Introduction
Dissemination of the primary tumor is the main cause
of death in breast cancer patients. The dissemination process
involves a series of distinct steps in which tumor cells migrate
from the primary tumor, spread through lymphatic and blood vessels,
and establish secondary tumors at distant sites (1). Accumulating evidence implicates
transforming growth factor beta (TGF-β) cytokines in the control of
tumor progression and dissemination. TGF-β cytokines repress tumor
growth at early phases of tumorigenesis, in part by inhibiting
cell-cycle progression and inducing cell death, but they are also
able to promote tumor invasion and metastatic dissemination in
late-stage tumors (2,3). Pro-tumorigenic TGF-β activity has been
linked to the induction of epithelial-mesenchymal transition (EMT),
cell motility and matrix-degrading enzymes. In addition, TGF-β may
promote tumor progression by repressing the immune response
(4), and by stimulating
angiogenesis via upregulation of the pro-angiogenic factors VEGF
and matrix metalloproteinase MMP-9 (5–7).
Blockade of the soluble TGF-β ligand impairs tumor invasion and
metastasis, further supporting the active role of TGF-β in cancer
progression (8,9). However, TGF-β receptors and Smad
transcription factors are frequently altered in cancer, and this
has been associated with poor prognosis (10,11).
The dual role of TGF-β in cancer complicates the development of
therapies targeting TGF-β (12).
Thus, unraveling the intracellular pathways and factors involved in
TGF-β pro-oncogenic activities is critical for the development of
putative anticancer TGF-β therapies.
TGF-β signal transduction is initiated by binding
TGF-β cytokines to TGF-β type I and type II receptors (TβRI and
TβRII), a complex of transmembrane glycoproteins with
serine-threonine kinase activity (3). Upon ligand binding, TβRII
phosphorylates TβRI, thus activating the TβRI kinase, which in turn
phosphorylates and activates Smad transcription factors.
Receptor-associated Smad2 and Smad3 (R-Smads) together with the
co-mediator Smad4 translocate to the nucleus, where they regulate
the transcription of TGF-β target genes. In addition, it has been
shown that TGF-β can activate MAP kinases as well as PI3K-Akt
signaling, contributing to the TGF-β effects on malignant tumor
cells (3).
Cell adhesion, motility and invasion which are
crucial for the metastatic process, depend on actin cytoskeleton
(13). The actin cytoskeleton
organization and dynamics are controlled by small-GTP-binding
proteins, protein kinases and phosphatases, which regulate a
multitude of actin cytoskeleton components, such as
actin-polymerizing proteins (Arp2/3 complex, formins),
actin-stabilizing proteins (α-actinin, filamins, tropomyosins),
actin-associated proteins (HSP27, MLC2), and actin-severing
proteins (gelsolin, cofilin). TGF-β promotes the disruption of
cell-cell contacts either by altering the actin cytoskeleton
(14) or by downregulating the
expression of E-cadherin (15).
Furthermore, TGF-β may positively or negatively control cell
motility and matrix-degrading enzymes via tropomyosin-stabilized
actin stress fibers (14,16). In carcinoma cells with low levels of
tropomyosins, the upregulation of matrix-degrading enzymes, such as
matrix metalloproteinases MMP-2 and MMP-9, participates in TGF-β
induction of invasive behavior (7,17). MAP
kinases have been involved in TGF-β regulation of the actin
cytoskeleton and cell motility (3,14).
Furthermore, oncogenic Ras-MAPK signaling interferes with the
induction of EMT by TGF-β-Smad pathway (18), indicating that MAP kinase signaling
may affect the outcome of TGF-β responses.
We have previously shown that highly invasive and
metastatic murine mammary adenocarcinoma LM3 cells express TGF-β
cytokines and receptors, and that they respond to TGF-β with
enhanced invasion and secretion of matrix-degrading enzymes
(19). The present study supports
an autocrine role of TGF-β signaling in tumor progression, and
explores mediators of the pro-oncogenic TGF-β activities in LM3
cells. Expression of kinase-inactive TGF-β receptors decreased both
basal and TGF-β-induced invasion. Furthermore, the evaluation of
signal-transduction mediators showed that p38MAPK and MEK
contribute to TGF-β stimulation of cell motility and invasion.
Experiments in syngeneic BALB/c mice showed that the expression of
kinase-inactive TGF-β receptors decreased the tumorigenic potential
of LM3 cells in vivo. Our study provides evidence for a role
of MAP kinases in the pro-oncogenic activities of TGF-β in mammary
tumor cells, including the regulation of the actin cytoskeleton,
cell motility and invasion.
Materials and methods
Antibodies and other reagents
Human recombinant TGF-β1 protein was obtained from
R&D Systems (#240-B). The following antibodies were used: Smad2
(34G6, #3107), phospho-Smad2 (#3101), phospho-ERK1/2 (#9101),
phospho-p38MAPK (#9215), phospho-MLC2 (#3675), from Cell Signaling
Technology; Smad4 for immunofluorescence (B-8, #sc-7966, Santa Cruz
Biotechnology); Smad4/DPC4 for immunoblotting (BD Biosciences);
β-actin (AC-40, #A4700), tropomyosin (TM311, #T-2780),
anti-mouse/TRITC (#T-6653), anti-sheep/HRP (#A-3415), from Sigma;
anti-rabbit/HRP (PI-1000) or anti-mouse/HRP (#PI-2000), from Vector
Laboratories; BB-94, from British Biotech Pharmaceuticals. Alexa
Fluor phalloidin was from Molecular Probes (#A-12379 or A-12380).
Inhibitors for p38MAPK (SB202190, #559388), MEK1/2 (PD98059,
#513000, or U0126, #662005), and Raf1
(5-iodo-3-[(3,5-dibromo-4-hydroxyphenyl)methylene]-2-indolinone,
#553008) were from Calbiochem. The dual luciferase reporter assay
system was from Promega (#E1910). The Arp2/3 complex kit,
containing Arp3 antibody, was from Cytoskeleton, Inc. (#BK009).
Cell lines and treatments
The mouse mammary hormone-independent adenocarcinoma
LM3 cell line has been previously described (20). Non-tumorigenic murine mammary gland
(NMuMG) cells were from ATCC (CRL-1636, ATCC). Mv1Lu cells were a
gift from Dr Harold Moses. NMuMG cells and Mv1Lu cells were used as
control for TGF-β response. LM3 and NMuMG cells were grown in
medium supplemented with fetal bovine serum (5% FBS MEM or 10% FBS
DMEM, respectively) with the addition of 80 μg/ml gentamycin. Mv1Lu
cells were grown in 10% FBS DMEM supplemented with 3.7 g/l sodium
bicarbonate. All cells were kept at 37°C in a humidified atmosphere
with 5% CO2. Cells in exponential growth phase were
treated with 2 ng/ml TGF-β1. For some assays, 1 ng/ml TGF-β1 was
used. Kinase inhibitors were added to cells 1 h prior to TGF-β
treatment, at the following doses: 10 μM SB202190; 5 μM PD98059; 5
μM U0126, and 5 μM c-Raf1 inhibitor.
Retroviral infection of cells
Retroviral vectors used in the study are described
in (21,22). Briefly, these vectors encode: EGFP
in pBMN-IRES-EGFP (control); dominant-negative TβRI-K232R mutant
and EGFP in pBMN-TβRI-K232R-IRES-EGFP; dominant-negative
TβRII-K277R and EGFP in pGABE-TβRII-K277R. Amphotropic retroviruses
were prepared as described in (21). Cells were infected with each
retrovirus using polybrene (10 μg/ml, Sigma), and EGFP-positive
cells were selected three consecutive times by FACS in order to
obtain cell populations with similar levels of EGFP expression.
Transcriptional analysis
Exponentially growing cells were transfected with
0.1 μg/ml of the following plasmids: pSBE-Lux, containing 12
repeats of Smad-binding sites (generously provided by J. M.
