-induced inflammation in H9c2 cells
The levels of IL-1β and IL-6 were measured in
response to DOX and PDTC (an inhibitor of NF-κB). Following the
exposure of H9c2 cells to 5 μmol/l DOX for 24 h, IL-1β (Fig. 4A) and IL-6 (Fig. 4B) levels were significantly
increased. Pretreatment with 100 μmol/l PDTC for 30 min prior to
DOX exposure markedly ameliorated IL-1β and IL-6 levels in H9c2
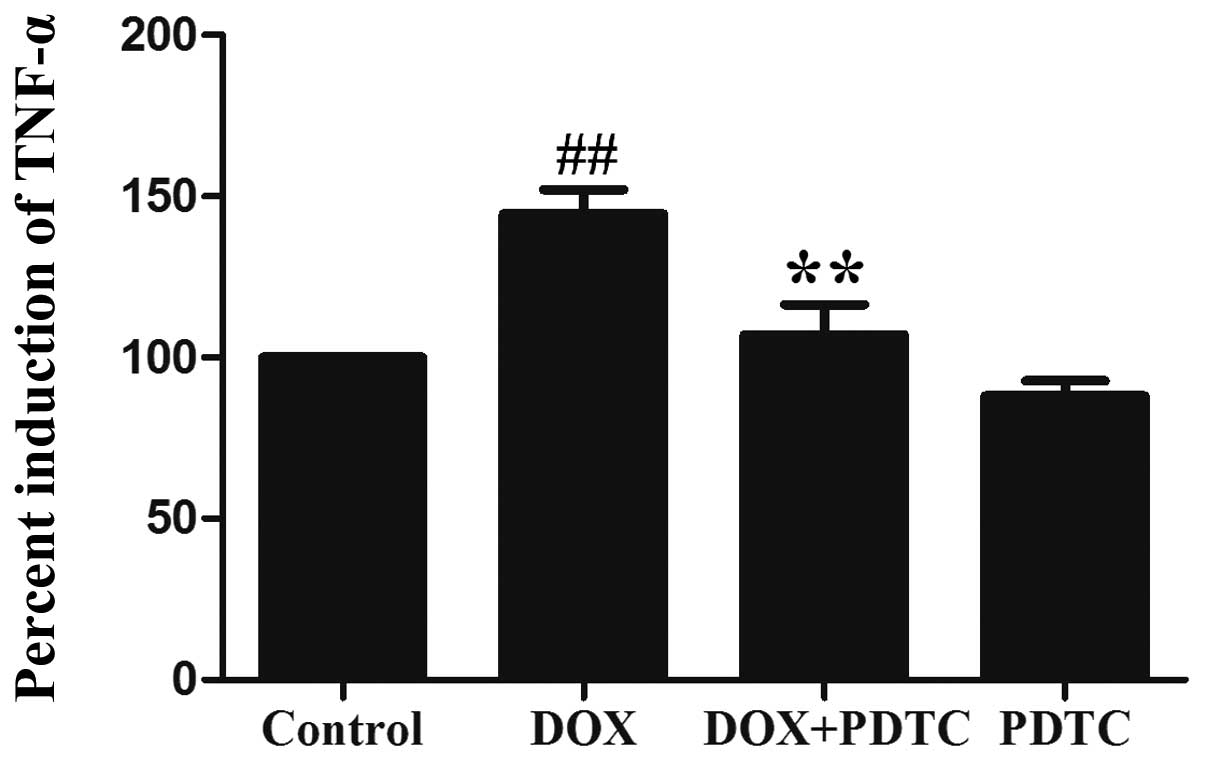
cells. Additionally, the exposure of H9c2 cells to 5 μmol/l DOX
significantly enhanced TNF-α (a proinflammatory cytokine)
production (Fig. 5), which was
reduced by pretreatment with PDTC. These results revealed that the
DOX-induced inflammatory response is associated with the activation
of NF-κB p65.
