Introduction
Multiple myeloma (MM) is a malignancy of terminally
differentiated B cells accounting for ~10% of all hematological
malignancies and affecting >20,000 patients each year in the
United States (1). Despite recent
advances in targeted therapies and regimens of high dose
chemotherapy with autologous stem cell transplant, there is still
no curative treatment. Relapse of disease and development of
resistance are major obstacles to overcome for improving treatment
response and patient survival in MM (2).
An expansive, highly developed rough endoplasmic
reticulum (ER) specialized for constant synthesis and secretion of
large amounts of immunoglobulin protein is a defining
characteristic of plasma cells. The innate biology of this class of
cells renders MM exquisitely sensitive to agents like the
proteasome inhibitor bortezomib (BTZ). By virtue of its proteasomal
inhibitory activity, BTZ causes accumulation of misfolded proteins
in the endoplasmic reticulum (ER), with the resultant ER stress
triggering activation of the unfolded protein response (UPR) and
apoptosis (3). BTZ has demonstrated
remarkable response rates in both relapsed and newly diagnosed MM,
but it carries the potential for development of resistance and
serious side effects. For example, >30% of patients receiving
BTZ treatment develop painful peripheral neuropathy (4).
ER stress triggers the unfolded protein response
(UPR), a cellular process activated when unfolded or misfolded
proteins accumulate in the lumen of the ER. In this capacity, the
UPR’s primary purpose is to restore normal cellular function by
halting protein translation and activating signaling pathways to
increase the production of molecular chaperones like HSP90 involved
in protein folding. If proteostasis is not restored in a timely
fashion, the aim of the UPR shifts to promote apoptosis (5). Key mediators of this process include
PERK (protein kinase RNA-like ER kinase), eIF2α (eukaryotic
translation initiation factor 2-α), XBP1 (X-box binding protein 1)
and CHOP (CCAAT/-enhancer-binding protein homologous protein). Upon
initiation of UPR activation, PERK undergoes phosphorylation and
oligomerization to cause translational attenuation by directly
phosphorylating the α subunit of the regulating initiator of the
mRNA translation machinery, eIF2 (6). Simultaneously, in a parallel arm of
UPR pathway activation, the mRNA of transcription factor XBP1 is
spliced. In its activated form XBP1 mRNA encodes for a
transcription factor that targets and induces expression of genes
containing an unfolded protein response element (UPRE). These genes
include ER chaperones, heat shock proteins and XBP1 itself
(7). Another effector of the UPR is
the transcription factor CHOP. Subsequent upregulation of certain
CHOP target genes promotes induction of ER-stress mediated
apoptosis (8).
Given its remarkable rates of induction of remission
and enhanced long-term survival in the treatment of acute
promyelocytic leukemia (9), the
utility of arsenic trioxide (ATO) (10) in the treatment of MM has recently
been evaluated. In vitro models as well as preclinical
studies suggest that ATO is able to induce apoptosis at clinically
achievable concentrations in drug-resistant MM cell lines and is
well tolerated (11,12). Since then, a number of clinical
trials have provided evidence for the efficacy of ATO in the
treatment of relapsed or refractory MM patients. However, like most
drugs used in the treatment of MM, >50% of patients with
refractory or relapsed disease eventually present with resistance
to ATO when it is used as a single agent (13). In addition to other reported
mechanisms of action, ATO has been shown to disrupt calcium stores
and promote ER stress-related signaling (14–16).
Sulforaphane (4-methylsulfinylbutyl isothiocyanate),
erysolin (4-methylsulfonylbutyl isothiocyanate) and erucin
(4-methythiobutyl isothiocyanate) are naturally occurring
isothiocyanates that account for the chemopreventative effects of
cruciferous vegetables such as broccoli and Brussels sprouts.
Sulforaphane is a well characterized inducer of several phase II
detoxification enzymes including glutathione-S-transferases and
quinone reductase (17). In
addition to its chemopreventative effects, sulforaphane has also
been reported to cause growth inhibition and induction of apoptosis
in a variety of human cancer cell lines (18,19).
More recently, proteasomal inhibitory activity has been attributed
to the isothiocyanates (20,21).
We have previously shown that ATO and sulforaphane
synergize to induce apoptosis in leukemic cells via a reactive
oxygen species (ROS)-dependent mechanism (22). Given previously published studies
demonstrating the synergistic relationship between ATO and BTZ as
well as sulforaphane’s purported proteasomal inhibitory activity
(21,23), we wanted to examine the efficacy of
sulforaphane in combination with ATO in MM cells. Our ultimate goal
is to identify effective combinations that could provide the
clinical benefit of BTZ by targeting similar pathways while
minimizing debilitating side effects or the emergence of
resistance. Here, we report that isothiocyanates block TNFα induced
degradation of Iκβ in MM cells in a manner similar to BTZ. Because
this inhibition is consistent with reported proteasomal inhibition
by isothiocyanates, we investigated potential synergy with ATO. We
show that sulforaphane synergizes with ATO in a panel of MM cell
lines. Combination treatment results in generation of ROS and ER
stress, culminating in enhanced UPR signaling which induces
apoptosis.
Materials and methods
Cell culture and reagents
Human multiple myeloma cell lines were cultured in
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10%
fetal bovine serum, 2 mmol/l L-glutamine, 5 U/ml penicillin, and 5
μg/ml streptomycin. PCNY-1, MM1.S, MM1.R, KMS-11 and ARP-1 cells
were kindly provided by Hearn Cho (New York University School of
Medicine, New York, NY, USA). All cells were maintained in an
incubator with a humidified atmosphere of 5% CO2 at
37°C. Sulforaphane, erysolin, erucin, arsenic trioxide and
N-acetyl-l-cysteine (NAC) were purchased from Sigma (St. Louis, MO,
USA). Bortezomib was acquired from LC Laboratories (Woburn, MA,
USA).
Cell proliferation assay
For dose response assays, 5,000 cells per well were
plated in 96-well culture plates. After overnight incubation, the
cells were treated with indicated compounds. Following a 72-h
incubation period, cellular proliferation was assessed using a
tetrazolium dye reduction assay (CellTiter 96 Aqueous
Non-Radioactive Cell Proliferation assay; Promega, Madison, WI,
USA) carried out according to the manufacturer’s instructions as
previously described (24).
Absorbance was recorded on a microplate reader at 495 nm. Cellular
proliferation was expressed as a percentage of vehicle-treated
cells which were defined as 100% viable. Drug interaction was
analyzed by Calcusyn software (Biosoft, Cambridge, UK). This
software determines the interaction of two drugs through
calculations of the combination index (25) based upon the multiple drug effect
equation of Chou and Talalay (26,27).
Denotation of CI values as follows: >1, antagonism; 1,
additivity; <1, synergy. GI50 values, which represent
the concentration necessary to cause 50% growth inhibition, were
calculated from dose-response curves generated from cell
proliferation assays.