Gauthier, Laboratoire Glaxo Wellcome, Les Ulis Cedex, France), or
p3TP-Lux, containing 3 AP-1 sites and a fragment of the human PAI-1
promoter (23). Cells were
co-transfected with 0.002 μg/ml pCMV-Renilla luciferase (Promega)
using FuGENE6 (Roche Molecular Biochemicals), according to the
manufacturer’s protocol. Cells were incubated in 0.5% FBS for 6 h
prior to 1 ng/ml TGF-β1 treatment for 16 h. Luciferase activity in
cell lysates was determined by the Dual Luciferase Reporter Assay
system, according to the manufacturer’s protocol, using a Monolight
2010 luminometer (Analytical Luminiscence Laboratory). Firefly
luciferase activity was normalized to Renilla activity, and
expressed as luciferase relative units (LRU).
RT-PCR analysis
Total RNA was prepared as previously described
(24). RT-PCR was performed using
One-Step RT-PCR system (Invitrogen). Amplification products were
separated on 1% agarose gels and visualized with ethidium bromide.
Primer sequences were mouse MMP-9 (NM_ 013599.2), forward
CGTCGTGATCCCCACTTACT and reverse, AGGAAGACGAAGGGGAAGAC;
α-tropomyosin (NM_024427.2): forward, GCTGGTGTCACTGCAAAAGA and
reverse, CCTGAGCCTCCAGTGACTTC; mouse β-actin (NM_007393): forward,
GCTGGTCGTCGACAACGGCTC and reverse, CAAACATGATCTGGGTCATCTTTTC.
Western blot analysis
Cells were treated with TGF-β1 for different periods
of time, and then lysed in buffer containing 20 mM Tris, pH 7.4,
137 mM NaCl, 1% NP-40, 10% glycerol, 20 mM NaF, 1 mM Na
orthovanadate, 1 mM PMSF, 2 μg/ml aprotinin, and 2 μg/ml leupeptin.
For signal transduction studies, cells were serum-starved for 4 h
prior to treatment with TGF-β. Immunoblot analysis of protein
extracts was performed as pre-viously described (19).
Actin cytoskeleton study
Cells were grown on glass coverslips for 24 h prior
to treatment with TGF-β1, and then fixed with 4% paraformaldehyde,
permeabilized with 0.05% Triton X-100 in PBS for 15 min, and
stained as described before (21).
Actin filaments (F-actin) were visualized with Alexa Fluor
phalloidin. Fluorescence images were captured using a Zeiss
Axiophot upright microscope and a Nikon TE2000-E inverted
microscope.
Affinity purification of Arp2/3 complex
activity
Activation of the Arp2/3 complex was examined using
a pull-down assay kit from Cytoskeleton, Inc., following the
manufacturer’s protocol. Briefly, cells were incubated for 4 h in
serum-free medium prior to treatment with TGF-β1, and then lysed.
Total proteins were incubated with either GST-VCA beads, in order
to precipitate active Arp2/3 complex, or with GST beads alone as a
control. Pellets containing the Arp2/3 complex were analyzed for
Arp2/3 activity by immunoblotting with anti-Arp3 antibody.
Supernatants were also examined, as a control.
Zymography for metalloproteinase (MMP)
activity
MMP-9 activity was measured by quantitative gelatin
zymography of conditioned media (CM) from cells treated with or
without TGF-β1, as previously described (19). Gelatinolytic bands were analyzed by
the GS-700 densitometer and the Molecular Analyst™ software
(Bio-Rad), and OD values were used as a measurement of total
cellular protein content.
Cell migration
Cell migration was studied in a wound healing assay,
as previously described (19).
Briefly, cells were cultured until confluency and wounds of ~400 μm
width were made on the monolayers with a plastic tip. Then, cells
were incubated with TGF-β1 for another 16 h. Photographs of the
same area were taken at ×400 magnification to determine wound
coverage due to cellular motility. Images were obtained and
evaluated by densitometry, using Image-Pro Plus 5.1 software.
Invasion assay
Cell invasion assays were performed using
Matrigel-coated Transwell chambers (8 μm filter pore, Corning), as
previously described (19). Cells
were seeded onto Transwell chambers and incubated with or without
TGF-β1 for 16 h (cytokine added in the plate well). Cells on the
bottom surface of the filter (those who had traversed the filter)
were stained with Hoechst 33258 (10 μg/ml, Sigma), and counted
under fluorescence microscope at ×600 magnification.
Tumor growth and metastatic ability
LM3 stably cells expressing TβRI-K232R, TβRII-K277R,
as well as control cells expressing EGFP, were harvested at the
exponential growth phase with trypsin/EDTA, washed and resuspended
in MEM. Cells (2×105) in 0.2 ml MEM were inoculated
subcutaneously into the flank of syngeneic BALB/c mice (10 female
mice per group). Tumor latency was defined as the time between
inoculation and detection of tumors by palpation. Tumor size was
measured with a caliper, in orthogonal directions, every 3 days.
Animals were euthanized on day 40 of tumor onset, time at which
spontaneous superficial lung metastases are detected in this tumor
model. The condition of every major organ was observed, the lungs
were removed, fixed with Bouin’s solution, and examined under a
magnifier to record the number and size of metastatic foci. Both
tumors and lungs were analyzed ex vivo under fluorescence
microscope to determine the presence of EGFP-positive cells.
Statistical analysis
In general, all experiments were performed at least
three times, and the mean value of triplicates in each comparable
group was analyzed using the Student’s t-test or the
ANOVA-Scheffé’s test. Differences in metastatic ability between the
groups were investigated using the non-parametric Mann-Whitney U
test. Results were considered of biological significance when
p<0.05.
Results
Expression and activation of Smads and
MAPK pathways
The regulation of MAP kinase and Smad pathways by
TGF-β in the mammary adenocarcinoma LM3 cells was evaluated by
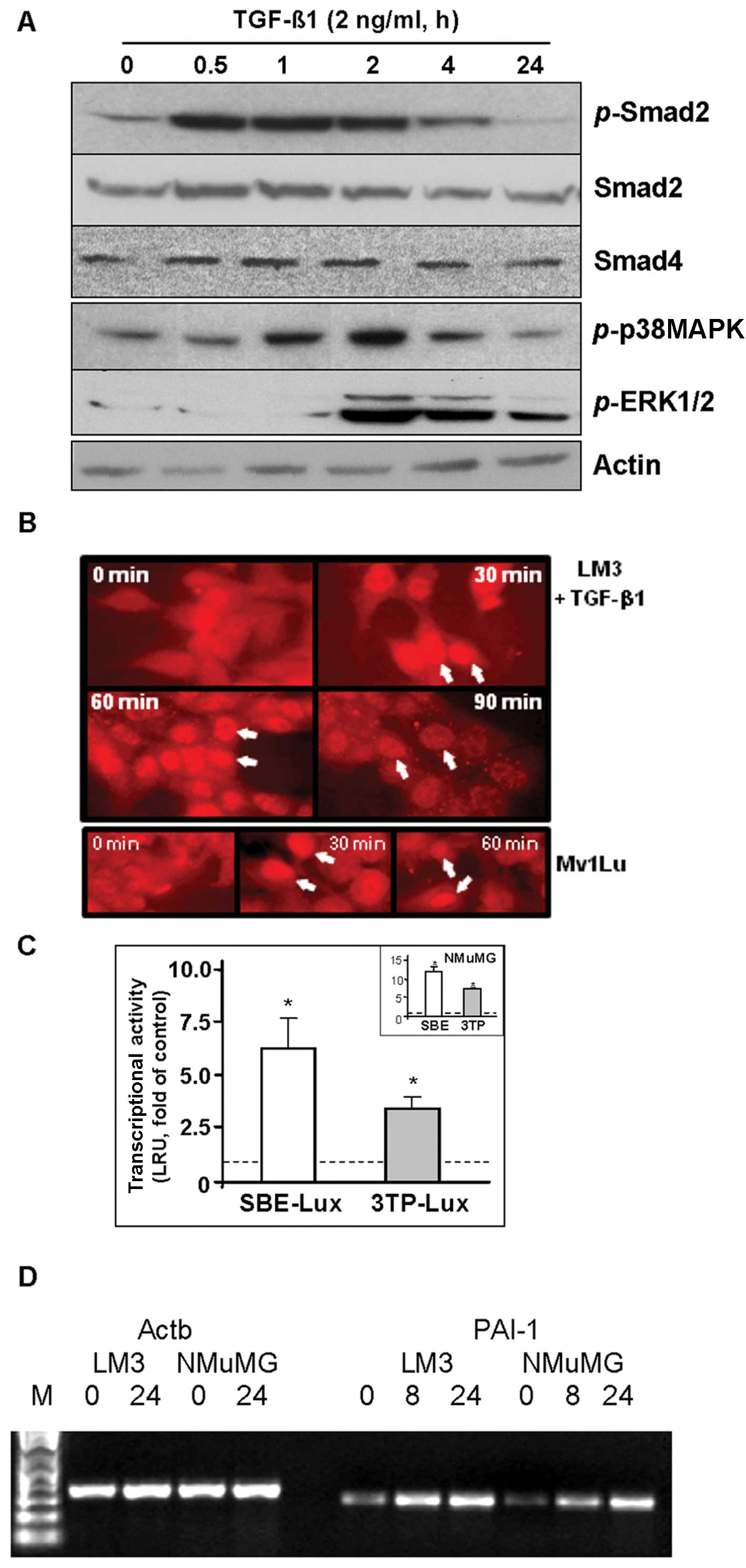
immunoblotting and immunofluorescence. Immunoblot analysis revealed
that TGF-β treatment increased phosphorylation of Smad2 between 30
min and 4 h, while total levels of Smad2 and Smad4 were not changed
for up to 24 h treatment (Fig. 1A).