The p38 MAPK/NF-κB pathway is involved in
DOX-induced cytotoxicity in H9c2 cells
To clarify the role of the p38 MAPK/NF-κB pathway in
DOX-induced cytotoxicity, H9c2 cells were pretreated with either
SB203580 (3 μmol/l) for 60 min or PDTC (100 μmol/l) for 30 min
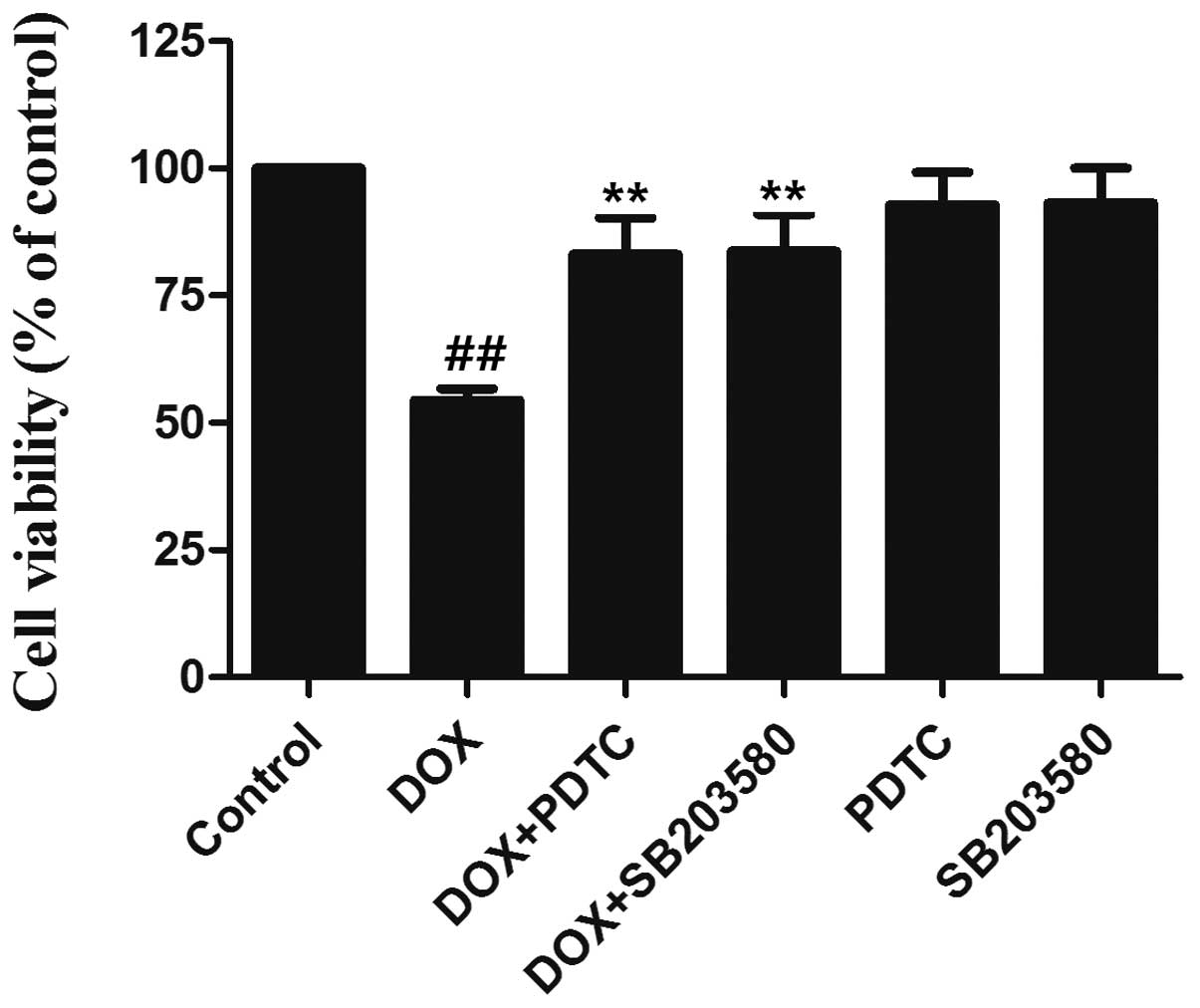
before exposure to 5 μmol/l DOX for 24 h. As shown in Fig. 6, the exposure of H9c2 cells to DOX
induced significant cytotoxicity, as indicated by the decrease in
cell viability. However, the decreased cell viability was markedly
inhibited by pretreatment with SB203580 or PDTC, indicating that
DOX-induced cytotoxicity is mediated, at least partially, by the
p38 MAPK/NF-κB pathway.
Discussion
Since dose-related adverse effects, in particular
cardiotoxicity, often limit the effectiveness of DOX in
chemotherapy, alternative strategies using pharmaceutical agents
have been investigated. Several of these agents, including
dexrazoxan (27),
angiotensin-converting enzyme (ACE) inhibitors (28), β-blockers (29) and vitamin E (30) have been tested in animal models and
clinical studies to prevent or reduce these dose-related clinical
events. However, to date, no single drug has clinically been
capable of fully preventing DOX cardiotoxicity. Additional basic
and clinical studies are required to validate the underlying
mechanism of action of these agents. Results of the present study
support the hypothesis that inflammatory responses to DOX treatment
are mediated, at least partially, by the activation of the p38
MAPK/NF-κB pathway, and that certain adverse inflammatory
consequences induced by DOX may be ameliorated by inhibiting the
p38 MAPK/NF-κB pathway.