Quantification of cellular glutathione
levels
Cellular levels of glutathione (GSH) levels were
determined using the HT Glutathione Assay kit from Trevigen
(Gaithersburg, MD, USA) as previously described (22). Briefly, 24 h after seeding MM cells
at a density of 1×106, cells were exposed to indicated
compounds for 3 h at 37°C. The cells were collected and washed with
PBS. An equal number of cells from each treatment group were
aliquoted for use in cellular GSH quantification per the
manufacturer’s instructions.
Protein extraction and western
blotting
Cells were harvested in extraction buffer [1% Triton
X-100, 50 mmol/l Tris, 2 mmol/l EDTA, 150 mmol/l NaCl (pH 7.5)]
containing protease inhibitor cocktail (Roche Applied Science,
Indianapolis, IN, USA) and phosphatase inhibitor cocktail (Sigma)
after two washes with ice-cold phosphate buffered saline (PBS). The
lysates were centrifuged at 10,000 × g at 4°C for 10 min in a
microcentrifuge. Protein supernatants were measured with a protein
assay kit (Bio-Rad, Hercules, CA, USA). Proteins were separated by
10% sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE) and transferred onto polyvinylidene difluoride membranes
(Polyscreen; Perkin-Elmer, Waltham, MA, USA). Antibodies against
cleaved forms of poly(ADP-ribose) polymerase (PARP), caspase-3 and
-4 were obtained from Cell Signaling Technology (Danvers, MA, USA).
Phospho-PERK, phospho-eIF2α, CHOP and HSP90 antibodies were
obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Anti-actin (Sigma) was used as a control. Immunoreactive bands were
visualized using enhanced chemiluminescence detection reagent
(Perkin-Elmer) and X-OMAT processing.
XBP1 splicing
Cells were treated with indicated concentrations of
ATO and/or sulforaphane for 24 h. Total RNA was isolated from lysed
cells with the RNeasy Mini kit (Qiagen, Germantown, MD, USA). XBP1
splicing was assessed by semi-quantitative RT-PCR as described
previously (28,29). cDNA was produced from total RNA
preps using ImProm-II Reverse Transcription system (Promega).
Primers spanning the fragment of XBP1 containing the intron
targeted by Ire1α were used: 5′-TACGGGAGAAAACTCACGGC-3′ and
5′-GGGTCCAACTTGTCCAGAATGC-3′. The thermal PCR cycling conditions
are as follows: 95°C for 5 min, 95°C for 1 min, 58°C for 30 sec,
72°C for 30 sec, and 72°C for 5 min with 35 cycles of
amplification. PCR products were digested with Pst1 (New England
Biolabs, Ipswich, MA, USA) before being separated on a 2.0%
agarose/1X TAE gel and visualized by ethidium bromide.
Gaussia luciferase secretion assay
Commercially available lentiviral particles obtained
from GenTarget (San Diego, CA, USA) expressing Gaussia
luciferase (Gluc) were introduced into KMS-11 and ARP-1 MM cells by
infection. For infection, cells were cultured in 6-well tissue
culture plates to a density of 3×106 cells/ml and then
diluted to 1×106 cells/ml in complete media. Lentiviral
particle was added at a ratio of 100 μl viral particles to 1 ml of
cells. After 24 h, equal parts of fresh media containing puromycin
selection were added. After 72 h, efficacy of transduction was
assessed by RFP fluorescence. After infection, cells were
maintained in media containing 3 μg/ml puromycin to establish
stable clones. For secretion assays, 100,000 cells/well were plated
in 96-well culture plates. Cells were immediately treated with
indicated concentrations of compounds. Following 24-h incubation,
expression and secretion of Gluc was monitored using The BioLux
Gaussia Luciferase Assay kit (New England Biolabs) according
to the manufacturer’s instructions through measurements of
luciferase activity as indicated by relative light units (RLU) on a
microplate luminometer (Molecular Devices, Sunnyvale, CA, USA).
Percent reduction in Gluc secretion was determined by the following
equation: (RLU of treated cells/RLU ratio of the untreated, DMSO
control cells) × 100.
Statistical analyses
Unless otherwise noted, experiments were performed
in triplicate. The data are presented as the average ± SEM.
P-values were determined by a two-sided Student’s t-test with
unequal variance, with P<0.05 considered significant.
Results
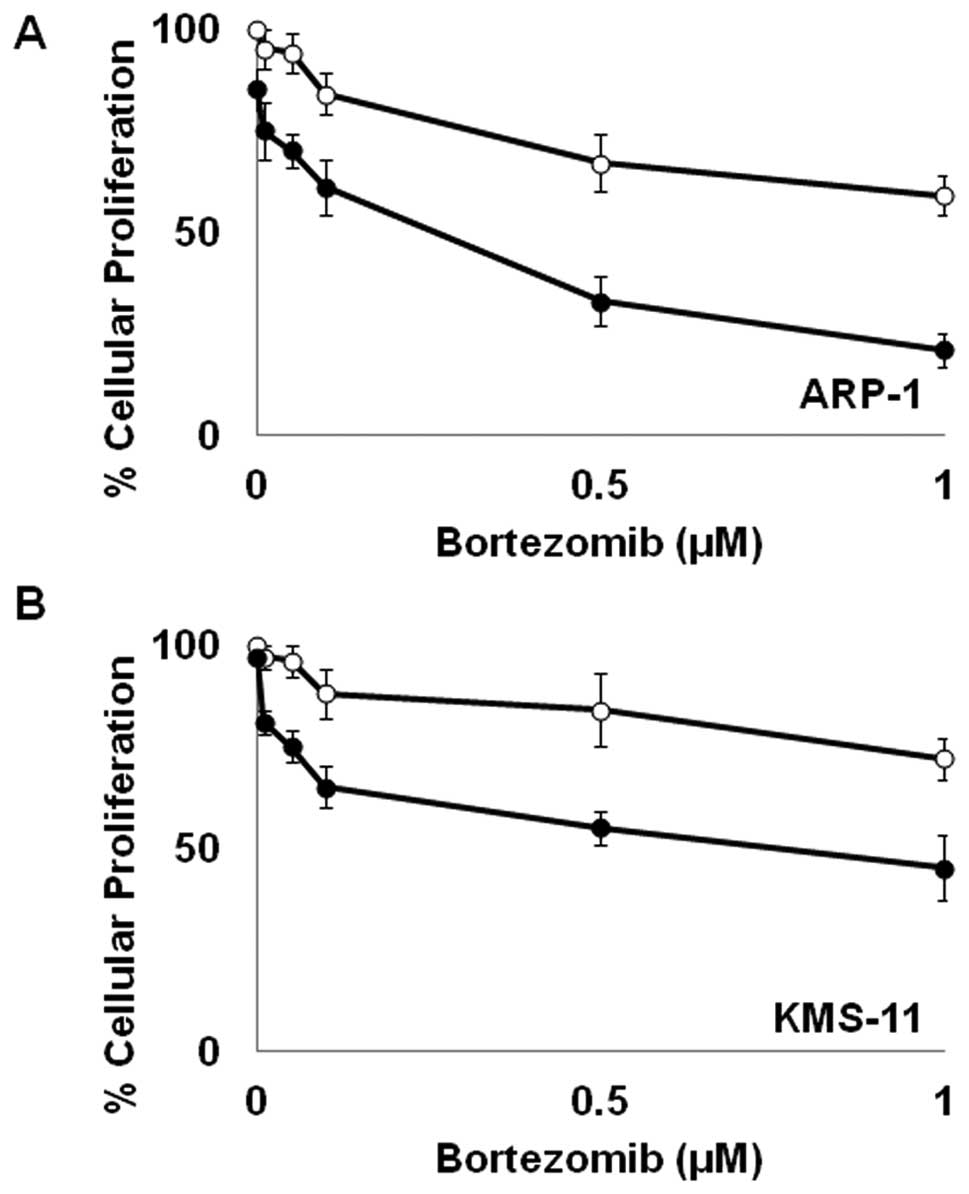
Bortezomib enhances ATO-mediated growth
inhibition
In order to enhance the potential clinical efficacy
of ATO in the treatment of MM, we wanted to assess its interactive
potential with BTZ, a current standard of care in the treatment of
MM. As shown in Fig. 1, low,
subclinical doses of BTZ and ATO as single agents have limited
effect on cellular proliferation in MM. However, when used in
combination, BTZ and ATO markedly inhibit cellular proliferation in
both KMS-11 and ARP-1 cells (Fig.