In Fig. 1B, immunofluorescence
showed nuclear translocation of Smad4 at 30 min of TGF-β treatment,
indicating activation of the Smad complex in response to TGF-β.
Concomitantly, TGF-β induced the phosphorylation of p38MAPK and
ERK1/2 (Fig. 1A).
TGF-β transcriptional responses were evaluated using
a luciferase reporter containing 12 repeats of Smad-binding sites
(SBE-Lux) and a reporter containing a fragment of the PAI promoter
and 3 repeats of AP1 sites (3TP-Lux) in LM3 cells. NMuMG cells,
which display a strong regulation by both reporters, were used as
the control (14). As shown in
Fig. 1C, TGF-β1 significantly
increased the activity of both reporters in LM3 cells. In addition,
RT-PCR analysis showed that TGF-β treatment upregulated endogenous
PAI-1 mRNA levels (Fig. 1D).
Together, these findings demonstrate that LM3 cells respond to
TGF-β with activation of the Smad and MAPK signaling pathways. As
evidenced by the modulation of downstream targets, such as PAI-1,
these pathways are functional.
TGF-β signaling enhances LM3 cells
invasive ability
Our previous studies have shown that the LM3 cell
line expresses TGF-β cytokines and TGF-β receptors, and is able to
respond to TGF-β with enhanced invasion in vitro(19). Here, LM3 cells were transduced with
retroviral vectors to express dominant negative (kinase-inactive)
forms of TGF-β type I and type II receptors (TβRI-K232R and
TβRII-K277R, respectively) by retroviral infection using
bicistronic EGFP-encoding vectors (21). These kinase-inactive receptors exert
dominant-negative effects on TGF-β signaling (7,21). An
EGFP-encoding vector was used as a control.
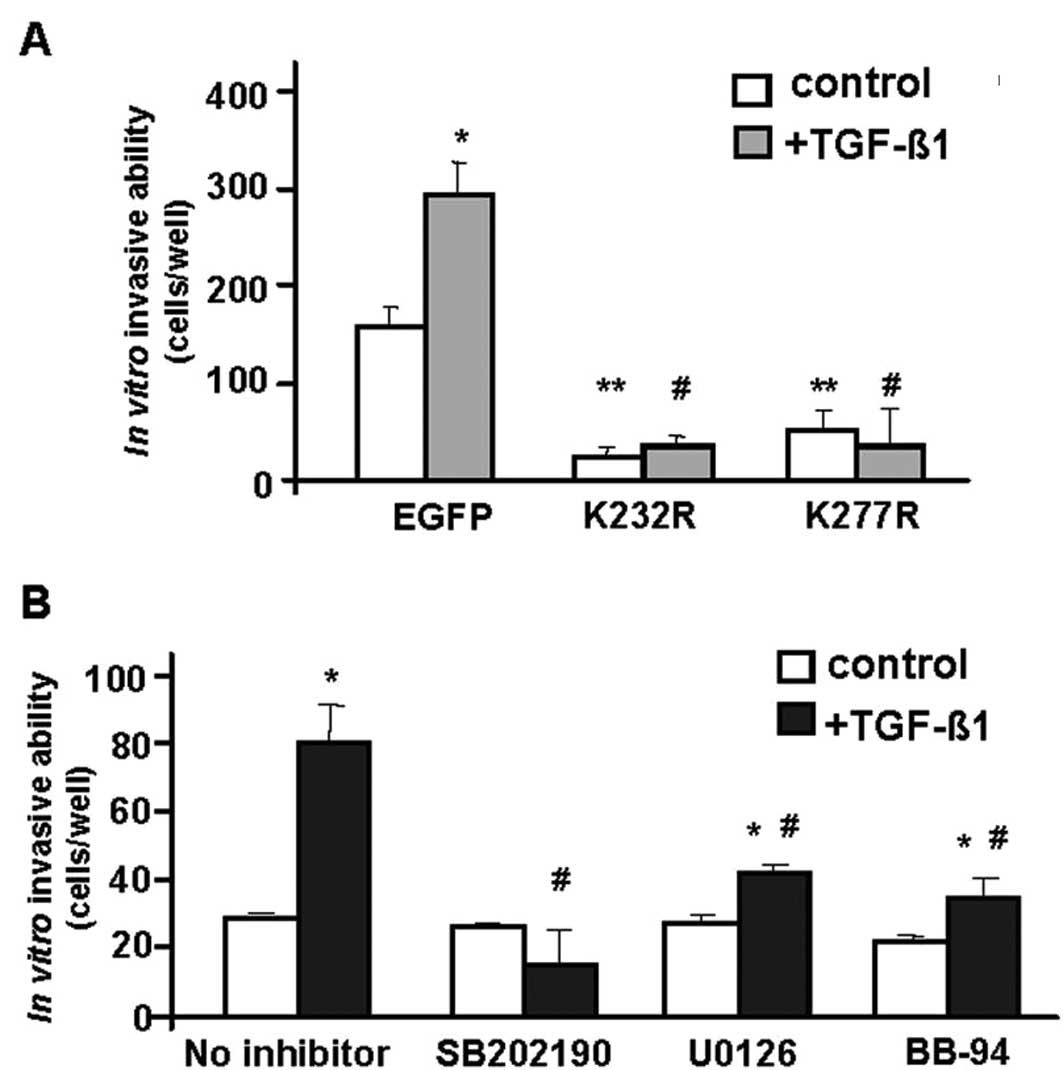
The invasive ability of cells expressing TGF-β
kinase-inactive receptors was significantly impaired in the absence
of exogenous ligand (compare to EGFP alone vector), indicating that
autocrine TGF-β signaling could contribute to the basal invasive
properties of LM3 cells (Fig. 2A).
In addition, while TGF-β1 stimulated the number of LM3-EGFP cells
invading the Matrigel-coated chambers by ~2-fold (Fig. 2A), kinase-inactive receptors blocked
TGF-β-induced invasion in LM3 cells.
This experiment was also performed in the presence
of pharmacological inhibitors of cell signaling pathways.
Inhibition of p38MAPK with SB202190, and of MEK-ERK signaling with
U0126, blocked the TGF-β1-induced invasive ability in LM3 cells
(Fig. 2B). Similar results were
obtained with a metalloproteinase inhibitor, BB-94 (Fig. 2B). These findings suggest that the
p38MAPK as well as the MEK-ERK signaling pathways are required for
TGF-β regulation of invasiveness in mammary tumor cells. Moreover,
the results indicate a putative synergistic role with
metalloproteinases.
