Introduction
Autophagy refers to a series of biochemical
processes in eukaryotic cells involving ‘self-digestion’ by
degrading their own cytoplasm and organelles. Under nutritional
deficiency, starvation or anoxia, autophagy is described as a
pathway capable of promoting cell survival (1). Under these conditions, it is
essential for autophagy to provide energy for cells. A previous
study indicated that autophagy is associated with the effects of
antitumor agents on tumor cells. Subsequently, tumor cells overcome
nutritional deficiency and hypoxia by autophagy and are therefore
protected from entering the apoptotic pathway (2).
One of the important factors for the poor prognosis
of tumors is their tolerance to radiotherapy and chemotherapy.
Recently, it was found that autophagy is one of the mechanisms
involved in the resistance of tumors to chemotherapy (3). Autophagy inhibitor, chloroquine,
improves the sensitivity of human lymphoma cells to apoptosis
inducers, and thus it is considered to treat tumors by combining
autophagy inhibitors and apoptosis inducers (4). Tumor cells grow in low-vascularized
environments by autophagy and the treatment of tumors may be
elucidated by inhibiting autophagic activities. Li et
al(5) found that the
inhibition of autophagy may improve the sensitivity of intestinal
cancer cells to fluorouracil (5-FU). Therefore, it is of great
importance to identify anti-tumor agents that target the molecular
mechanisms of autophagy.
According to the concept of bioinformatics and
network pharmacology, theoretically, if the inhibition of
autophagic genes (or gene knockout) prevents autophagy, agents
inhibiting the same genes may also suppress autophagy (6). At present, there are ~500 biological
targets for therapeutic agents and it is estimated that human
genome data may contain ~5,000–10,000 novel targets, which is 10-
to 20-fold higher than currently known targets. It is not difficult
to obtain a number of novel agents, which may be 10-fold higher
than the currently available drugs if genomic data are sufficiently
mined (7). The present study
screened differentially expressed protein kinases during autophagy
using bioinformatics and constructed the kinase-kinase inhibitor
regulatory network. The aim therefore was to explore kinase
inhibitors capable of arresting autophagy with potential research
values.
Materials and methods
Data resources
Data of gene expression chip analysis of
starve-induced autophagy was obtained from the Gene Expression
Omnibus (GEO) database at the intersection set of GSE2435
(http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE2345)
and GSE31040 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31040)
as shown in Table I. The CEL data
compression package of microarray was downloaded in the
supplementary file of GEO and decompressed into another folder for
further use. Moreover, the original data for the sample in TXT
format was also downloaded.
 | Table ISummary of the two data series from
the GEO database. |
Table I
Summary of the two data series from
the GEO database.
| Series | Samples | Group | Experimental
design | Array platform | References |
|---|
| GSE2435 | GSM45796 | Autophagy-6 h-1 | Human
B-lymphoblastoid cell line after 6 h and 24 h of starvation | GPL570
HG-U133-Plus-2
Affymetrix Human
Genome U133
Plus 2.0 Array | Proc Natl Acad Sci
USA, 2005 PMID: 15894616 (8) |
| GSM45797 | Autophagy-24 h-1 |
| GSM45798 | Autophagy-24 h-2 |
| GSM45799 | Control-6 h-1 |
| GSM45800 | Control-24 h-1 |
| GSM45801 | Control-24 h-2 |
| GSE31040 | GSM769043 | Autophagy-48 h-1 | Human lymphoblastoid
cell lines after 48 h of starvation | GPL570
HG-U133-Plus-2
Affymetrix Human
Genome U133
Plus 2.0 Array | Age (Dordr) 2011
PMID: 21904824 (9) |
| GSM769045 | Autophagy-48 h-2 |
| GSM769049 | Autophagy-48 h-3 |
| GSM769042 | Control-48 h-1 |
| GSM769044 | Control-48 h-2 |
| GSM769048 | Control-48 h-3 |
Microarray data processing
mRNA microarray in the present study was performed
as follows: Three samples from Homo sapiens were prepared
under ‘Control’ and ‘Autophagy’ conditions, RMA algorithm was used
to calculate the expression level and MAS algorithm was used to
calculate the detection call. The probe numbers showing low
expression levels were filtered using a P-value for a minimum of
two detection calls to preserve any group of samples, and human
HGNC gene abbreviation was used to unify the gene names. LIMMA
differential gene screening algorithm was used to screen for the
upregulated and downregulated genes under ‘Autophagy’ conditions
relative to ‘Control’ conditions, and differential multiples,
P-value and FDR values were calculated. P<0.05 was considered to
indicate differentially expressed genes.
The differentially expressed genes were screened by
selecting the intersection set of GSE2435 and GSE31040, and the
differentially expressed protein kinases were screened according to
a ‘protein kinase database’ (http://kinasource.co.uk/Database/substrates.html).
Gene ontology (GO) enrichment analysis
and cluster analysis on differentially expressed kinases
GO-BP (biological process) enrichment analysis was
performed using DAVID software (10). Gene sets for further analysis were
submitted to the DAVID database (http://david.abcc.ncifcrf.gov/), corresponding gene
designators were simultaneously selected, human whole genome was
ticked off as the background genes, the ‘Functional Annotation
Tool’ was used as the analytical tool and the results for GO-BP
enrichment analysis were obtained (P<0.05). Each GO-BP term and
corresponding differentially expressed kinases were selected, and
cytoscape software (version 2.6.3) was used to construct a
visualized network to connect the kinases with their corresponding
functions.
The differentially expressed protein kinases were
identified to serve as the candidates for hierarchical cluster
analysis using Cluster2.2 and TreeView3.2 software (11). The ratios of the coexpressed
candidate kinases were transformed into log2%, the
distance was measured by means of Pearson correlation and cluster
analysis was performed with single linkage clustering.
Construction of the kinase-gene
interaction network
The protein-protein interaction (PPI) network was
obtained from the HPRD database (http://www.hprd.org/), with 36,874 lines and 9,453
nodes being included in the network. The differentially expressed
genes for analysis were projected into the PPI network and the
correlation pairs in which the two interacting genes were
differentially expressed were reserved, thus the sub-network was
obtained. The differentially expressed kinase genes for further
analysis were projected into the sub-network, the correlation pairs
in which the differentially expressed kinases and the
differentially expressed genes were interacting were reserved and
thus the kinase-gene interaction network was obtained.
Screening of kinase inhibitors that
regulate autophagy
An Excel file of 194 kinase inhibitors was
established based on a database (http://www.selleckbio.com/servlet/DownloadServlet?fileName=Selleck-Kinase-Inhibitor-Library.xlsx).