1).
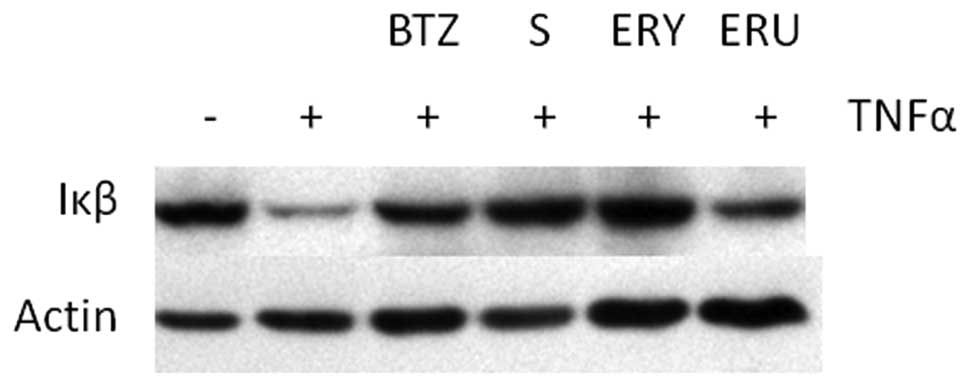
Sulforaphane and erysolin inhibit
TNFα-induced degradation of Iκβ
Given that recent studies indicate that
isothiocyanates possess anti-proteasomal activity, we hypothesized
that isothiocyanates should have biological effects similar to BTZ.
In order to test this hypothesis, we examined the effect of
isothiocyanates on TNFα induced proteasomal degradation of Iκβ.
Upstream activation signaling by TNFα-stimulation causes
phosphorylation of Iκβ which is ultimately targeted for degradation
by the 26S proteasome (30). As
previously reported (31) and shown
in Fig. 2, proteasome inhibitors
such as BTZ prevent TNFα-induced Iκβ proteasomal degradation.
Similarly, sulforaphane and erysolin pretreatment also prevent Iκβ
degradation after TNFα stimulation. Erucin pretreatment only
minimally inhibits Iκβ degradation.
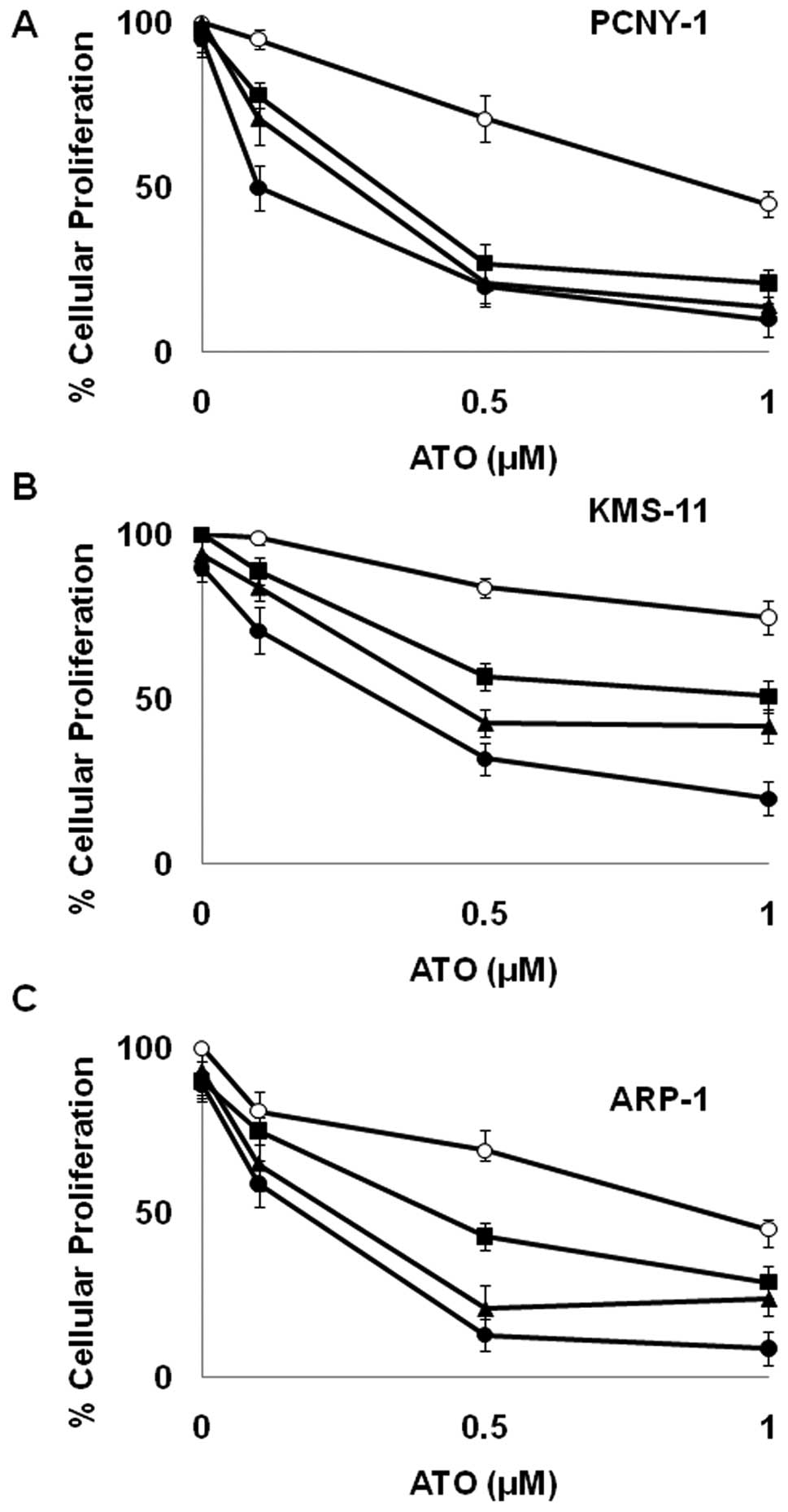
Sulforaphane and erysolin enhance ATO
growth inhibition in a synergistic fashion
Because these data along with other published
studies suggest that isothiocyanates inhibit proteasomal mediated
protein degradation, we wanted to investigate their ability to
potentiate the growth inhibitory effects of ATO. As shown in
Tables I and II, sulforaphane and erysolin display a
greater than additive effect (i.e., synergize) with ATO in 3 MM
cell lines. Consistent with its inability to inhibit TNFα induced
degradation of Iκβ (Fig. 2), erucin
interaction with ATO was found to be antagonistic (Table II). Due to its potency, we focused
further experiments on sulforaphane.