TGF-β modulation of matrix
metalloproteinase 9/gelatinase-B (MMP-9)
The invasive ability of cells depends on cell
motility and on the activity of matrix-degrading enzymes. Our
previous studies have demonstrated that LM3 cells express MMP-9 and
that TGF-β markedly enhances the secreted MMP-9 activity (19). Here, we explored the mechanism
underlying this TGF-β effect.
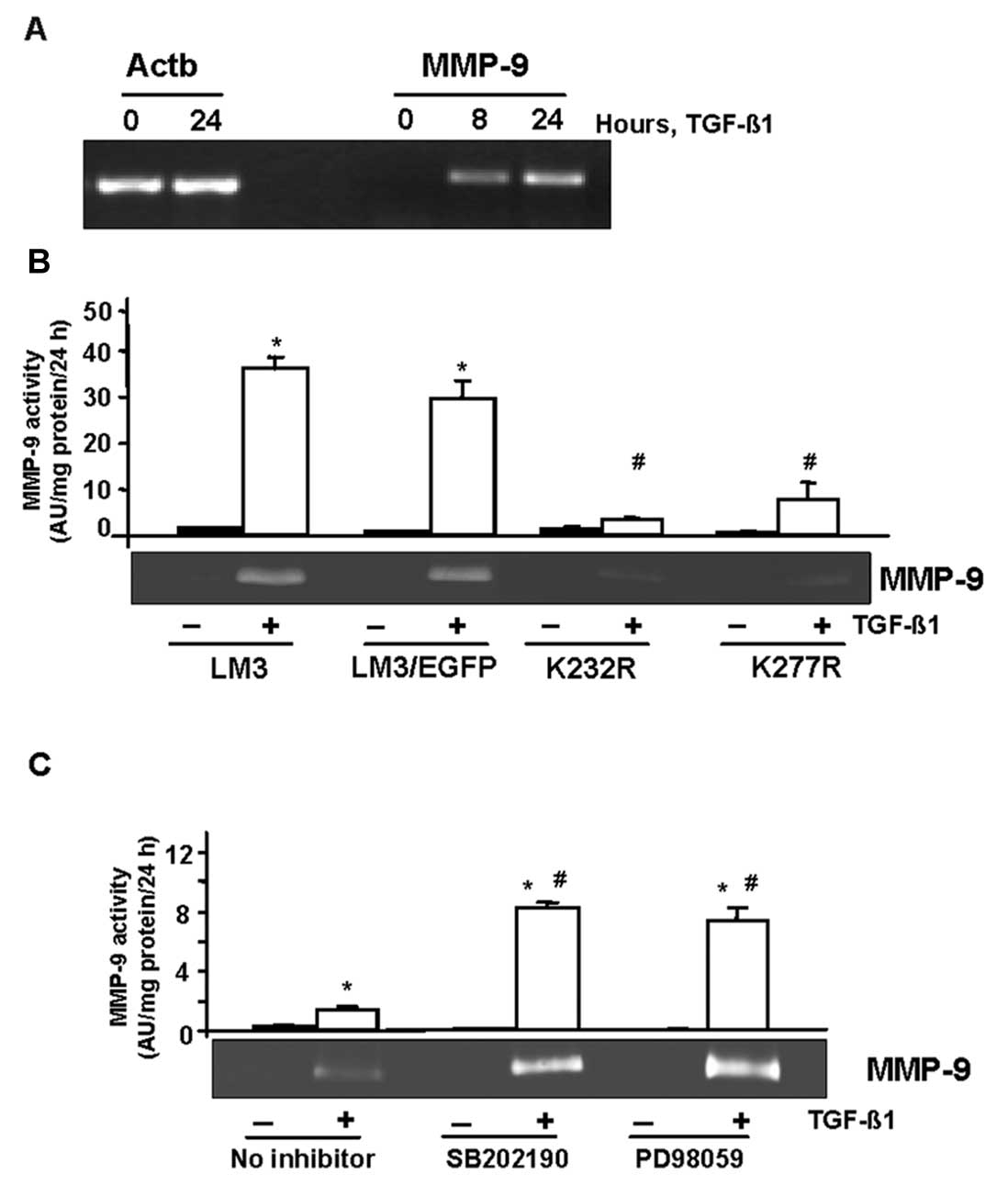
RT-PCR analysis showed that TGF-β1 upregulated MMP-9
mRNA level within 8 h of treatment (Fig. 3A). Gelatin zymography assays
revealed that dominant-negative TGF-β receptors blocked the
stimulation of secreted MMP-9 by TGF-β in LM3 and LM3-EGFP
(control) cells (Fig. 3B). In order
to identify signaling pathways involved in MMP-9 induction by
TGF-β, the same experiment was performed in the presence of kinase
inhibitors. Surprisingly, we observed that both the p38MAPK
inhibitor SB202190 and the MEK inhibitor PD098059 enhanced the
TGF-β-induced MMP-9 activity (Fig.
3C), suggesting the antagonic effects between the p38MAPK and
MEK pathways and the TGF-β pathways on MMP-9 secretion by LM3
cells.
Modulation of cell motility by TGF-β
requires MAP kinases
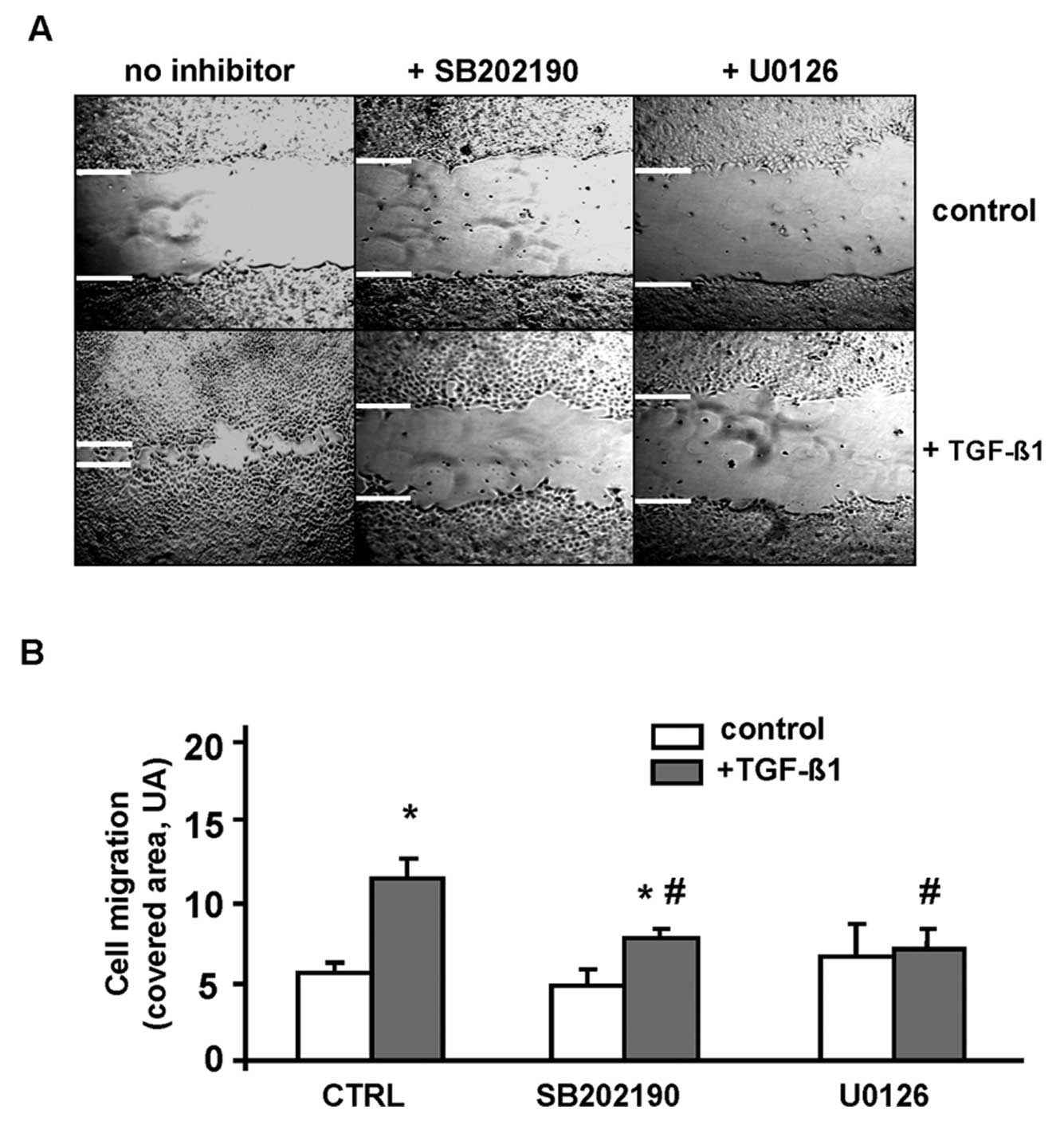
The effect of TGF-β on cell motility was evaluated
by wound assay. Incubation of LM3 cells with 2 ng/ml TGF-β1
significantly accelerated the healing of wounds in cell monolayers,
indicating that TGF-β1 enhanced cell motility (Fig. 4A). To assess the role of MAPK
signaling in TGF-β-induced motility in these cells, wound assays
were performed in the presence of p38MAPK and MEK inhibitors. The
inhibition of either kinase significantly abrogated TGF-β induction
of wound healing (Fig. 4B),
suggesting that both p38MAPK and MEK-ERK pathways are involved in
the regulation of LM3 cell motility by TGF-β1.