In the literature mining approach, Perl language software
(www.perl.com) was used to write a program of
literature mining. The Excel files of ‘differentially expressed
kinases’ and ‘kinase inhibitors’ were imported to the ActivePerl
5.16.2 software, and the literature information was obtained from
the National Library of Medicine’s PubMed database (http://www.ncbi.nlm.nih.gov/pubmed/). The scope
of the retrieval included titles and abstracts of all the articles
in the PubMed database database. Valuable articles were identified
by manual screening and we constructed the regulatory network by
Cytoscape (version 2.6.3).
The name of each screened kinase inhibitor and
‘Autophagy’ were input into Pubmed for searching and the kinase
inhibitors were divided into 3 groups (0, 1–10 and >11)
according to the number of retrieved references.
Results
Differentially expressed kinases and
their function
In total 13,234 genes were detected in GSE2435 and
2,783 differentially expressed genes were screened. A total of
14,442 genes were detected in GSE31040 and 2,369 differentially
expressed genes were screened. A total of 544 genes were allocated
in the intersection set for the differentially expressed genes
screened out from GSE2435 and GSE31040.
Among the 544 differentially expressed genes, 19
genes encoding protein kinases were screened out (as shown in
Table II), of which 11 genes were
upregulated and 8 genes were downregulated. The 11 upregulated
genes were used as targets for further intervention by kinase
inhibitors.
| Table IIDifferentially expressed protein
kinase of starve-induced autophagic cells. |
Table II
Differentially expressed protein
kinase of starve-induced autophagic cells.
| | | P-value | |
|---|
| | |
| |
|---|
| Gene symbol | Gene ID | Gene name | GSE2435 | GSE31040 | Regulation |
|---|
| CLK4 | 826442 | CDC-like kinase
4 | 0.006853 | 0.016876591 | Up |
| NEK9 | 821660 | NIMA (never in
mitosis gene a)-related kinase 9 | 0.047605 | 0.018157559 | Down |
| TLK2 | 809721 | Tousled-like kinase
2 | 0.010894 | 0.007442178 | Up |
| RIOK3 | 815805 | RIO kinase 3
(yeast) | 0.016673 | 0.028184311 | Up |
| TBCK | 811953 | TBC
domain-containing protein kinase-like | 0.01651 | 0.007265638 | Down |
| BRD2 | 773220 | Bromodomain
containing 2 | 0.010431 | 0.004012674 | Up |
| PRKCH | 774932 | Protein kinase C,
eta | 0.005525 | 0.008714029 | Down |
| CSNK1A1 | 791771 | Casein kinase
1α1 | 0.007338 | 0.042396721 | Up |
| IRAK4 | 807982 | Interleukin-1
receptor-associated kinase 4 | 0.024849 | 0.029144612 | Down |
| HIPK1 | 787073 | Homeodomain
interacting protein kinase 1 | 0.005074 | 0.005330583 | Up |
| TRIB3 | 822826 | Tribbles homolog 3
(Drosophila) | 0.010471 | 1.33E-05 | Up |
| BRD4 | 799732 | Bromodomain
containing 4 | 0.040358 | 0.012603665 | Up |
| EIF2AK2 | 824980 | Eukaryotic
translation initiation factor 2-α kinase 2 | 0.002633 | 0.00920921 | Down |
| CDK2 | 803206 | Cyclin-dependent
kinase 2 | 0.030362 | 0.011442217 | Down |
| BRAF | 821298 | V-raf murine
sarcoma viral oncogene homolog B1 | 0.01214 | 0.016176288 | Up |
| PASK | 811281 | PAS domain
containing serine/threonine kinase | 0.003506 | 0.00239591 | Down |
| CLK1 | 823347 | CDC-like kinase
1 | 0.000355 | 0.000302227 | Up |
| TXK | 810008 | TXK tyrosine kinase
v-akt murine thymoma | 0.00019 | 0.001451441 | Up |
| AKT1 | 791692 | Viral oncogene
homolog 1 | 0.033324 | 0.011637129 | Down |
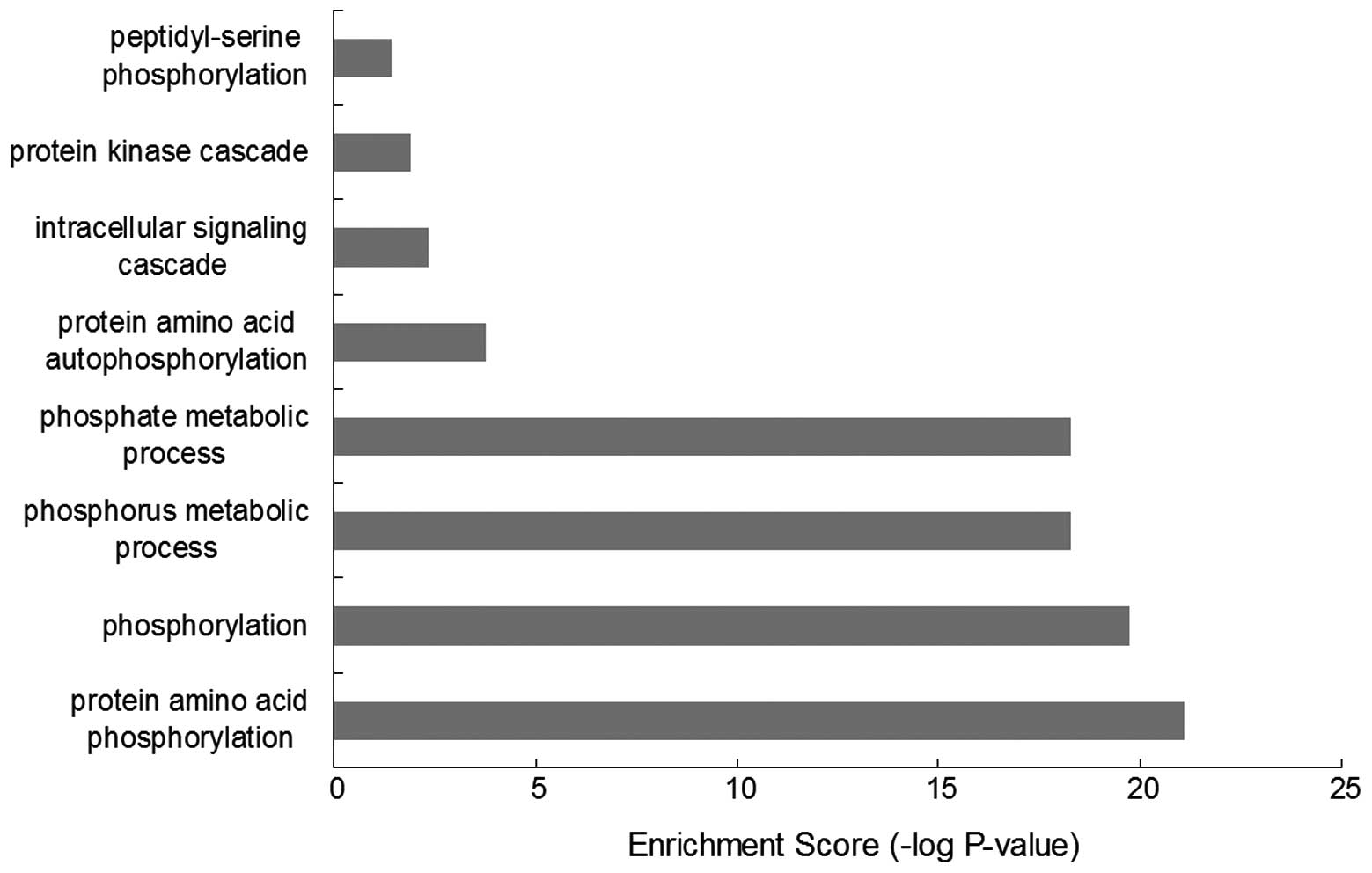
Results of the GO-BP enrichment analysis (Fig. 1) revealed 19 kinases were mainly
associated with functions, such as protein amino acid
phosphorylation (GO:0006468), phosphorylation (GO:0016310),
phosphorus metabolic process (GO:0006793), phosphate metabolic
process (GO:0006796), protein amino acid autophosphorylation
(GO:0046777, intracellular signaling cascade (GO:0007242), protein
kinase cascade (GO:0007243) and peptidyl-serine phosphorylation
(GO:0018212).