Recently, inflammation has been shown to play a role
in DOX cardiotoxicity. DOX induces a significant increase in the
levels of specific inflammatory cytokines and chemokines, including
IL-1β (31), IL-6, TNF-α (5–8,31),
COX-2 (9) and CCL2/MCP-1 (31). COX-2 inhibitors are capable of
improving left ventricular function and mortality in murine models
of DOX-induced heart failure (10). Studies with IL-1β-deficient mice
have demonstrated that IL-1β signaling is critical in DOX-induced
increases in IL-6 and granulocyte colony stimulating factor (GCSF)
levels (31). Furthermore, DOX is
able to induce the activation of NF-κB (a positive regulator of
COX-2 expression) (13–15), which contributes to cardiac
inflammation and necrosis (32).
Since the signaling pathway that induces the expression of the 35
kDa pro-IL-1β is mediated by the activation of NF-κB and p38 MAPK
(33), we hypothesize that the
activation of p38 MAPK and NF-κB may modulate the inflammatory
response in DOX-treated cardiomyocytes. The results of the present
study confirmed our hypothesis. In agreement with previous studies
(18–21), we demonstrated that the expression
of p-p38 MAPK was markedly enhanced in DOX-treated H9c2 cardiac
cells. In addition, the exposure of H9c2 cells led to DOX-induced
activation of NF-κB, which is consistent with previous studies
(13–15). Notably, we observed that the
pretreatment of cells with SB203580, a specific inhibitor of p38
MAPK, attenuated the increased activation of NF-κB p65 by DOX,
suggesting a modulatory effect of the p38 MAPK pathway on
DOX-induced NF-κB activation. Furthermore, our results showed that
DOX significantly induced inflammatory responses, as indicated by
an increase in the levels of IL-1β, IL-6 and TNF-α. However,
whether there is an association between the p38 MAPK/NF-κB pathway
and DOX-induced inflammatory markers is unclear.
To clarify the modulatory effects of NF-κB
activation on the levels of inflammatory markers, the H9c2 cells
were pretreated with PDTC, a selective inhibitor of NF-κB, prior to
exposure to DOX treatment. Firstly, we demonstrated that
pretreatment with PDTC significantly reduced the levels of IL-1β
and IL-6 induced by DOX, highlighting the modulatory role of the
NF-κB pathway in the DOX-induced secretion of IL-1β and IL-6 from
H9c2 cells. IL-1β is an initiator cytokine that is important in the
regulation of the immune and inflammatory responses (34), and contributes to the DOX-induced
increase in the levels of IL-6 and GCSF (31). Thus, elucidating the role of NF-κB
in the IL-1β-mediated inflammatory response may present
opportunities to inhibit the inflammatory consequences of DOX. This
study also demonstrated that the activation of NF-κB is necessary
for the induction of IL-1β and IL-6 by DOX in H9c2 cells. In
addition, we observed that PDTC pretreatment had a notable
inhibitory effect on the induction of TNF-α by DOX treatment,
revealing the involvement of the NF-κB pathway in the modulation of
TNF-α induction. TNF-α, a proinflammatory cytokine, may cause
apoptotic cell death, cellular proliferation, differentiation,
inflammation, tumorigenesis and viral replication (35). Recent studies have demonstrated
that DOX increases TNF-α expression (7,31,36,37).
Notably, TNF-α is capable of activating NF-κB (14). Daunorubicin, a DOX analogue, was
demonstrated to strongly affect the potential ability of TNF-α to
activate NF-κB, suggesting a synergy between these two agents in
this response (38). Based on our
results and those of previous studies (7,14,31,36–38),
we suggest that a cross-talk between the NF-κB pathway and TNF-α
exists, which may be important in DOX-induced inflammation. Further
studies are required to confirm this hypothesis.
Additionally, we examined the role of the p38
MAPK/NF-κB pathway in DOX-induced cytotoxicity. The findings of
this study showed that the pretreatment of H9c2 cells with either
SB203580 or PDTC prior to exposure to DOX markedly inhibited
DOX-induced cytotoxicity, leading to an increase in cell viability.
The results suggest that the induction of cardiac cytotoxicity and
inflammation by DOX may share common mechanisms, including the p38
MAPK/NF-κB pathway.