Effect of TGF-β on LM3 cytoskeleton
The regulation of cell motility by TGF-β in normal
and tumor cells has been linked to EMT, which involves the
disruption of cell-cell junctions as well as actin remodeling
(15) and, in some cases, it also
involves actin-stabilizing proteins such as high molecular-weight
(HMW) tropomyosins. Therefore, in order to investigate the
mechanism of TGF-β-mediated cell migration in LM3 cells, we
assessed the regulation of actin cytoskeleton and
HMW-tropomyosins.
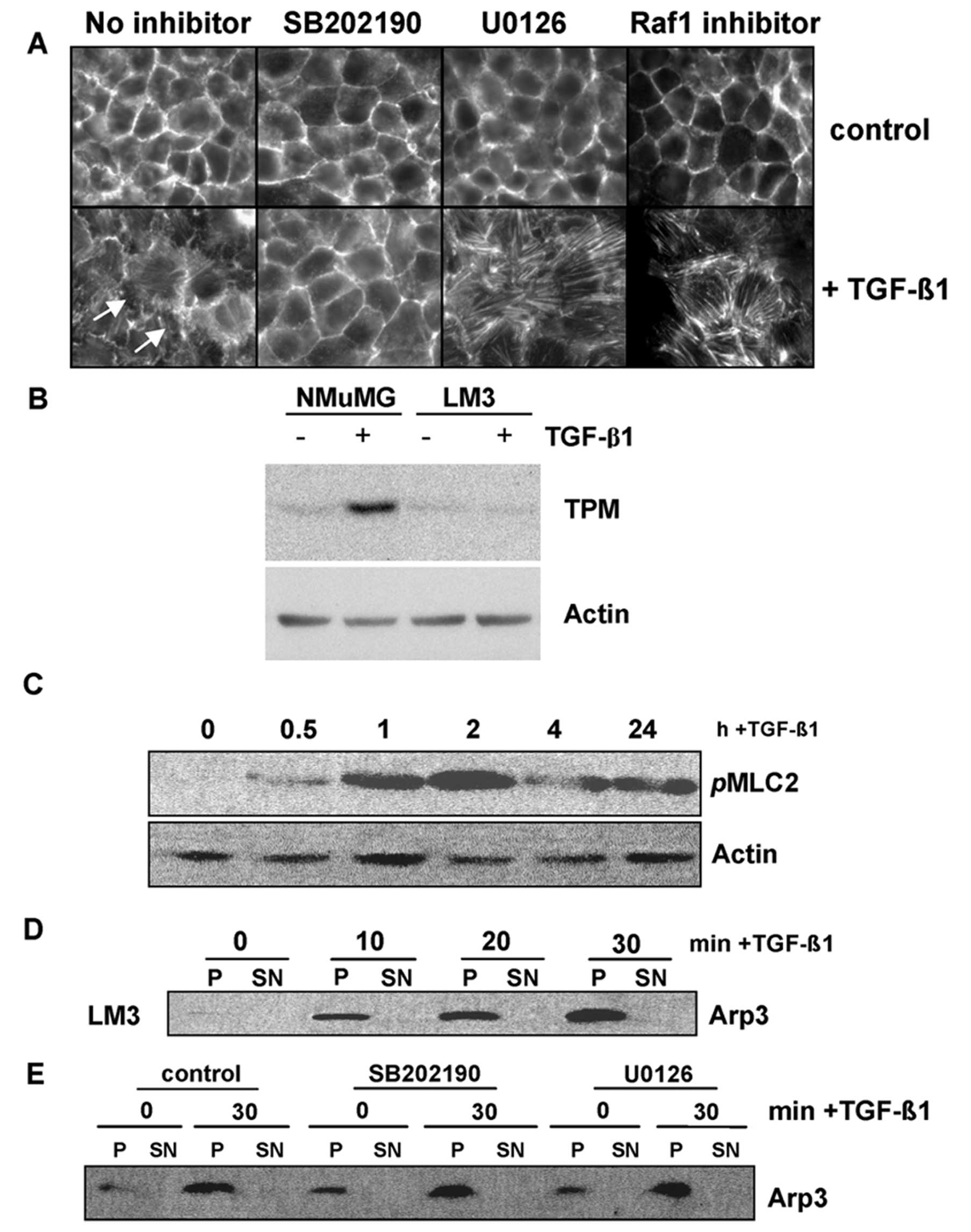
Immunofluorescence microscopy showed the
organization of actin filaments in adhesion belt-like structures in
control LM3 cells, typical of epithelial cells (Fig. 5A). Treatment with TGF-β1 for 24 h
disrupted these structures and increased linear actin filaments
(Fig. 5A). Co-incubation with a
p38MAPK inhibitor blocked actin remodeling in response to TGF-β,
whereas co-incubation with a MEK inhibitor (U0126) markedly
enhanced the TGF-β-induced increase of actin stress fibers
(Fig. 5A). Similar results were
obtained with a Raf1 inhibitor, corroborating that the inhibition
of another molecule of the Ras/MAPK/ERK pathway positively
stimulates the formation of stress fibers in the presence of
TGF-β.
On the other hand, immunoblotting showed that TGF-β
did not modulate the expression of HMW-tropomyosins (TPMs),
contrasting our findings in NMuMG cells, employed as a positive
control (Fig. 5B). Moreover, the
mRNA levels of TPM-α and TPM-β genes were not regulated by TGF-β in
LM3 cells (data not shown).
We further assessed TGF-β modulation of the actin
cytoskeleton by analyzing the activity of myosin II regulatory
light chain (MLC2), which regulates actomyosin contractility and
cell migration (13). The
immunoblots in Fig. 5C show the
induction of MLC2 phosphorylation and activation within 30 min of
TGF-β treatment, which persisted for 24 h, indicating that TGF-β
increased actomyosin contractility in LM3 cells.
Arp2/3 protein complex mediates de novo actin
filament nucleation during polymerization of branched actin
structures (13). We thus analyzed
whether the function of this complex is affected by TGF-β. The
Arp2/3 activity complex was assessed by a pull-down assay using
GST-VCA fusion proteins in which the C-terminal VCA domain of WASP
was linked to GST. A conserved VCA domain of WASP contains a
verprolin homology segment (V), a cofilin homology segment (C) and
an acidic region (A). This domain interacts and activates the
Arp2/3 complex (13). We found that
TGF-β increased the association of the Arp2/3 complex with the VCA
domain within 10–30 min of treatment in LM3 cells (Fig. 5D). This response was not affected by
p38MAPK or MEK kinase inhibitors (Fig.