 | Table ICombinatorial effects of ATO and
sulforaphane in a panel of MM cell lines. |
Table I
Combinatorial effects of ATO and
sulforaphane in a panel of MM cell lines.
| Cell line | ATO IC50
(μM) | ATO + 3 μM
sulforaphane IC50 (μM) | CI | Interpretation |
|---|
| PCNY-1 | 0.94 | 0.26 | 0.432 | Synergistic |
| KMS-11 | 1.41 | 0.54 | 0.659 | Synergistic |
| ARP-1 | 1.05 | 0.33 | 0.594 | Synergistic |
| MM1.S | 0.89 | 0.49 | 0.641 | Synergistic |
| MM1.R | 2.14 | 2.02 | 1.21 | Antagonistic |
| Table IICombinatory effects of
isothiocyanates and ATO in a panel of MM cell lines. |
Table II
Combinatory effects of
isothiocyanates and ATO in a panel of MM cell lines.
| Cell line | Combination | CI | Interpretation |
|---|
| PCNY-1 | 3 μM erysolin, 1 μM
ATO | 0.646 | Synergistic |
| ARP-1 | 3 μM erysolin, 1 μM
ATO | 0.714 | Synergistic |
| KMS-11 | 3 μM erysolin, 1 μM
ATO | 0.597 | Synergistic |
| PCNY-1 | 3 μM erucin, 1 μM
ATO | 1.02 | Antagonistic |
| ARP-1 | 3 μM erucin, 1 μM
ATO | 0.994 | Antagonistic |
| KMS-11 | 3 μM erucin, 1 μM
ATO | 1.13 | Antagonistic |
As shown in Fig. 3,
ATO caused modest growth inhibition in PCNY-1 MM cells when
employed as a single agent. Similarly, concentrations up to 5 μM
sulforaphane had a minimal effect on the proliferation of MM cells
(Fig. 3). However, when used in
combination, the ability of these compounds to reduce proliferative
capacity was dramatically enhanced. Combination index analysis
demonstrates the relationship between 0.5 μM ATO and 3 μM
sulforaphane to be strongly synergistic (Table I). Similar effects were observed in
4 out of 5 MM cell lines examined (Fig.
3 and Table I). The synergistic
relationship was not observed in MM1.R cells which are a subclone
of the MM.1 human MM cell line selected for resistance to
glucocorticoid therapy (32).
Sulforaphane and ATO in combination
enhance apoptotic induction through an ROS-dependent mechanism
As we previously demonstrated in leukemic cells
(22), sulforaphane also acts as an
ATO sensitizing agent through depletion of intracellular GSH in MM
cells (Fig. 4A). In this capacity,
cellular depletion of glutathione by sulforaphane renders cells
largely incapable of protection against ATO’s ability to generate
ROS. In order to understand the biological consequences of enhanced
ROS, we evaluated the induction of apoptosis via cleavage of
effector caspase-3 and poly(ADP-ribose) polymerase (PARP),
indicators of apoptosis. As shown in Fig. 4B, higher levels of cleaved caspase-3
and PARP protein were present in KMS-11 cells exposed to the
combination treatment of 3 μM sulforaphane and 1 μM ATO than with
either agent alone. Similar results were observed in ARP-1 cells
(data not shown). These data indicate that combinatorial
sulforaphane/ATO treatment enhances the apoptotic response.
Interestingly, cleavage of the ER stress specific caspase-4 is also
enhanced in response to combination treatment (Fig. 4B).
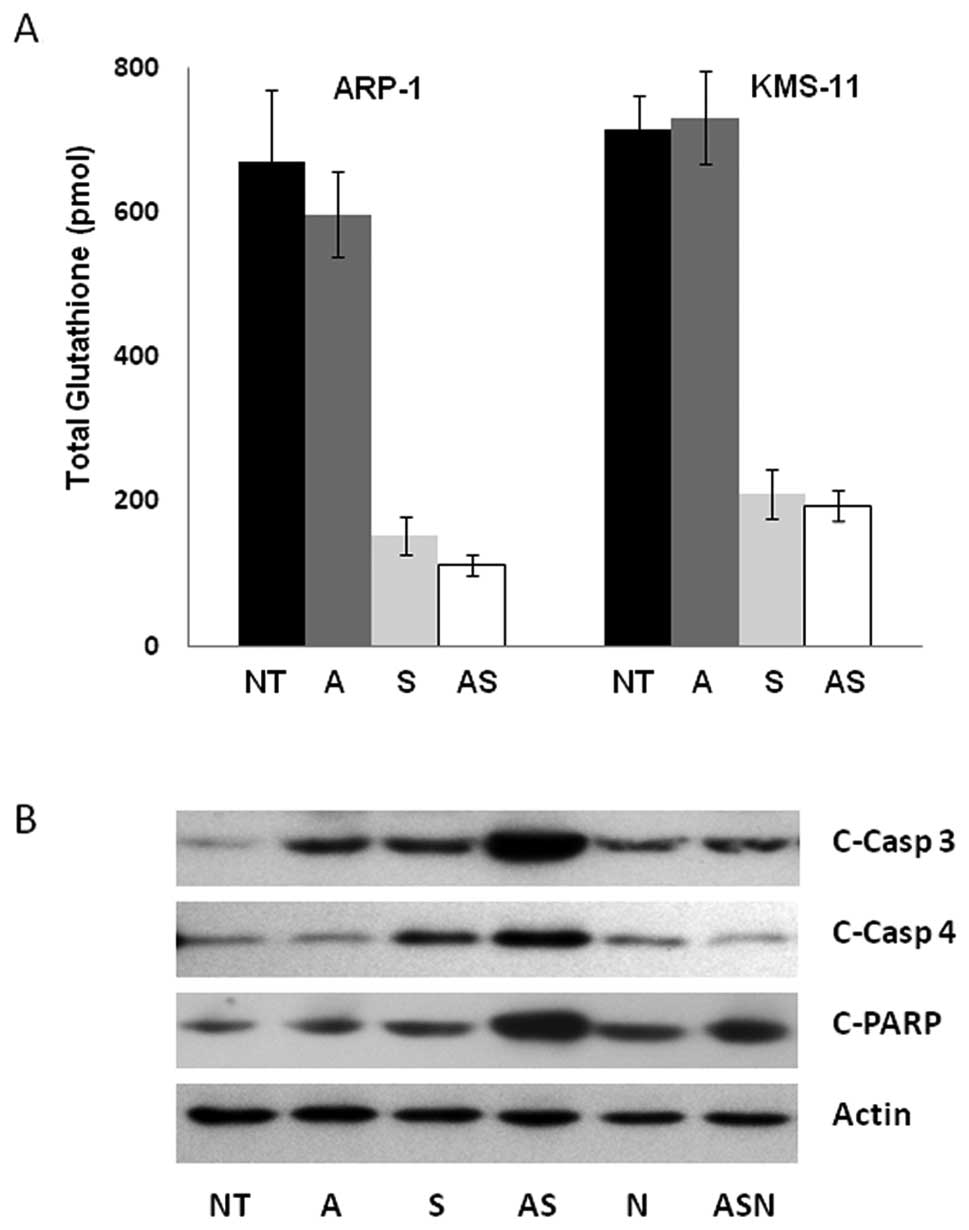 | Figure 4The combination ATO/sulforaphane
treatment induces apoptosis partially mediated by ROS generation.