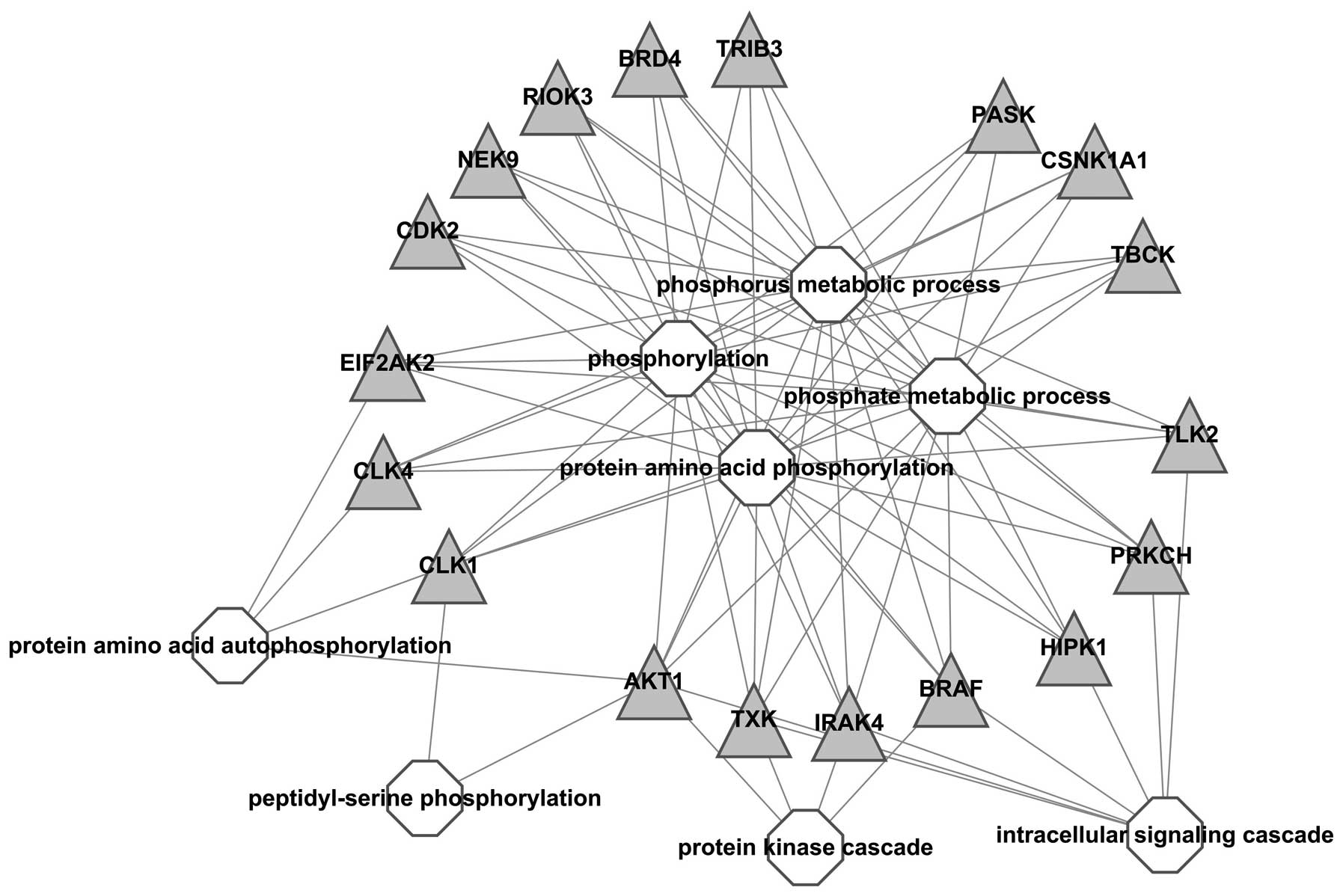
A visualized network was constructed to exhibit the
correlation between kinases and GO-BP terms, results showed all
kinases were associated with ‘phosphorus metabolic process’,
‘phosphorylation’ and ‘protein amino acid phosphorylation’, which
indicated that these kinases were important in the process of
phosphorylation. In addition, BRAF, IRAK, TXK and AKT1 were also
associated with ‘protein kinase cascade’ and BRAF, IRAK, TXK, AKT1,
HIPK1, PRKCH and TLK2 with intracellular signaling cascade;
suggesting that these kinases should be further studied (Fig. 2).