In conclusion, to the best of our knowledge, this is
the first study to demonstrate the role of the p38 MAPK/NF-κB
pathway in the DOX-induced inflammatory response in H9c2 cells. A
clearer understanding of the functional significance of this
pathway may constitute a potential new therapeutic option to
prevent DOX-induced cardiotoxicity. However, further clinical
studies are required to verify whether this hypothesis is valid in
patients.
Acknowledgements
This study was supported by the Science and
Technology Planning Project of Guangdong Province in China
(2010B080701035 and 2009B080701014).
References
|
1
|
Danesi R, Fogli S, Gennari A, Conte P and
Del Tacca M: Pharmacokinetic-pharmacodynamic relationships of the
anthracycline anticancer drugs. Clin Pharmacokinet. 41:431–444.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hrdina R, Gersl V, Klimtová I, Simůnek T,
Machácková J and Adamcová M: Anthracycline-induced cardiotoxicity.
Acta Medica (Hradec Kralove). 43:75–82. 2000.
|
|
3
|
Scully RE and Lipshultz SE: Anthracycline
cardiotoxicity in long-term survivors of childhood cancer.
Cardiovasc Toxicol. 7:122–128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zucchi R and Danesi R: Cardiac toxicity of
antineoplastic anthracyclines. Curr Med Chem Anticancer Agents.
3:151–171. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Morsi MI, Hussein AE, Mostafa M, El-Abd E
and El-Moneim NA: Evaluation of tumour necrosis factor-alpha,
soluble P-selectin, gamma-glutamyl transferase, glutathione
S-transferase-pi and alpha-fetoprotein in patients with
hepatocellular carcinoma before and during chemotherapy. Br J
Biomed Sci. 63:74–78. 2006.
|
|
6
|
Mukherjee S, Banerjee SK, Maulik M, Dinda
AK, Talwar KK and Maulik SK: Protection against acute
adriamycin-induced cardiotoxicity by garlic: role of endogenous
antioxidants and inhibition of TNF-alpha expression. BMC Pharmacol.
3:162003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Riad A, Bien S, Westermann D, et al:
Pretreatment with statin attenuates the cardiotoxicity of
Doxorubicin in mice. Cancer Res. 69:695–699. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zordoky BN, Anwar-Mohamed A, Aboutabl ME
and El-Kadi AO: Acute doxorubicin toxicity differentially alters
cytochrome P450 expression and arachidonic acid metabolism in rat
kidney and liver. Drug Metab Dispos. 39:1440–1450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang CC, Chen PC, Huang CW and Yu J:
Aristolochic Acid induces heart failure in zebrafish embryos that
is mediated by inflammation. Toxicol Sci. 100:486–494. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Delgado RM 3rd, Nawar MA, Zewail AM, et
al: Cyclooxygenase-2 inhibitor treatment improves left ventricular
function and mortality in a murine model of doxorubicin-induced
heart failure. Circulation. 109:1428–1433. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Huang CY, Fujimura M, Noshita N, Chang YY
and Chan PH: SOD1 down-regulates NF-kappaB and c-Myc expression in
mice after transient focal cerebral ischemia. J Cereb Blood Flow
Metab. 21:163–173. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kang YJ, Wingerd BA, Arakawa T and Smith
WL: Cyclooxygenase-2 gene transcription in a macrophage model of
inflammation. J Immunol. 177:8111–8122. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lin X, Li Q, Wang YJ, et al: Morphine
inhibits doxorubicin-induced reactive oxygen species generation and
nuclear factor kappaB transcriptional activation in neuroblastoma
SH-SY5Y cells. Biochem J. 406:215–221. 2007. View Article : Google Scholar
|
|
14
|
Riganti C, Doublier S, Costamagna C, et
al: Activation of nuclear factor-kappa B pathway by simvastatin and
RhoA silencing increases doxorubicin cytotoxicity in human colon
cancer HT29 cells. Mol Pharmacol. 74:476–484. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu HG, Ai YW, Yu LL, et al:
Phosphoinositide 3-kinase/Akt pathway plays an important role in
chemoresistance of gastric cancer cells against etoposide and
doxorubicin induced cell death. Int J Cancer. 122:433–443. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang C, Ling H, Zhang M, et al: Oxidative
stress mediates chemical hypoxia-induced injury and inflammation by
activating NF-κb-COX-2 pathway in HaCaT cells. Mol Cells.