(A) KMS-11 cells were incubated with 0.3 μM ATO, 3 μM sulforaphane,
or both. After 3 h of treatment, total cellular GSH values were
assessed. Experiments were performed in triplicate and error bars
were calculated using SEM. (B) KMS-11 cells were incubated with 1
μM ATO, 3 μM sulforaphane, or in combination for 24 h. Proteins
were extracted from the cells for analysis by immunoblotting with
antibodies to actin, cleaved caspase-3 (c-Casp-3), cleaved
caspase-4 (c-Casp 4) and cleaved PARP (c-PARP). Representative
blots shown from 3 independent experiments. NT, no treatment; A,
ATO; S, sulforaphane; N, NAC. |
ROS play a critical role in the apoptotic response
of combined ATO and sulforaphane treatment as demonstrated by the
fact that preincubation with the ROS scavenger N-acetyl cysteine
(NAC) partially attenuated the apoptotic induction of combination
treatment (Fig. 4B). Pretreatment
with NAC had no effect on apoptosis alone. However, in combination
with ATO and sulforaphane, NAC partially inhibited PARP, caspase-3
and -4 cleavage to levels comparable to treatment with ATO
alone.
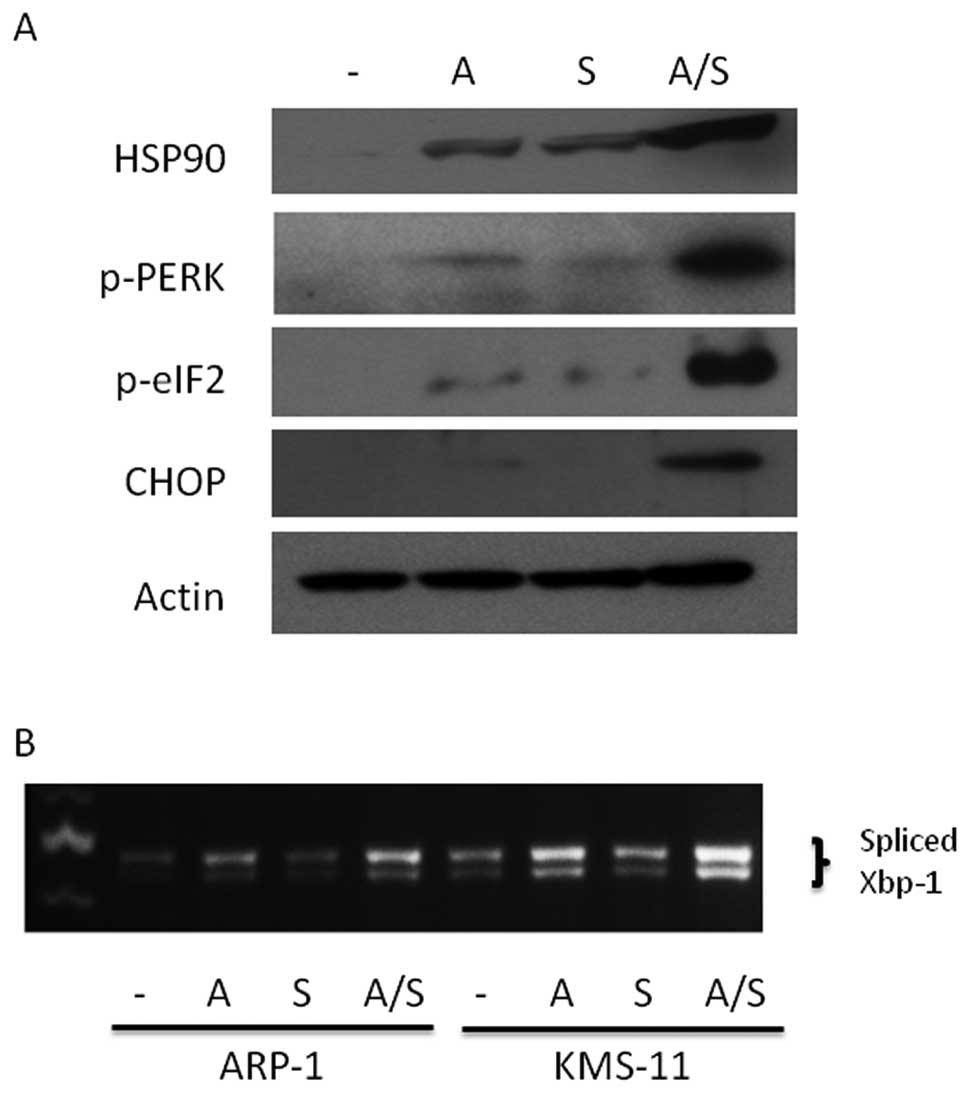
Combination sulforaphane and ATO
treatment promotes ER stress
Hypothesizing that sulforaphane’s anti-proteasomal
activity also is integral to its ability to augment ATO
cytotoxicity in MM cells, we investigated whether combination
treatment resulted in enhanced ER stress due to perturbations in
protein processing. Consistent with this notion, we observed
upregulation of HSP90, a general marker for ER stress (33), in KMS-11 cells co-treated with ATO
and sulforaphane (Fig. 5A).