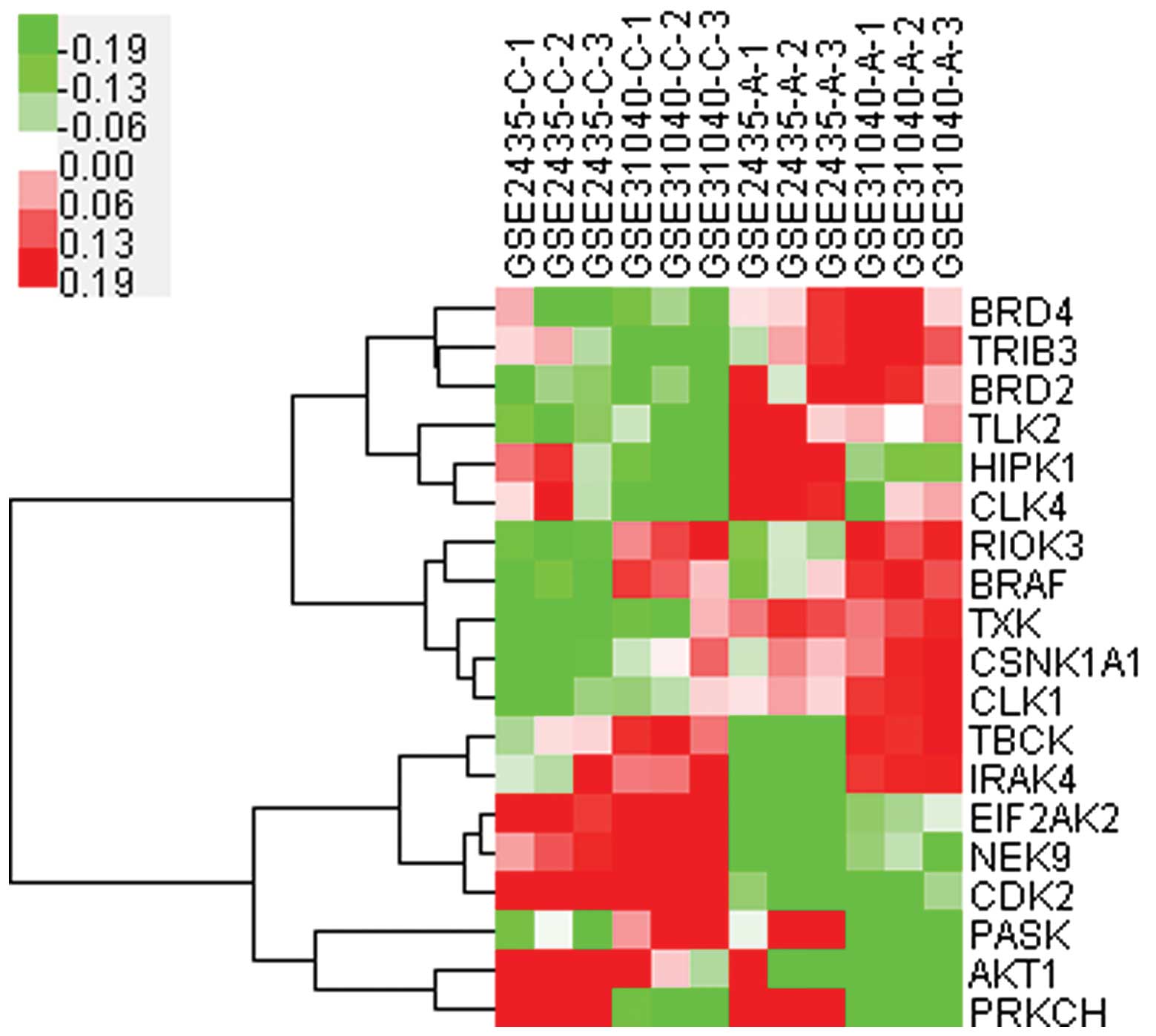
Cluster analysis (Fig.
3) of these 19 kinases indicated that the expression of 11
kinases were upregulated (BRD4, TRIB3, BRD2, TLK2, HIPK1, CLK4,
RIOK3, BRAF, TXK, CSNK1A1 and CLK1) and these should be potential
targets for further study. Among them, BRAF, was closely associated
with RIOK3, and HIPK1 with CLK4, therefore regulation of one kinase
may induce synchronic changes of its correlated kinase.
Role and function of BRAF in regulating
autophagy
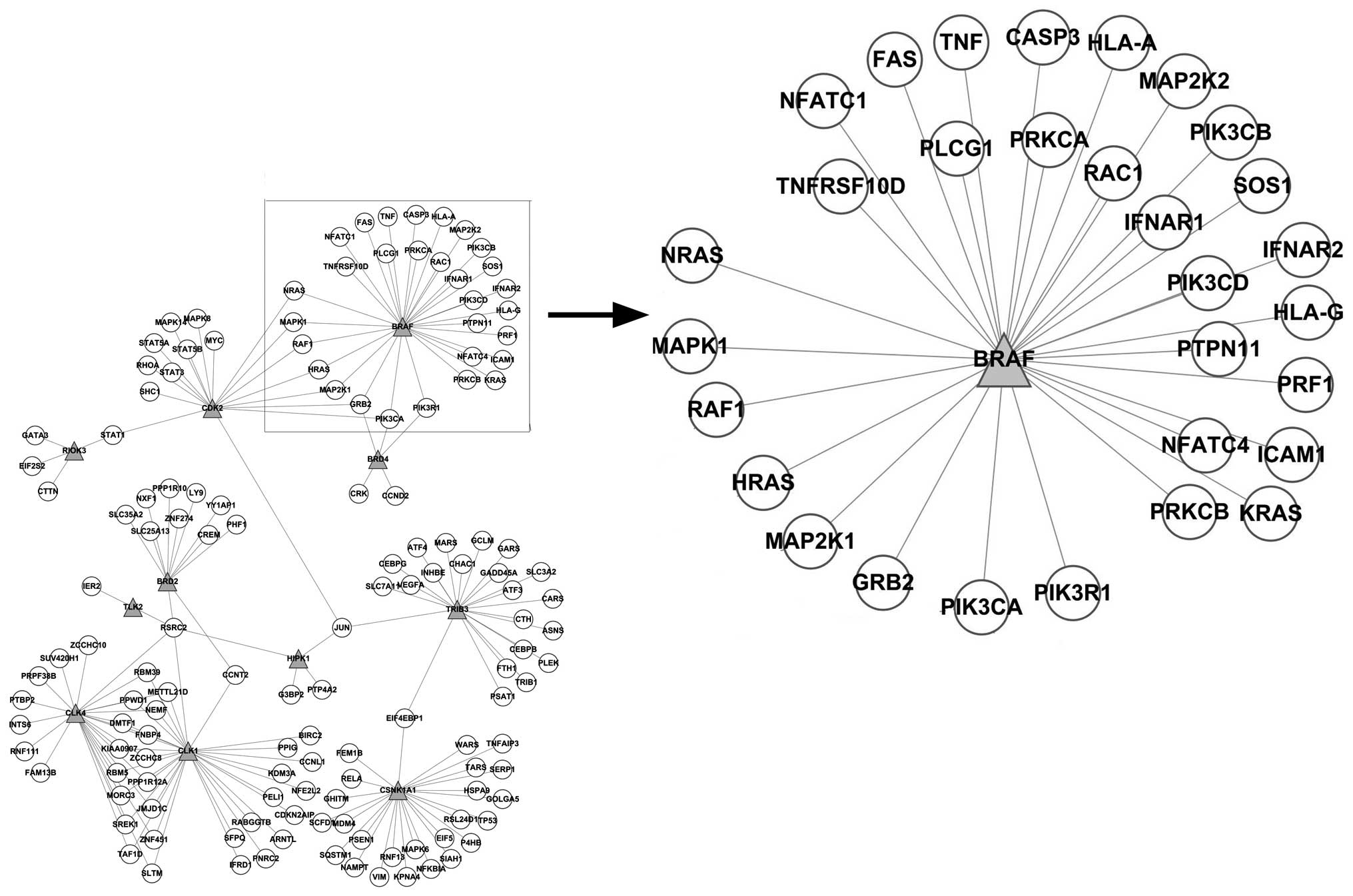
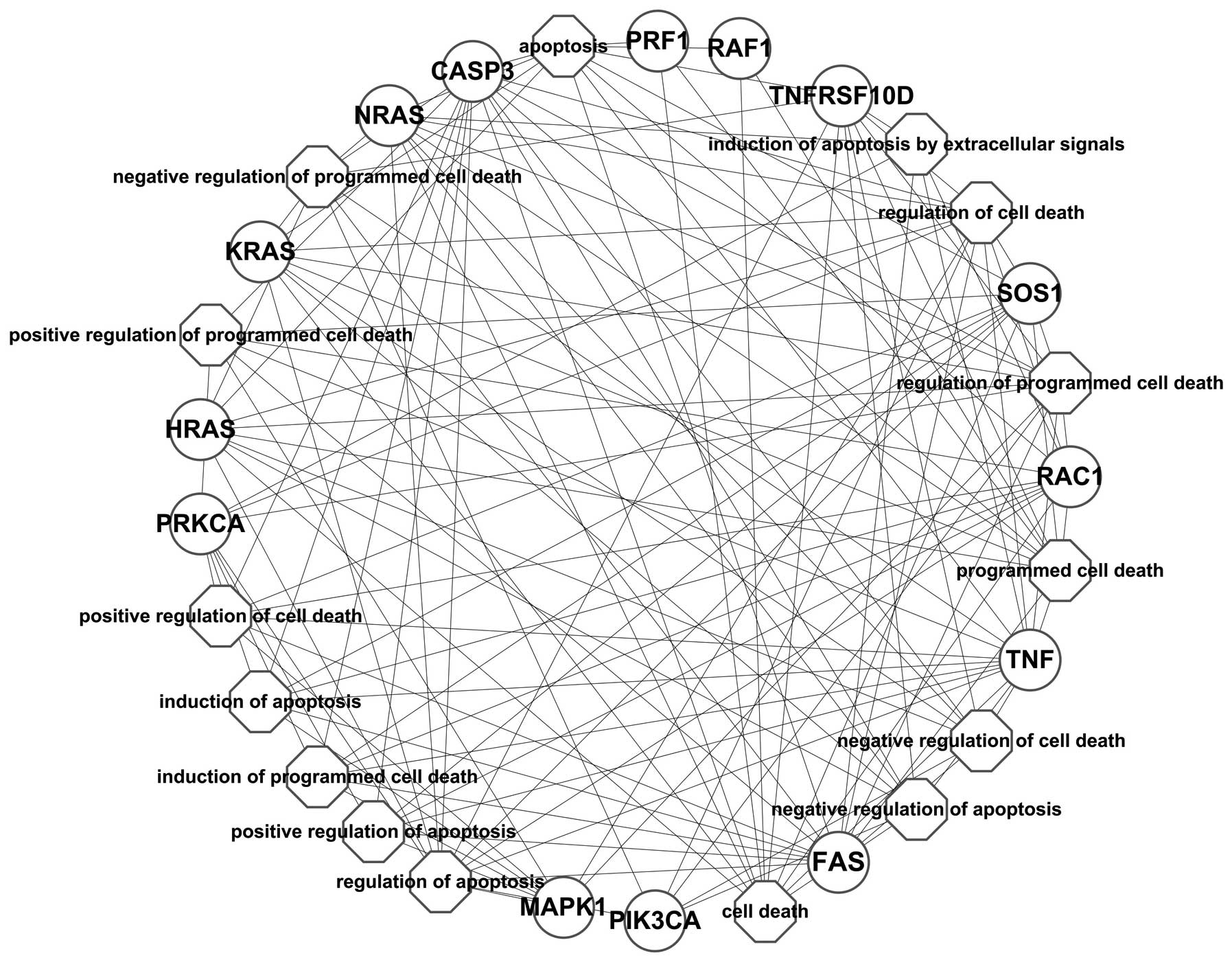
As shown in Fig. 4,
the network constructed based on the PPI network showed that BRAF
interacted with the differentially expressed genes, such as CASP3,
FAS, GRB2, HLA-A, HLA-G, HRAS, ICAM1, IFNAR1, IFNAR2, KRAS, MAP2K1,
MAP2K2, MAPK1, NFATC1, NFATC4, NRAS, PIK3CA, PIK3CB, PIK3CD,
PIK3R1, PLCG1, PRF1, PRKCA, PRKCB, PTPN11, RAC1, RAF1, SOS1, TNF
and TNFRSF10D. The enrichment analysis of these differentially
expressed genes annotated the function of BRAF; however, it may be
presumed that the inhibition of BRAF may have caused the
synchronized alteration of these genes.