5E).
These findings suggest that the mechanism of TGF-β
stimulation of cell motility in LM3 cells may involve actin
remodeling, actomyosin contractility, and Arp2/3 complex
activity.
TGF-β signaling in tumor development and
progression
To examine the role of TGF-β signaling pathway in
tumor growth and spontaneous metastasis, we employed syngeneic
BALB/c mice. LM3 cells expressing TβRI-K232R (LM3/K232R),
TβRII-K277R (LM3/K277R) or EGFP-control (LM3/EGFP) were injected
subcutaneously into the flank of syngeneic BALB/c mice. All three
groups of mice developed palpable tumors within a week of tumor
cell inoculation, and showed comparable tumor incidence (90–100%)
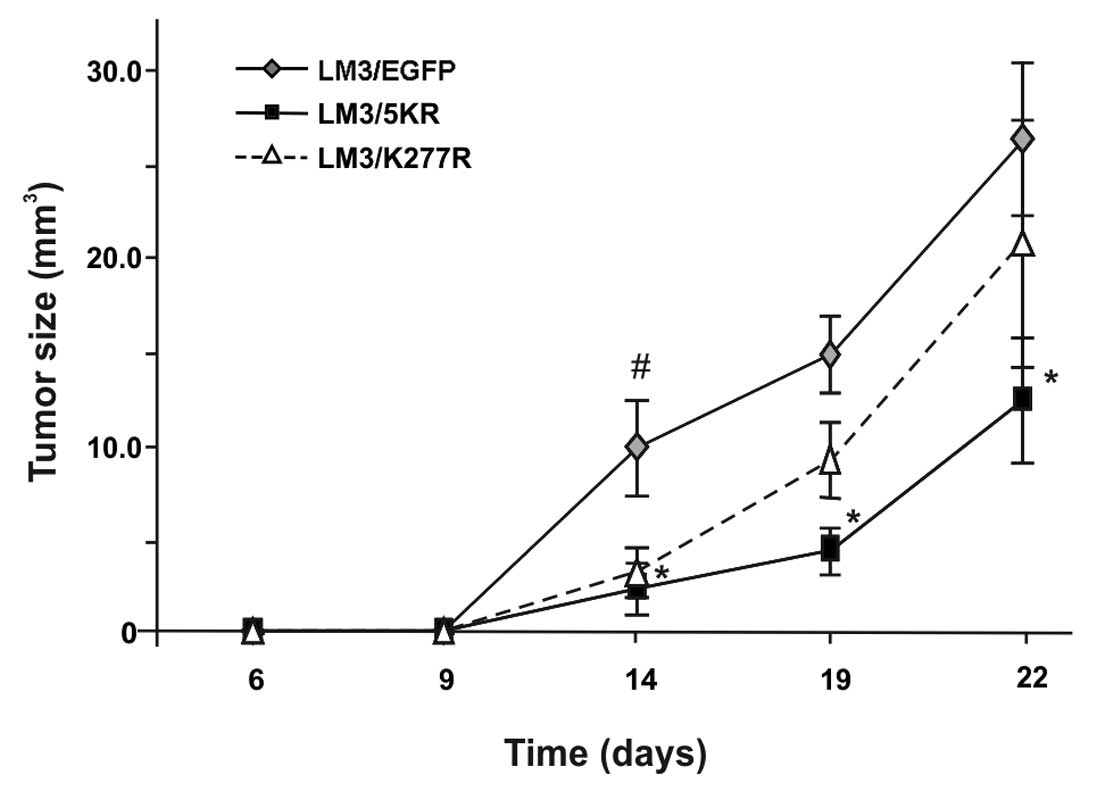
as well as latency period. However, we observed that tumor growth
during the first ~25 days was significantly reduced in both
dominant-negative TGF-β receptor groups (Fig. 6). We continued our observations on
tumor growth until day 40, at which time the animals were
sacrificed. The dominant negative TβR bearing-LM3 tumors were
smaller in size than the control tumors (LM3/EGFP), although the
values did not reach statistical significance (data not shown). In
addition, no difference was observed in the number of lung foci at
the latest time point analyzed (40 days after tumor cell
inoculation). The median number of metastases were 5 (range, 2–32)
in the control group; 5 (0–11) in the K232R group, and 9 (0–56) in
the K277R group.
Discussion
In our model of murine mammary LM3 adenocarcinoma
cells, TGF-β triggered the activation of Smad and non-Smad
signaling pathways, with the upregulation of downstream targets,
such as PAI-1, confirming the functionality of the TGF-β/TβR system
in these cells. We demonstrate that TGF-β enhances LM3 cell
motility, inducing actin remodeling, actomyosin contractility, and
the activation of the Arp2/3 complex. Our results show that the
non-Smad downstream effectors p38MAPK and MEK/ERK regulate actin
remodeling and cell motility, but do not contribute to the
regulation of the Arp2/3 complex. In addition, TGF-β also induced
the matrix-degrading ability of LM3 cells mediated by MMP-9
secretion. Expression of kinase-inactive dominant-negative TGF-β
receptors markedly reduced the invasive potential of LM3 cells,
indicating that an autocrine TGF-β signaling loop may contribute to
the invasive phenotype. Moreover, we show that TGF-β signaling may
be a determinant of initial tumor growth in vivo.
In order to invade and spread to distant organs,
carcinoma cells must lose polarity, cell-cell contacts, and acquire
fibroblastic-like properties. In this process of
epithelial-mesenchymal transition (EMT), cells become highly motile
and invasive, which allows survival in an anchorage-independent
environment and provides them with stem cell-like properties. The
activity of proteases such as MMPs, which leads to the degradation
of extracellular matrix proteins, may render tumor cells with a
migratory and invasive advantage. Our results show that TGF-β
presents pro-invasive and pro-migratory effects on LM3 cells.
Similar results were observed in other murine and human mammary
carcinoma cell lines (8,25–27).
It has been established that even though Smad
signaling is required for the majority of TGF-β-mediated signals,
not all responses to TGF-β are solely dependent on the Smad
complex. In fact, the TGF-β response implies alternative signaling
modules acting in parallel with Smads. As an example, it was
demonstrated that TGF-β signaling is engaged in RhoA-ROCK
signaling, required for the regulation of cell shape and movement
(28,29). In addition, TβRI activates ERK-MAPK
signaling through direct phosphorylation of Shc, and TβRII can
signal independently of TβRI by directly phosphorylating Par6, an
EMT-associated biomarker that enhances proliferation, migration and
invasiveness in cells in vitro(30). Thus, the signaling pathways
triggered by TGF-β/TβR signaling are pliable and diverse. Here, we
demonstrate that intracellular signaling by p38MAPK and MEK is
involved in both the pro-invasive and the pro-migratory activities
of TGF-β in LM3 cells. Dumont et al have also found that
these two signaling pathways mediate the pro-invasive effect of
TGF-β on the human ER-negative MDA-MB-231 cell line (26).
On the other hand, we found that p38MAPK and MEK
have distinct effects on TGF-β-induced disruption of the epithelial
actin cytoskeleton and cell-cell junctions, and on the formation of
actin fibers, which are key aspects of EMT (15). Our findings indicate that p38MAPK is
required for the disruption of epithelial organization of actin
filaments and cell-cell junctions. This result is in agreement with
previous studies showing that p38MAPK is required for the
TGF-β-induced EMT process, and for actin remodeling on the
non-tumorigenic NMuMG cells (21,31).
Surprisingly, we found that blocking MEK1/2 significantly increased
the formation of stress fibers, similarly to a Raf1 inhibitor, a
MEK upstream molecule. These results indicate that Raf-MEK-ERK
signaling suppresses TGF-β-induced actin stress fibers formation in
LM3 cells. Other authors, employing a similar approach,
demonstrated that TGF-β1 induces the activation of the ERK
signaling pathway in NMuMG cells, which is required for
TGF-β1-mediated EMT in vitro(32). Therefore, it appears that the
signaling pathways activated by the TβR are highly dependent on
cell properties.