Additionally, activation of the PERK pathway, a key component of
the unfolded protein response (UPR), was enhanced upon
co-treatment. As shown in Fig. 5A,
PERK phosphorylation was elevated after treatment with ATO and
sulforaphane with pathway activation demonstrated by increased
expression of downstream mediators CHOP as well as phosphorylation
of eIF2. Consistent with upregulation of the PERK arm of the UPR
response, parallel activation of the IRE1 arm of the UPR response
was observed through enhanced splicing of the UPR transcription
factor XBP1 upon treatment with ATO and sulforaphane (Fig. 5B). Similar results were observed in
ARP-1 MM cells (data not shown).
Combination sulforaphane and ATO
treatment disrupts protein secretion in MM cells
B cells synthesize and secrete immunoglobulin
protein. Blocking or decreasing protein processing in the secretory
pathway is another hallmark of ER stress that is specifically
relevant to the biology of MM cells (34). In order to assess the effects of ATO
and sulforaphane on protein secretion, we employed the reporter
protein Gaussia luciferase (Gluc). Gluc is a naturally
secreted luciferase that can be easily monitored through
extracellular release of luciferase activity in real time, and has
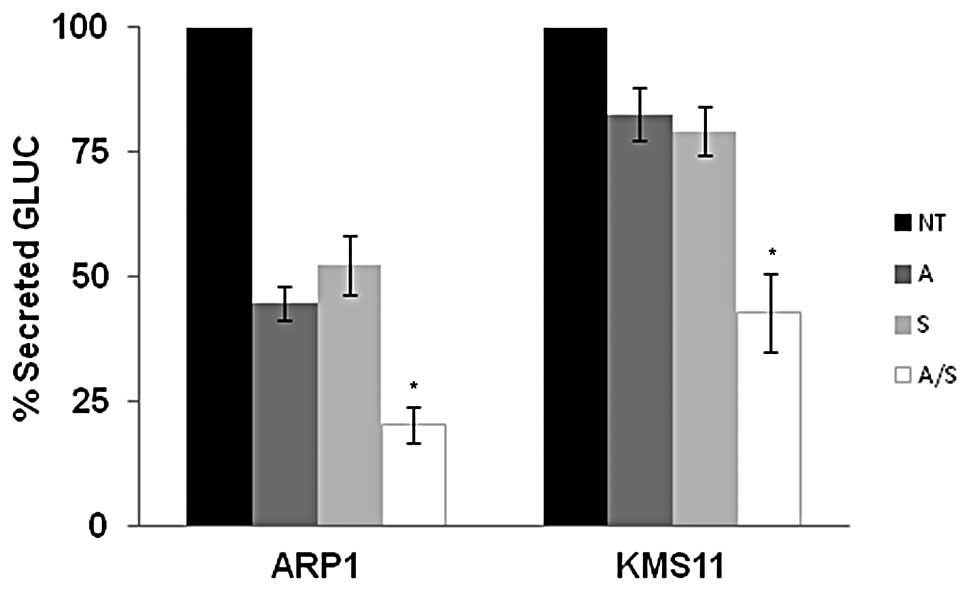
been developed as a sensor of ER stress (35). As shown in Fig. 6, clones of KMS-11 and ARP-1 stably
expressing Gluc show a notable reduction in Gluc secretion when
treated with 1 μM ATO or 3 μM sulforaphane alone. Consistent with
our studies of additional ER stress markers, combination ATO and
sulforaphane treatment inhibits protein secretion in a fashion that
is greater than treatment with single agent alone. Altogether,
these data suggest that when ATO and sulforaphane are administered
together, ER stress mediated pathways are enhanced.
Discussion
In the present study, we demonstrated that the
naturally occurring dietary compound sulforaphane inhibited TNFα
induced proteasomal degradation of Iκβ in a manner similar to BTZ
(Fig. 2). Similarly to BTZ,
sulforaphane is a potent ATO sensitizer (Figs. 1 and 3). In 4 out of 5 MM cell lines examined, a
synergistic relationship between the compounds was observed when
combined with 3 μM sulforaphane (Table
I). The synergistic growth inhibition was due to enhanced
induction of apoptosis (Fig. 4), in
keeping with the the combination’s ability to generate ROS and ER
stress, activate UPR signaling and inhibit protein secretion
(Figs. 4–6).
The combinatorial effects of ATO and BTZ have been
reported in a variety of leukemic cell lines as well as in MM cell
lines (25,36). More recently, combination ATO/BTZ
regimens have demonstrated synergistic activity against MM in both
preclinical and clinical studies (37,38).
Indeed, our results show a similar effect of ATO/BTZ in KMS-11 and
ARP-1 MM cells (Fig. 1). Previous
studies have implicated a variety of mechanisms for the combined
anti-proliferative activity of these agents including p38 MAPK
activation and proteolytic activation of protein kinase C delta
(PKCδ) (23,36). The similarities between ATO/BTZ and
ATO/sulforaphane combined growth inhibition along with BTZ and
sulforaphane’s described anti-proteasomal activity caused us to
hypothesize that induction of ER stress could be implicated as a
mechanism of action with the data presented herein supporting that
notion. This activity is consistent with the previously described
mechanisms of action for BTZ/ATO synergy, as both p38 MAPK and PKCδ
are downstream signaling components of ER stress mediated apoptotic
pathways (39,40).
We previously demonstrated that ATO and sulforaphane
were an effective combination in a panel of non-acute promyelocytic
leukemia hematological malignancies (22). In our studies using leukemic cells,
we demonstrated that sulforaphane depleted intracellular
glutathione levels causing enhanced ROS generation upon combination
treatment. Here, we also demonstrated a dependence on ROS for
enhanced apoptotic induction in MM cells (Fig. 4), and that combination treatment by
sulforaphane and ATO effectively induced ER stress mediated
responses such as upregulation of HSP90, activation of the UPR and
inhibition of protein secretion (Figs.
5 and 6). Recent studies have
suggested that the ER also may play an important role in response
to oxidative stress (41,42). Moreover, the ER is exquisitely
sensitive to oxidative damage (43). Therefore, given our data suggesting
involvement of both ROS and ER stress pathways in response to
combination ATO and sulforaphane treatment, we examined any
potential interplay between these 2 critical cellular stress
responses. Indeed, the antioxidant NAC attenuated ER
stress-mediated apoptosis as measured by a reduction in cleavage of
the ER specific caspase-4, suggesting ROS involvement in the
induction of ER stress specific apoptosis (Fig. 4). Because depletion of glutathione
is already observed after only 3 h of treatment, it is possible
that ROS act as upstream signaling molecules to initiate UPR
pathways and ER stress apoptosis. Nevertheless, further studies to
elucidate the specific links between ROS and ER-stress are
needed.
Interestingly, the synergistic effects of
isothiocyanates with ATO were limited to sulforaphane and erysolin
(Table II). The structurally
related isothiocyanate erucin did not display synergy with ATO in
MM cells (Table II). Moreover, the
effect of each isothiocyanate on inhibition of TNFα-induced Iκβ
degradation appears to parallel each compound’s combinatorial
effect with ATO. For example, both sulforaphane and erysolin
synergize with ATO and inhibit Iκβ degradation in a manner similar
to BTZ. In contrast, erucin had minimal effect on Iκβ degradation
and did not synergize with ATO. Our studies with isothiocyanates in
leukemic cells also demonstrated ATO synergism only with
sulforaphane and erysolin (22).