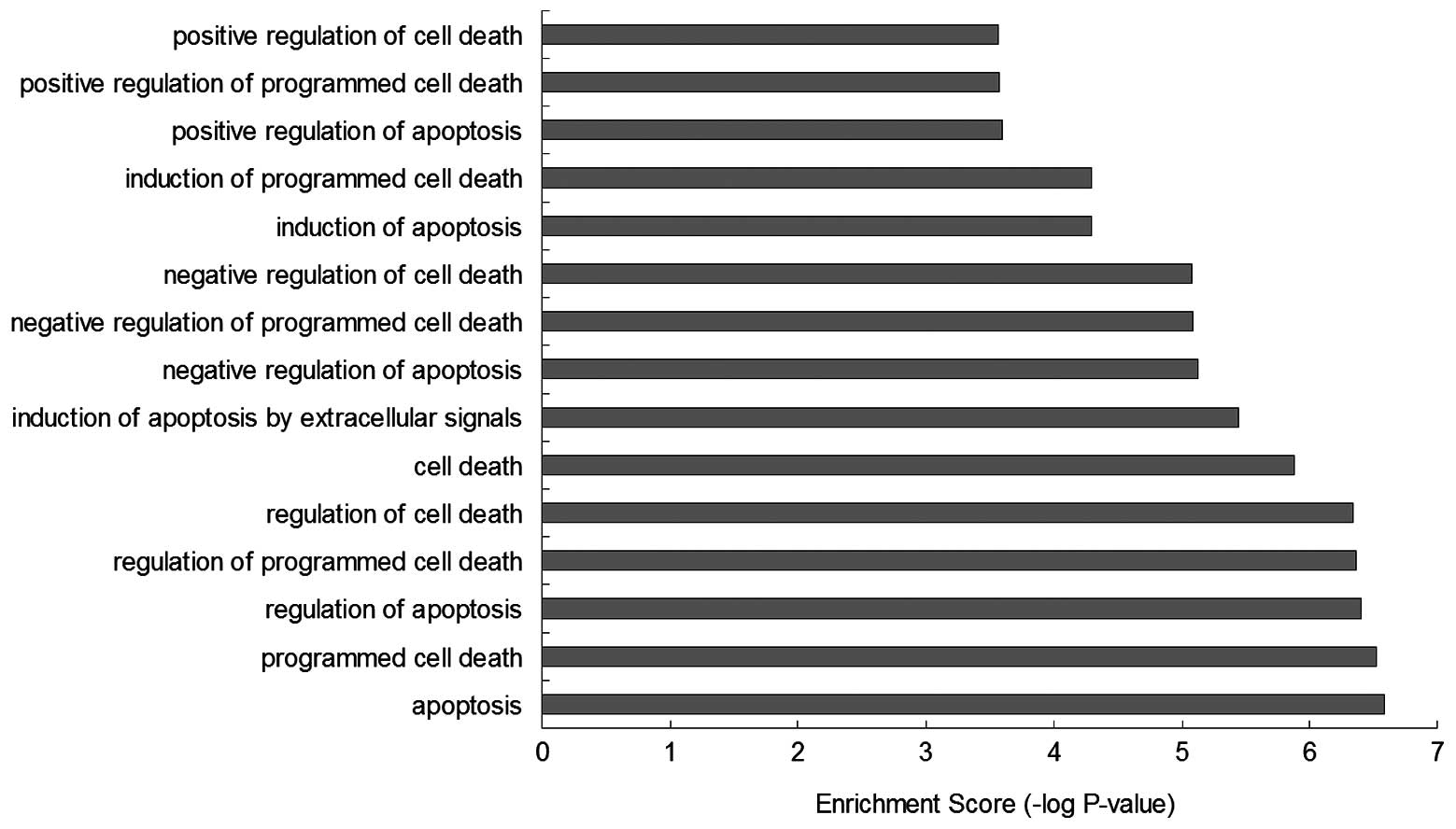
GO-BP enrichment analysis showed that the function
of these BRAF-associated genes were mainly involved in biological
processes, such as ‘apoptosis’ (GO:0006915), ‘programmed cell
death’ (GO:0012501), ‘regulation of apoptosis’ (GO:0042981),
‘regulation of programmed cell death’ (GO:0043067), ‘regulation of
cell death’ (GO:0010941), ‘cell death’ (GO:0008219), ‘induction of
apoptosis by extracellular signals’ (GO:0008624), ‘negative
regulation of apoptosis’ (GO:0043066), ‘negative regulation of
programmed cell death’ (GO:0043069), ‘negative regulation of cell
death’ (GO:0060548), ‘induction of apoptosis’ (GO:0006917),
‘induction of programmed cell death’ (GO:0012502), ‘positive
regulation of apoptosis’ (GO:0043065 ), ‘positive regulation of
programmed cell death’ (GO:0043065) and ‘positive regulation of
cell death’ (GO:0010942), which infer that the regulation of BRAF
may effect the mechanisms of cell death (Figs. 5 and 6).
Screening result of BRAF-targeting kinase
inhibitors
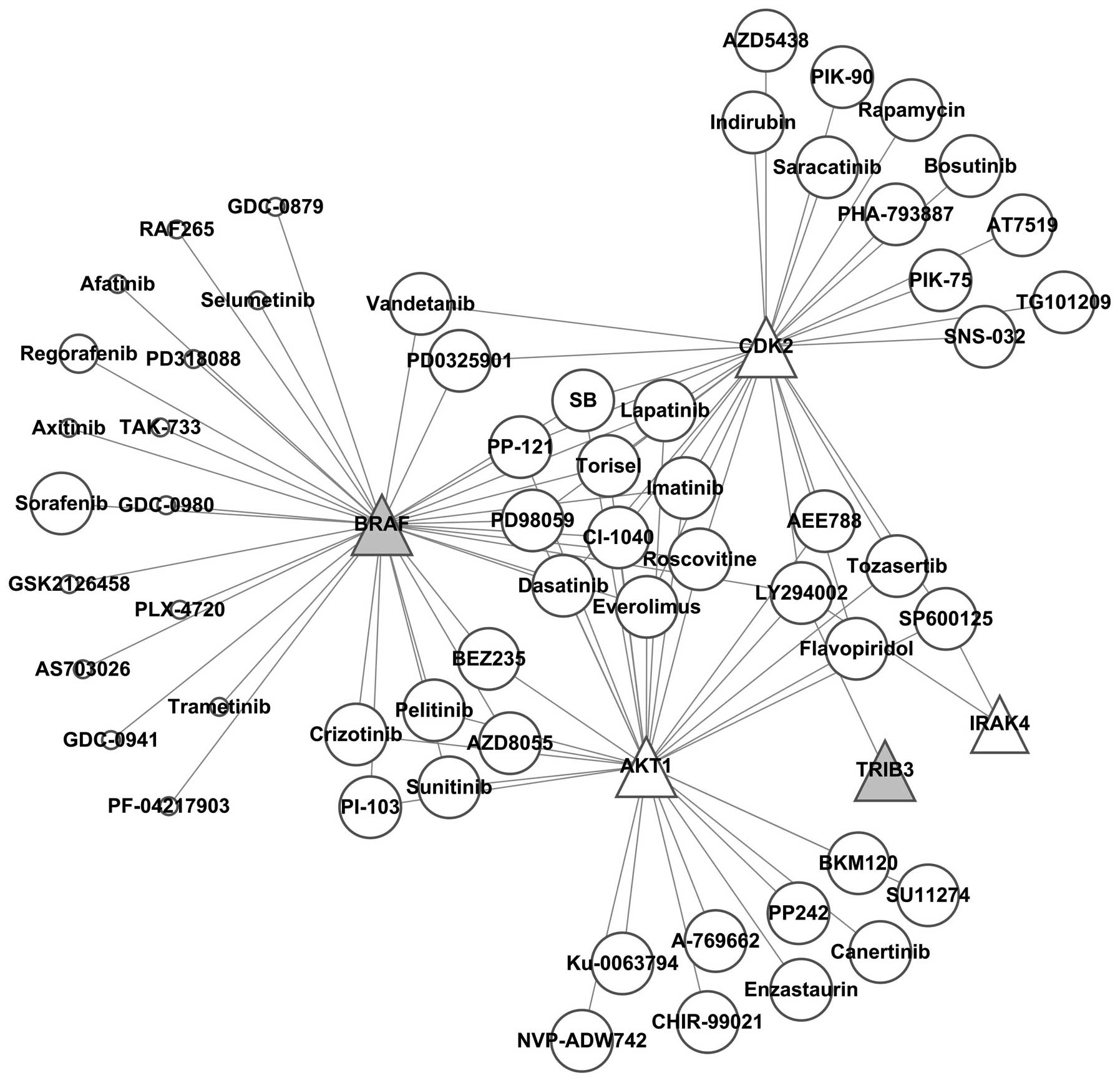
It was found by constructing the kinase-kinase
inhibitor regulatory network (Fig.