Stimulation of cell motility by TGF-β is a complex
process involving several factors. In our model, in addition to the
disruption of the actin filaments architecture, TGF-β increased
phosphorylation of the regulatory subunit of the actomyosin
contractility complex MLC2, thus enhancing actomyosin
contractility, and ultimately contributing to cell motility and
cell-matrix adhesion (33,34). MLC2 phosphorylation is controlled by
RhoA-ROCK signaling, Pak1, MLC kinase and phosphatase (34) and TGF-β may be regulating MLC2
phosphorylation via RhoA-ROCK and Pak1 signaling (29,35,36).
Moreover, in our study we made the novel observation that TGF-β
regulates the Arp2/3 complex, which is also related to cell
motility and invasion (13,34). The Arp2/3 complex mediates actin
nucleation enabling de novo polymerization of actin
filaments (13,37). We found that TGF-β rapidly activates
Arp2/3 complex in LM3 cells, and that the p38MAPK or MAPK-ERK
pathways are not involved in this TGF-β effect. To the best of our
knowledge, this is the first report of Arp2/3 complex activation by
TGF-β. It is known that Arp2/3 complex activation may involve other
proteins, such as WASP (Wiskott–Aldrich syndrome protein)/WAVE3
proteins, which are activated by Rac and CDC42 GTPases (13). On the other hand, TGF-β can rapidly
activate the Rho-family GTPases, Rac1, CDC42, and RhoA, in normal
and tumor cells, although the mechanism is still unknown (21,35,36,38).
WAVE3 is frequently upregulated in mammary carcinomas and it may
contribute to the regulation of p38MAPK (39). Thus, the activation of the Arp2/3
complex by TGF-β may involve Rac1/CDC42 and WASP/WAVE3 proteins.
Further studies may help elucidate the mechanisms of this TGF-β
response, but our observations suggest that TGF-β may have a more
profound effect on the actin machinery than previously thought.
The formation of actin fibers also requires Smad-
and p38MAPK-dependent expression of HMW-tropomyosins (14). In the non-tumorigenic NMuMG cells,
TGF-β upregulates HMW-tropomyosins, and it inhibits cell invasion
(18), whereas in LM3 tumor cells,
TGF-β does not modulate HMW-tropomyosins but stimulates cell
invasion. It appears that the difference may be linked to MEK-ERK
signaling. Active Ras-MEK-ERK signaling inhibits Smad activity
(40,41), and overexpression of oncogenic
RasV12 in NMuMG cells represses the TGF-β-Smad-mediated induction
of HMW-tropomyosins and actin fibers but enhances cell motility and
invasion (18). Furthermore,
siRNA-mediated suppression of HMW-tropomyosins inhibits formation
of actin stress fibers during the EMT process (14), whereas expression of tropomyosin
enhances actin fibers and inhibits Ras-mediated cell transformation
(18). Similar observations have
been reported for breast cancer MDA-MB-231 and murine carcinoma 4T1
cell lines, which express high levels of active Ras-MEK-ERK
signaling (8,14,25,27).
Smad4 knock-out accelerates the development of pancreatic ductal
carcinomas and metastases in the context of K-RasG12D transgenic
mice (42,43). Together, these findings suggest that
TGF-β pro-oncogenic activities in tumor cells are associated with
reduced Smad-dependent responses and elevated levels of MEK-ERK
signaling.
We found that while TGF-β signaling enhances cell
invasion and secretion of matrix metalloproteinase-9/gelatinase-B
(MMP-9) in LM3 cells, the MMP inhibitor BB-94 decreases LM3
invasiveness. Solid evidence implicates MMPs in tumor invasion and
metastasis, and the link between TGF-β and MMP-9 has been
extensively studied (44,45). Interestingly, MMP-9 serves as both a
downstream target of TGF-β as well as an activator of latent TGF-β.
In another breast carcinoma model, MMP-9 regulation by TGF-β did
not require p38MAPK (7,46). However, by using kinase inhibitors,
we observed an antagonic effect between the p38MAPK and MEK
pathways and TGF-β signaling on MMP-9 secretion in LM3 cells. More
studies are currently in progress to elucidate this mechanism.
Studies in both animal and human tumors have
suggested an active role for TGF-β during in vivo tumor
dissemination. Moreover, some breast cancer metastases have higher
TGF-β immunostaining than primary tumors (11). Our in vivo studies further
support the important role of TGF-β in tumorigenesis, since the
expression of dominant-negative TGF-β receptors, which disrupts
TGF-β signaling, significantly delayed initial LM3 tumor growth in
syngeneic mice. Even though this effect was diluted with tumor
evolution, our results allow us to speculate the TGF-β implication
in tumor development. Thus, in our mammary cancer model, LM3 cells
seem to be dependent on a functional TGF-β signaling, together with
p38MAPK and MEK, in order to acquire migratory and invasive
abilities, which allows tumor growth in vivo. However, once
the tumor is established and reaches log-phase growth, further
tumor progression appears to become independent of TGF-β. The
determinant signals during later steps of tumor growth as well as
during tumor progression, remain to be unraveled.
In summary, our studies demonstrate the important
role of TGF-β signaling, together with other intracellular
pathways, in the invasive and migratory properties of LM3 mammary
adenocarcinoma cells. TGF-β pro-tumorigenic activities were
apparent through the regulation of the actin cytoskeleton, an
increase in migratory and invasive abilities, and through the
induction of tumor growth in vivo. Since the LM3 cell line
is derived from a spontaneous mammary adenocarcinoma in BALB/c
mice, it represents a useful and novel model for investigating the
pro-oncogenic activities of cytokines.
Acknowledgements
E. Bal de Kier Joffe and Lydia Puricelli are members
of the National Council of Scientific and Technical Research
(CONICET). This study was supported by PICT 00417 y 01296 (ANCYPT)
and M003 y M243 (UBACYT) awarded to E.B.K.J and L.P.; by PHS grant
R01 CA095263 and USAMRMC grant DAMD17-02-01-0602 awarded to A.V.B,
and by a fellowship of the Bunge y Born Foundation, Argentina,
awarded to M.C.D.
References
|
1
|
Hunter KW, Crawford NP and Alsarraj J:
Mechanisms of metastasis. Breast Cancer Res. 10(Suppl 1): S22008.
View Article : Google Scholar
|
|
2
|
Roberts AB and Wakefield LM: The two faces
of transforming growth factor beta in carcinogenesis. Proc Natl
Acad Sci USA. 100:8621–8623. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:8621–8623.
2007. View Article : Google Scholar
|
|
4
|
Li Mo, Wan YY, Sanjabi S, Robertson AK and
Flavell RA: Transforming growth factor-β regulation of immune
responses. Annu Rev Immunol. 24:99–146. 2006.
|
|
5
|
Enholm B, Paavonen K, Ristimaki A, et al:
Comparison of VEGF, VEGF-B, VEGF-C and Ang-1 mRNA regulation by
serum, growth factors, oncoproteins and hypoxia. Oncogene.
14:2475–2483. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Christofori G: New signals from the
invasive front. Nature. 441:444–450. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Safina A, Vandette E and Bakin AV: LK5
promotes tumor angiogenesis by upregulating matrix
metalloproteinase-9 in tumor cells. Oncogene. 26:2407–2422. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Muraoka RS, Dumont N, Ritter CA, et al:
Blockade of TGF-β inhibits mammary tumor cell viability, migration,
and metastases. J Clin Invest. 109:1551–1559. 2002.
|
|
9
|
Yang YA, Dukhanina O, Tang B, et al:
Lifetime exposure to a soluble TGF-β antagonist protects mice
against metastasis without adverse side effects. J Clin Invest.
109:1607–1615. 2002.PubMed/NCBI
|
|
10
|
Elliott RL and Blobe GC: Role of
transforming growth factor beta in human cancer. J Clin Oncol.