These data suggest the importance of proteasomal inhibition as a
mechanism for disrupting MM cellular proliferation.
One limitation of this combination is the
observation that ATO/sulforaphane is not synergistic in MM1.R
cells. MM1.R cells are a subclone of the parental MM1 human MM cell
line selected for resistance to glucocortocoid therapy through loss
of the glucocortocoid receptor. MM1.S cells also used in this study
are subclones selected for sensitivity to glucocortocoid therapy.
Interestingly, MM1.S displayed a synergism between ATO and
sulforaphane, whereas the relationship in MM1.R cells was
classified as antagonistic (Table
I). According to the literature as well as our own studies,
MM1.R cells are not resistant to other proteasome inhibitors like
BTZ or carfilzomib (44,45). These data could potentially suggest
that the reason for antagonism in MM1.R cells is not based upon a
mechanism rooted in ER stress. Interestingly, the glucocorticoid
receptor itself has been implicated in NFκβ inactivation through
tethering processes which disrupt critical interaction with
translational machinery (32). In
our study, ATO and sulforaphane are synergistic in MM1.S cells
expressing glucocorticoid receptors (GCRs), but antagonistic in
MM1.R cells which lack GCRs, which may point to an additional
mechanism for regulation of NFκβ in response to cytotoxic
stressors. Although not specifically addressed in this study, this
is an area of further interest.
Given the clinically validated importance of
targeting ER stress pathways in the treatment of MM as exemplified
by BTZ (46), our data suggest that
the combination ATO and sulforaphane may hold therapeutic
potential. It is important to note that BTZ carries the potential
for serious side effects with >30% of patients reporting painful
peripheral neuropathy (4). In
contrast, sulforaphane is a natural product with a well documented
safety profile. Moreover, the concentrations used in these studies
are clinically achievable after dietary consumption (47). Similarly, the effective
concentrations of ATO are clinically relevant (48). Therefore, ATO and sulforaphane
combination is deserving of further investigation as a potentially
well tolerated yet effective treatment for MM.
Acknowledgements
This study was supported in part by research funding
from a NYU Cancer Center Core Grant (to A.M.).
Abbreviations:
|
MM
|
multiple myeloma
|
|
ER
|
endoplasmic reticulum
|
|
ROS
|
reactive oxygen species
|
|
BTZ
|
bortezomib
|
|
ATO
|
arsenic trioxide
|
|
CI
|
combination index
|
|
TNFα
|
tumor necrosis factor alpha
|
|
Iκβ
|
inhibitor of kappa beta
|
|
UPR
|
unfolded protein response
|
|
PARP
|
poly(ADP-ribose) polymerase
|
|
PERK
|
protein kinase RNA-like endoplasmic
reticulum kinase
|
|
eIF2α
|
eukaryotic translation initiation
factor 2α
|
|
XBP1
|
X-box binding protein 1
|
|
NAC
|
N-acetyl-cysteine
|
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics. CA Cancer J Clin. 59:225–249. 2009.
|
|
2
|
Kumar S: Multiple myeloma - current issues
and controversies. Cancer Treat Rev. 36(Suppl 2): S3–S11. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chari A, Mazumder A and Jagannath S:
Proteasome inhibition and its therapeutic potential in multiple
myeloma. Biologics. 4:273–287. 2010.PubMed/NCBI
|
|
4
|
Cavaletti G: Bortezomib-induced peripheral
neuropathy: facts and genes. Lancet Oncol. 12:120–121. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lai E, Teodoro T and Volchuk A:
Endoplasmic reticulum stress: signaling the unfolded protein
response. Physiology. 22:193–201. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Calfon M, Zeng H, Urano F, Till JH,
Hubbard SR, Harding HP, Clark S and Ron D: IRE1 couples endoplasmic
reticulum load to secretory capacity by processing the XBP-1 mRNA.
Nature. 415:92–96. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Redman BG, Flaherty L, Chou TH, al-Katib
A, Kraut M, Martino S, Chen B, Kaplan J and Valdivieso M: A phase I
trial of recombinant interleukin-2 combined with recombinant
interferon-gamma in patients with cancer. J Clin Oncol.
8:1269–1276. 1990.PubMed/NCBI
|
|
10
|
Funato T, Ishii T, Kanbe M, Scanlon KJ and
Sasaki T: Reversal of cisplatin resistance in vivo by an anti-fos
ribozyme. In Vivo. 11:217–220. 1997.PubMed/NCBI
|
|
11
|
Munshi NC: Arsenic trioxide: an emerging
therapy for multiple myeloma. Oncologist. 6(Suppl 2): S17–S21.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rousselot P, Larghero J, Labaume S, Poupon
J, Chopin M, Dosquet C, Marolleau JP, Janin A, Brouet JC and
Fermand JP: Arsenic trioxide is effective in the treatment of
multiple myeloma in SCID mice. Eur J Haematol. 72:166–171. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Munshi NC, Tricot G, Desikan R, Badros A,
Zangari M, Toor A, Morris C, Anaissie E and Barlogie B: Clinical
activity of arsenic trioxide for the treatment of multiple myeloma.
Leukemia. 16:1835–1837. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Binet F, Chiasson S and Girard D: Arsenic
trioxide induces endoplasmic reticulum stress-related events in
neutrophils. Int Immunopharmacol. 10:508–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Duncan G, Wang L, Liu P, Cui H,
Reddan JR, Yang BF and Wormstone IM: Arsenic trioxide initiates ER
stress responses, perturbs calcium signalling and promotes
apoptosis in human lens epithelial cells. Exp Eye Res. 85:825–835.
2007. View Article : Google Scholar
|
|
16
|
Tang CH, Chiu YC, Huang CF, Chen YW and
Chen PC: Arsenic induces cell apoptosis in cultured osteoblasts
through endoplasmic reticulum stress. Toxicol Appl Pharmacol.
241:173–181. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brooks JD, Paton VG and Vidanes G: Potent
induction of phase 2 enzymes in human prostate cells by
sulforaphane. Cancer Epidemiol Biomarkers Prev. 10:949–954.
2001.PubMed/NCBI
|
|
18
|
Gamet-Payrastre L, Li P, Lumeau S, Cassar
G, Dupont MA, Chevolleau S, Gasc N, Tulliez J and Terce F:
Sulforaphane, a naturally occurring isothiocyanate, induces cell
cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer
Res. 60:1426–1433. 2000.PubMed/NCBI
|
|
19
|
Fimognari C, Nusse M, Cesari R, Iori R,
Cantelli-Forti G and Hrelia P: Growth inhibition, cell-cycle arrest
and apoptosis in human T-cell leukemia by the isothiocyanate
sulforaphane. Carcinogenesis. 23:581–586. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Balasubramanain S, Chew YC and Eckert RL:
Sulforaphane sup-presses polycomb group protein level via a
proteasome-dependent mechanism in skin cancer cells. Mol Pharmacol.