7) that SB, PP-121, Torisel, PD98059, imatinib, CI-1040,
roscovitine, everolimus, dasatinib and lapatinib simultaneously
regulated BRAF, AKT4 and CDK2, while AKT4 and CDK2 were
downregulated; thus these kinase inhibitors were excluded.
Vandetanib and PD0325901 simultaneously inhibited BRAF and CDK2,
therefore these two kinase inhibitors were also excluded.
Crizotinib, pelitinib, PI-103, BEZ235, sunitinib and AZD8055
simultaneously inhibited BRAF and AKT1, thus these 6 kinase
inhibitors were also excluded. The remaining 16 kinase inhibitors
specifically suppressed BRAF, among which 39 references and 2
references reported that sorafenib and regorafenib regulated
autophagy, respectively, while there was no report for GDC-0879,
RAF265, afatinib, selumetinib, PD318088, axitinib, TAK-733,
GDC-0980, GSK2126458, PLX-4720, AS703026, trametinib, GDC-0941 or
PF-04217903 of regulating autophagy. This finding indicated that
these kinase inhibitors are potential targets for further
investigation on the regulations of autophagy.
Discussion
A number of anti-tumor treatments may induce
autophagy (12). It was found that
in breast cancer cells, tamoxifen and other estrogen receptor
antagonists induced autophagy (13). Chemotherapy at varied doses may
result in autophagy at different degrees in breast cancer cells,
colorectal cancer cells and prostate cancer cells (14). Autophagy of tumor cells may be
induced during treatment on spongiocytoma using arsenic trioxide,
temozolomide and rapamycin (15),
treatments on ovarian cancer using resveratrol (16) and treatments on cervical cancer
using histone deacetylase (17).
Therefore, it has been estimated that the therapeutic alliance of
autophagic inhibitors may improve therapeutic efficacy and
prognosis (18). Mathew et
al(19) demonstrated from
animal testing that the inhibition of autophagy may aid anti-tumor
therapy. They indicated that a large concentration of tumor cells
died following cytotoxic treatment, while autophagy promoted
resilience and hibernation of tumor cells, and autophagy inhibitors
may directly eliminate these cells. When the autophagy inhibitor,
chloroquine, was used in combination with alkylating agents,
satisfactory therapeutic efficacy was obtained (20). The phenomenon of the induction of
apoptosis by autophagy inhibitors was observed in previously
mentioned studies (21);
therefore, sensitization may be induced in anti-tumor treatments if
autophagy inhibitors were used in combination to block the
autophagic pathway. However, only a limited number of autophagy
inhibitors are available for current clinical application,
including bafilomycin and chloroquine (22).
Numerous molecules are involved in autophagy and the
regulatory mechanisms are extremely complex. The majority of
regulatory mechanisms remain unclear; however, a number of efforts
have been made for exploration and many signaling molecules are
shared in regulating autophagy and apoptosis. The determined
signaling molecules involved in regulating autophagy include
cytokine, MAPK and mitochondrial signaling for which most of the
signals exert regulatory functions by mTOR kinase (23).
Molecular switches accurately control proteins
during cascade reactions for a series of signaling transduction
processes in cells by activation or deactivation mechanisms. Among
the cascade reactions for a series of signaling transduction
processes, positive and negative feedback mechanisms complementing
each other are essential for accurate control, thus the function of
molecule switches is influential (24). The proteins involved in signaling
transduction processes include protein kinases and GTP-binding
proteins. Protein kinases control the activities of 30% of the
proteins in cells, which are associated with almost all of the cell
functions (25). Protein kinases
are particularly important for intracellular signaling transduction
and implementation of complex cell functions, such as cell division
and death. The core function of kinases in controlling cell
behaviors subsequently caused them to be considered as targets for
treating multiple diseases and thus they have been extensively
investigated. In addition, in contrast to gene transfection and RNA
interference, a number of small molecular compounds are capable of
effectively regulating kinase activities (26). They have been commercialized and it
is of practical significance to introduce them to switch autophagy,
which may realize the transformation of laboratory achievements to
clinical application, and meets with the concepts of translational
medicine. Therefore, the present study examined the factors
regulating autophagy. In total, 544 differentially expressed genes
were screened using the genome-wide expression profile microarray
in 2 groups of autophagy models induced by starvation, among which
11 upregulated protein kinases were used as the targets for
intervention by kinase inhibitors. Among the upregulated kinases,
BRAF and TRIB3 were found to have corresponding specific inhibitors
in the kinase-kinase inhibitor regulatory network. Moreover,
numerous inhibitors were available for BRAF (27) and thus BRAF was analyzed.
BRAF is one of the three isoforms of RAF and it is
an important transduction factor in the MAPK pathway (28). The MAPK signaling pathway is one of
the most important signaling pathways in cells, it exists in most
cells and transmits extracellular stimuli into cells or even
nuclei. It has important functions in inducing proliferation,
differentiation, transformation, apoptosis and other biological
reactions (29). Extracellular
signals stimulate ERK and RAS, and RAS further activates RAF and
phosphorylates MAPK kinase (MEK1/MEK2). ERKs are translocated into
the nucleus, phosphorylated cytosolic proteins and certain nuclear
transcription factors, such as c-FOS, c-JUN, ELK-1, c-MYC and ATF2;
therefore, they are involved in the regulation of cell
proliferation and differentiation. ERKl/2 is activated by the MAPK
pathway by survival signals and promotes autophagy by PI3K via mTOR
(30). Shimada et
al(31) indicated that
endoplasmic reticulum stress may induce autophagy by activating the
MAPK signaling pathway. To determine the functions of BRAF, the
present study constructed a correlation network between BRAF and
the differentially expressed genes regulated by BRAF. GO-BP
enrichment analysis on these differentially expressed genes showed
that their major functions were associated with cell death and
apoptosis; therefore, regulation of BRAF may result in significant
effects on the mechanisms of cell death. Results of the present
study have shown that RIOK3 was closely associated with BRAF by
cluster analysis. RIOK3 was very important for maintaining normal
physiological function of cells, and RIOK3 depletion may result in
cell death or significant abnormality in physiological function.
Shan et al(32) reported
that RIOK3 may be involved in the regulation of apoptosis by
coordinating caspase-10, caspase-2 and NF-κB activation, indicating
that RIOK3 may be closely associated with cell death.
The present study has demonstrated by constructing
the kinase-kinase inhibitor regulatory network, that certain kinase
inhibitors had multiple targets, and may inhibit downregulated
genes (AKT4 and CDK2) and simultaneously inhibit upregulated genes
(BRAF), thus off-target side-effects may occur and should be
excluded. Of the 16 included specific kinase inhibitors functioning
on BRAF, sorafenib was previously confirmed to be capable of
regulating autophagy, while regorafenib had also been reported.