31:531–538. 2011.PubMed/NCBI
|
|
17
|
Yang C, Yang Z, Zhang M, et al: Hydrogen
sulfide protects against chemical hypoxia-induced cytotoxicity and
inflammation in HaCaT cells through inhibition of ROS/NF-κB/COX-2
pathway. PLoS One. 6:e219712011.PubMed/NCBI
|
|
18
|
Kang YJ, Zhou ZX, Wang GW, Buridi A and
Klein JB: Suppression by metallothionein of doxorubicin-induced
cardiomyocyte apoptosis through inhibition of p38 mitogen-activated
protein kinases. J Biol Chem. 275:13690–13698. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lou H, Danelisen I and Singal PK:
Involvement of mitogen-activated protein kinases in
adriamycin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol.
288:H1925–H1930. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lou H, Kaur K, Sharma AK and Singal PK:
Adriamycin-induced oxidative stress, activation of MAP kinases and
apoptosis in isolated cardiomyocytes. Pathophysiology. 13:103–109.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poizat C, Puri PL, Bai Y and Kedes L:
Phosphorylation-dependent degradation of p300 by
doxorubicin-activated p38 mitogen-activated protein kinase in
cardiac cells. Mol Cell Biol. 25:2673–2687. 2005. View Article : Google Scholar
|
|
22
|
Lechner C, Zahalka MA, Giot JF, Møller NP
and Ullrich A: ERK6, a mitogen-activated protein kinase involved in
C2C12 myoblast differentiation. Proc Natl Acad Sci USA.
93:4355–4359. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Young PR, McLaughlin MM, Kumar S, et al:
Pyridinyl imidazole inhibitors of p38 mitogen-activated protein
kinase bind in the ATP site. J Biol Chem. 272:12116–12121. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Puri PL and Sartorelli V: Regulation of
muscle regulatory factors by DNA-binding, interacting proteins, and
post-transcriptional modifications. J Cell Physiol. 185:155–173.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lan AP, Xiao LC, Yang ZL, et al:
Interaction between ROS and p38MAPK contributes to chemical
hypoxia-induced injuries in PC12 cells. Mol Med Report. 5:250–255.
2012.PubMed/NCBI
|
|
27
|
Hensley ML, Hagerty KL, Kewalramani T, et
al: American Society of Clinical Oncology 2008 clinical practice
guideline update: use of chemotherapy and radiation therapy
protectants. J Clin Oncol. 27:127–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cardinale D, Colombo A, Sandri MT, et al:
Prevention of high-dose chemotherapy-induced cardiotoxicity in
high-risk patients by angiotensin-converting enzyme inhibition.
Circulation. 114:2474–2481. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kalay N, Basar E, Ozdogru I, et al:
Protective effects of carvedilol against anthracycline-induced
cardiomyopathy. J Am Coll Cardiol. 48:2258–2262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Berthiaume JM, Oliveira PJ, Fariss MW and
Wallace KB: Dietary vitamin E decreases doxorubicin-induced
oxidative stress without preventing mitochondrial dysfunction.
Cardiovasc Toxicol. 5:257–267. 2005. View Article : Google Scholar
|
|
31
|
Sauter KA, Wood LJ, Wong J, Iordanov M and
Magun BE: Doxorubicin and daunorubicin induce processing and
release of interleukin-1beta through activation of the NLRP3
inflammasome. Cancer Biol Ther. 11:1008–1016. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shi Y, Moon M, Dawood S, McManus B and Liu
PP: Mechanisms and management of doxorubicin cardiotoxicity. Herz.
36:296–305. 2011. View Article : Google Scholar
|
|
33
|
Bankers-Fulbright JL, Kalli KR and McKean
DJ: Interleukin-1 signal transduction. Life Sci. 59:61–83. 1996.
View Article : Google Scholar
|
|
34
|
Dinarello CA: IL-1: discoveries,
controversies and future directions. Eur J Immunol. 40:599–606.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
MacEwan DJ: TNF ligands and receptors - a
matter of life and death. Br J Pharmacol. 135:855–875. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gilliam LA, Moylan JS, Ferreira LF and
Reid MB: TNF/TNFR1 signaling mediates doxorubicin-induced diaphragm
weakness. Am J Physiol Lung Cell Mol Physiol. 300:L225–L231. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Riad A, Bien S, Gratz M, et al: Toll-like
receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy
in mice. Eur J Heart Fail. 10:233–243. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boland MP, Foster SJ and O’Neill LA:
Daunorubicin activates NFkappaB and induces kappaB-dependent gene
expression in HL-60 promyelocytic and Jurkat T lymphoma cells. J
Biol Chem. 272:12952–12960. 1997. View Article : Google Scholar : PubMed/NCBI
|