23:2078–2093. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stover DG, Bierie B and Moses HL: A
delicate balance: TGF-β and the tumor microenvironment. J Cell
Biochem. 101:851–861. 2007.
|
|
12
|
Saunier EF and Akhurst RJ: TGF beta
inhibition for cancer therapy. Curr Cancer Drug Targets. 6:565–578.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pollard TD and Borisy GG: Cellular
motility driven by assembly and disassembly of actin filaments.
Cell. 112:453–465. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bakin AV, Safina A, Rinehart C, Daroqui C,
Darbary H and Helfman DM: A critical role of tropomyosins in TGF-β
regulation of the actin cytoskeleton and cell motility in
epithelial cells. Mol Biol Cell. 15:4682–4694. 2004.
|
|
15
|
Zavadil J and Bottinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2006. View Article : Google Scholar
|
|
16
|
Zheng Q, Safina A and Bakin AV: Role of
high-molecular weight tropomyosins in TGF-β-mediated control of
cell motility. Int J Cancer. 122:78–90. 2008.
|
|
17
|
Farina AR, Coppa A, Tiberio A, et al:
Transforming growth factor-β1 enhances the invasiveness of human
MDA-MB-231 breast cancer cells by up-regulating urokinase activity.
Int J Cancer. 75:721–730. 1998.
|
|
18
|
Safina A, Varga AE, Bianchi A, Zheng Q,
Kunnev D, Liang P and Bakin AV: RAS alters epithelial-mesenchymal
transition in response to TGF-β by reducing actin fibers and
cell-matrix adhesion. Cell Cycle. 8:284–298. 2009.PubMed/NCBI
|
|
19
|
Daroqui CM, Puricelli LI, Urtreger AJ, Bal
de Kier Joffé E, Elizalde PV and Lanuza GM: Involvement of
TGF-β/TβRs system in tumor progression of murine mammary
adenocarcinomas. Breast Cancer Res Treat. 80:287–301. 2003.
|
|
20
|
Urtreger AJ, Ghiso JAA, Werbajh SE,
Puricelli LI, Muro AF and Bal de Kier Joffé E: Involvement of
fibronectin in the regulation of urokinase production and binding
in murine mammary tumor cells. Int J Cancer. 82:748–753. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bakin AV, Rinehart C, Tomlinson AK and
Arteaga CL: p38 mitogen-activated protein kinase is required for
TGFβ-mediated fibroblastic transdifferentiation and cell migration.
J Cell Sci. 115:3193–3206. 2002.
|
|
22
|
Oft M, Heider KH and Beug H: TGFβ
signaling is necessary for carcinoma cell invasiveness and
metastasis. Curr Biol. 8:1243–1252. 1998.
|
|
23
|
Dennler S, Itoh S, Vivien D, ten Dijke P,
Huet S and Gauthier JM: Direct binding of Smad3 and Smad4 to
critical TGF beta-inducible elements in the promoter of human
plasminogen activator inhibitor-type 1 gene. EMBO J. 17:3091–3100.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chomczynski P and Sacchi N: Single-step
method of RNA isolation by acid guanidinium
thiocyanate-phenol-chloroform extraction. Anal Biochem.
162:156–159. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McEarchern JA, Kobie JJ, Mack V, et al:
Invasion and metastasis of a mammary tumor involves TGF-β
signaling. Int J Cancer. 91:76–82. 2001.
|
|
26
|
Dumont N, Bakin AV and Arteaga CL:
Autocrine transforming growth factor-beta signaling mediates
Smad-independent motility in human cancer cells. J Biol Chem.
278:3275–3285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for TGFβ-mediated epithelial to mesenchymal transition and
cell migration. J Biol Chem. 275:36803–36810. 2000.
|
|
28
|
Hutchison N, Hendry BM and Sharpe CC: Rho
isoforms have distinct and specific functions in the process of
epithelial to mesenchymal transition in renal proximal tubular
cells. Cell Signal. 21:1522–1531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhowmick NA, Ghiassi M, Bakin A, et al:
Transforming growth factor-β1 mediates epithelial to mesenchymal
transdifferentiation through a RhoA-dependent mechanism. Mol Biol
Cell. 12:27–36. 2001.
|
|
30
|
Valkov A, Sorbye SW, Kilvaer TK, Donnem T,
Smeland E, Bremnes RM and Busund LT: The prognostic impact of
TGF-β1, fascin, NF-κB and PKC-ζ expression in soft tissue sarcomas.
PLoS One. 6:e175072011.
|
|
31
|
Yu L, Hebert MC and Zhang YE: TGF-beta
receptor-activated p38 MAP kinase mediates Smad-independent TGF-β
responses. EMBO J. 21:3749–3759. 2002.PubMed/NCBI
|
|
32
|
Xie L, Law BK, Chytil AM, Brown KA, Aakre
ME and Moses HL: Activation of the Erk pathway is required for
TGF-β1-induced EMT in vitro. Neoplasia. 6:603–610. 2004.
|
|
33
|
Zaidel-Bar R, Cohen M, Addadi L and Geiger
B: Hierarchical assembly of cell-matrix adhesion complexes. Biochem
Soc Trans. 32:416–420. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pellegrin S and Mellor H: Actin stress
fibres. J Cell Sci. 120:3491–3499. 2007. View Article : Google Scholar
|
|
35
|
Zhou H and Kramer RH: Integrin engagement
differentially modulates epithelial cell motility by RhoA/ROCK and
PAK1. J Biol Chem. 280:10624–10635. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ueda Y, Wang S, Dumont N, Yi JY, Koh Y and
Arteaga CL: Overexpression of HER2 (erbB2) in human breast
epithelial cells unmasks TGF-induced cell motility. J Biol Chem.
279:24505–24513. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Welch MD: The world according to Arp:
regulation of actin nucleation by the Arp2/3 complex. Trends Cell
Biol. 9:423–427. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Edlund S, Landstrom M, Heldin CH and
Aspenstrom P: Transforming growth factor-beta-induced mobilization
of actin cytoskeleton requires signaling by small GTPases Cdc42 and
RhoA. Mol Biol Cell. 13:902–914. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sossey-Alaoui K, Safina A, Li X, Vaughan
MM, Hicks DG, Bakin AV and Cowell JK: Down-regulation of WAVE3, a
metastasis promoter gene, inhibits invasion and metastasis of
breast cancer cells. Am J Pathol. 170:2112–2121. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saha D, Datta PK and Beauchamp RD:
Oncogenic ras represses transforming growth factor-beta/Smad
signaling by degrading tumor suppressor Smad4. J Biol Chem.
276:29531–29537. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kretzschmar M, Doody J, Timokhina I and
Massague J: A mechanism of repression of TGFbeta/ Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kojima K, Vickers SM, Adsay NV, et al:
Inactivation of Smad4 accelerates KrasG12D-mediated pancreatic
neoplasia. Cancer Res. 67:8121–8130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ijichi H, Chytil A, Gorska AE, Aakre ME,
Fujitani Y, Fujitani S, Wright CV, Moses HL, et al: Aggressive
pancreatic ductal adenocarcinoma in mice caused by
pancreas-specific blockade of transforming growth factor-beta
signaling in cooperation with active Kras expression. Genes Dev.
20:3147–3160. 2006. View Article : Google Scholar
|
|
44
|
Van den Steen P, Dubois B, Nelissen I,
Rudd P, Dwek R and Opdenakker G: Biochemistry and molecular biology
of gelatinase B or matrix metalloproteinase-9 (MMP-9). Crit Rev
Biochem Mol Biol. 37:375–536. 2002.
|
|
45
|
Bergers G, Brekken R, McMahon G, et al:
Matrix metalloproteinase-9 triggers the angiogenic switch during
carcinogenesis. Nat Cell Biol. 2:737–744. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Safina A, Ren M-Q, Vandette E and Bakin
AV: TAK1 is required for TGF-β1-mediated regulation of matrix
metalloproteinase-9 and metastasis. Oncogene. 27:1198–1207.
2008.
|