However, no report was available on the regulation of autophagy by
afatinib, selumetinib, PD318088, axitinib, TAK-733, GDC-0980,
GSK2126458, PLX-4720, AS703026, trametinib, GDC-0941 and
PF-04217903. Thus, these kinase inhibitors are potential targets
for investigating the regulation of autophagy in the future.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81370566).
References
|
1
|
Efeyan A, Zoncu R, Chang S, et al:
Regulation of mTORC1 by the Rag GTPases is necessary for neonatal
autophagy and survival. Nature. 493:679–683. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang ZB, Peng XZ, Chen SS, et al: High p53
and MAP1 light chain 3A co-expression predicts poor prognosis in
patients with esophageal squamous cell carcinoma. Mol Med Rep.
8:41–46. 2013.PubMed/NCBI
|
|
3
|
Yao F, Wang G, Wei W, Tu Y, Tong H and Sun
S: An autophagy inhibitor enhances the inhibition of cell
proliferation induced by a proteasome inhibitor in MCF-7 cells. Mol
Med Rep. 5:84–88. 2012.PubMed/NCBI
|
|
4
|
Amaravadi RK, Yu D, Lum JJ, et al:
Autophagy inhibition enhances therapy-induced apoptosis in a
Myc-induced model of lymphoma. J Clin Invest. 117:326–336. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yildirim MA, Goh KI, Cusick ME, Barabási
AL and Vidal M: Drug-target network. Nat Biotechnol. 25:1119–1126.
2007. View
Article : Google Scholar
|
|
7
|
Hopkins AL: Network pharmacology: the next
paradigm in drug discovery. Nat Chem Biol. 4:682–690. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dengjel J, Schoor O, Fischer R, et al:
Autophagy promotes MHC class II presentation of peptides from
intracellular source proteins. Proc Natl Acad Sci USA.
102:7922–7927. 2005.PubMed/NCBI
|
|
9
|
Matarrese P, Tinari A, Ascione B, et al:
Survival features of EBV-stabilized cells from centenarians:
morpho-functional and transcriptomic analyses. Age (Dordr).
34:1341–1359. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong MG, Pawitan Y, Magnusson PK and
Prince JA: Strategies and issues in the detection of pathway
enrichment in genome-wide association studies. Hum Genet.
126:289–301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Juan HF and Huang HC: Bioinformatics:
microarray data clustering and functional classification. Methods
Mol Biol. 382:405–416. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu T, Li Y, Gong L, et al: Multi-step
process of human breast carcinogenesis: a role for BRCA1, BECN1,
CCND1, PTEN and UVRAG. Mol Med Rep. 5:305–312. 2012.PubMed/NCBI
|
|
13
|
Scarlatti F, Bauvy C, Ventruti A, et al:
Ceramide-mediated macroautophagy involves inhibition of protein
kinase B and up-regulation of beclin 1. J Biol Chem.
279:18384–18391. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
15
|
Kanzawa T, Zhang L, Xiao L, Germano IM,
Kondo Y and Kondo S: Arsenic trioxide induces autophagic cell death
in malignant glioma cells by upregulation of mitochondrial cell
death protein BNIP3. Oncogene. 24:980–991. 2005. View Article : Google Scholar
|
|
16
|
Opipari AW Jr, Tan L, Boitano AE, Sorenson
DR, Aurora A and Liu JR: Resveratrol-induced autophagocytosis in
ovarian cancer cells. Cancer Res. 64:696–703. 2004. View Article : Google Scholar
|
|
17
|
Takeuchi H, Kondo Y, Fujiwara K, et al:
Synergistic augmentation of rapamycin-induced autophagy in
malignant glioma cells by phosphatidylinositol 3-kinase/protein
kinase B inhibitors. Cancer Res. 65:3336–3346. 2005.PubMed/NCBI
|
|
18
|
Jin S and White E: Role of autophagy in
cancer: management of metabolic stress. Autophagy. 3:28–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar
|
|
20
|
Carew JS, Nawrocki ST, Kahue CN, et al:
Targeting autophagy augments the anticancer activity of the histone
deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug
resistance. Blood. 110:313–322. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hashimoto K and Sakagami H: Induction of
apoptosis by epigallocatechin gallate and autophagy inhibitors in a
mouse macrophage-like cell line. Anticancer Res. 28:1713–1718.
2008.
|
|
22
|
Amaravadi RK and Thompson CB: The roles of
therapy-induced autophagy and necrosis in cancer treatment. Clin
Cancer Res. 13:7271–7279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nazio F, Strappazzon F, Antonioli M, et
al: mTOR inhibits autophagy by controlling ULK1 ubiquitylation,
self-association and function through AMBRA1 and TRAF6. Nat Cell
Biol. 15:406–416. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang ZJ and Wang LX: Phosphorylation: a
molecular switch in opioid tolerance. Life Sci. 79:1681–1691. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pelkmans L, Fava E, Grabner H, et al:
Genome-wide analysis of human kinases in clathrin- and
caveolae/raft-mediated endocytosis. Nature. 436:78–86. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bellodi C, Lidonnici MR, Hamilton A, et
al: Targeting autophagy potentiates tyrosine kinase
inhibitor-induced cell death in Philadelphia chromosome-positive
cells, including primary CML stem cells. J Clin Invest.
119:1109–1123. 2009. View
Article : Google Scholar
|
|
27
|
Liu D, Liu Z, Jiang D, Dackiw AP and Xing
M: Inhibitory effects of the mitogen-activated protein kinase
kinase inhibitor CI-1040 on the proliferation and tumor growth of
thyroid cancer cells with BRAF or RAS mutations. J Clin Endocrinol
Metab. 92:4686–4695. 2007. View Article : Google Scholar
|
|
28
|
Derdas SP, Soulitzis N, Balis V, Sakorafas
GH and Spandidos DA: Expression analysis of B-Raf oncogene in
V600E-negative benign and malignant tumors of the thyroid gland:
correlation with late disease onset. Med Oncol. 30:3362013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu Y, Zhang J and Dong WG:
Indole-3-carbinol (I3C)-induced apoptosis in nasopharyngeal cancer
cells through Fas/FasL and MAPK pathway. Med Oncol. 28:1343–1348.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Thorburn A: Apoptosis and autophagy:
regulatory connections between two supposedly different processes.
Apoptosis. 13:1–9. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shimada Y, Kobayashi H, Kawagoe S, et al:
Endoplasmic reticulum stress induces autophagy through activation
of p38 MAPK in fibroblasts from Pompe disease patients carrying
c.546G>T mutation. Mol Genet Metab. 104:566–573. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shan J, Wang P, Zhou J, Wu D, Shi H and
Huo K: RIOK3 interacts with caspase-10 and negatively regulates the
NF-kappaB signaling pathway. Mol Cell Biochem. 332:113–120. 2009.
View Article : Google Scholar : PubMed/NCBI
|