80:870–878. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mi L, Gan N and Chung FL: Isothiocyanates
inhibit proteasome activity and proliferation of multiple myeloma
cells. Carcinogenesis. 32:216–223. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Doudican NA, Bowling B and Orlow SJ:
Enhancement of arsenic trioxide cytotoxicity by dietary
isothiocyanates in human leukemic cells via a reactive oxygen
species-dependent mechanism. Leuk Res. 34:229–234. 2010. View Article : Google Scholar
|
|
23
|
Yan H, Wang YC, Li D, Wang Y, Liu W, Wu YL
and Chen GQ: Arsenic trioxide and proteasome inhibitor bortezomib
synergistically induce apoptosis in leukemic cells: the role of
protein kinase Cdelta. Leukemia. 21:1488–1495. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Doudican N, Rodriguez A, Osman I and Orlow
SJ: Mebendazole induces apoptosis via Bcl-2 inactivation in
chemoresistant melanoma cells. Mol Cancer Res. 6:1308–1315. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Canestraro M, Galimberti S, Savli H,
Palumbo GA, Tibullo D, Nagy B, Guerrini F, Piaggi S, Cine N,
Metelli MR and Petrini M: Synergistic antiproliferative effect of
arsenic trioxide combined with bortezomib in HL60 cell line and
primary blasts from patients affected by myeloproliferative
disorders. Cancer Genet Cytogenet. 199:110–120. 2010. View Article : Google Scholar
|
|
26
|
Chou TC and Talaly P: A simple generalized
equation for the analysis of multiple inhibitions of
Michaelis-Menten kinetic systems. J Biol Chem. 252:6438–6442.
1977.PubMed/NCBI
|
|
27
|
Chou TC and Talaly P: Analysis of combined
drug effects: a new look at a very old problem. Trends Pharmacol
Sci. 4:450–454. 1983. View Article : Google Scholar
|
|
28
|
Lin JH, Li H, Yasumura D, Cohen HR, Zhang
C, Panning B, Shokat KM, Lavail MM and Walter P: IRE1 signaling
affects cell fate during the unfolded protein response. Science.
318:944–949. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Manga P, Bis S, Knoll K, Perez B and Orlow
SJ: The unfolded protein response in melanocytes: activation in
response to chemical stressors of the endoplasmic reticulum and
tyrosinase misfolding. Pigment Cell Melanoma Res. 23:627–634. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Feinman R, Siegel DS and Berenson J:
Regulation of NF-κB in multiple myeloma: therapeutic implications.
Clin Adv Hematol Oncol. 2:162–166. 2004.
|
|
31
|
Maniatis T: A ubiquitin ligase complex
essential for the NF-kappaB, Wnt/Wingless, and Hedgehog signaling
pathways. Genes Dev. 13:505–510. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Greenstein S, Krett NL, Kurosawa Y, Ma C,
Chauhan D, Hideshima T, Anderson KC and Rosen ST: Characterization
of the MM. 1 human multiple myeloma (MM) cell lines: a model system
to elucidate the characteristics, behavior, and signaling of
steroid-sensitive and -resistant MM cells. Exp Hematol. 31:271–282.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Marcu MG, Doyle M, Bertolotti A, Ron D,
Hendershot L and Neckers L: Heat shock protein 90 modulates the
unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol.
22:8506–8513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang K and Kaufman RJ: The unfolded
protein response: a stress signaling pathway critical for health
and disease. Neurology. 66:S102–S109. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Badr CE, Hewett JW, Breakefield XO and
Tannous BA: A highly sensitive assay for monitoring the secretory
pathway and ER stress. PLoS One. 2:e5712007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wen J, Feng Y, Huang W, Chen H, Liao B,
Rice L, Preti HA, Kamble RT, Zu Y, Ballon DJ and Chang CC: Enhanced
antimyeloma cytotoxicity by the combination of arsenic trioxide and
bortezomib is further potentiated by p38 MAPK inhibition. Leuk Res.
34:85–92. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Campbell RA, Sanchez E, Steinberg JA,
Baritaki S, Gordon M, Wang C, Shalitin D, Chen H, Pang S, Bonavida
B, Said J and Berenson JR: Antimyeloma effects of arsenic trioxide
are enhanced by melphalan, bortezomib and ascorbic acid. Br J
Haematol. 138:467–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Berenson JR, Matous J, Swift RA, Mapes R,
Morrison B and Yeh HS: A phase I/II study of arsenic
trioxide/bortezomib/ascorbic acid combination therapy for the
treatment of relapsed or refractory multiple myeloma. Clin Cancer
Res. 13:1762–1768. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Qi X and Mochly-Rosen D: The PKCdelta-Abl
complex communicates ER stress to the mitochondria - an essential
step in subsequent apoptosis. J Cell Sci. 121:804–813. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hung JH, Su IJ, Lei HY, Wang HC, Lin WC,
Chang WT, Huang W, Chang WC, Chang YS, Chen CC and Lai MD:
Endoplasmic reticulum stress stimulates the expression of
cyclooxygenase-2 through activation of NF-kappaB and pp38
mitogen-activated protein kinase. J Biol Chem. 279:46384–46392.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hayashi T, Saito A, Okuno S, Ferrand-Drake
M, Dodd RL and Chan PH: Damage to the endoplasmic reticulum and
activation of apoptotic machinery by oxidative stress in ischemic
neurons. J Cereb Blood Flow Metab. 25:41–53. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Walter L and Hajnoczky G: Mitochondria and
endoplasmic reticulum: the lethal interorganelle cross-talk. J
Bioenerg Biomembr. 37:191–206. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Brewster JL, Linseman DA, Bouchard RJ,
Loucks FA, Precht TA, Esch EA and Heidenreich KA: Endoplasmic
reticulum stress and trophic factor withdrawal activate distinct
signaling cascades that induce glycogen synthase kinase-3 beta and
a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol
Cell Neurosci. 32:242–253. 2006. View Article : Google Scholar
|
|
44
|
Kuhn DJ, Chen Q, Voorhees PM, Strader JS,
Shenk KD, Sun CM, Demo SD, Bennett MK, van Leeuwen FW, Chanan-Khan
AA and Orlowski RZ: Potent activity of carfilzomib, a novel,
irreversible inhibitor of the ubiquitin-proteasome pathway, against
preclinical models of multiple myeloma. Blood. 110:3281–3290. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mitsiades CS, Mitsiades NS, McMullan CJ,
Poulaki V, Kung AL, Davies FE, Morgan G, Akiyama M, Shringarpure R,
Munshi NC, Richardson PG, Hideshima T, Chauhan D, Gu X, Bailey C,
Joseph M, Libermann TA, Rosen NS and Anderson KC: Antimyeloma
activity of heat shock protein-90 inhibition. Blood. 107:1092–1100.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shah JJ and Orlowski RZ: Proteasome
inhibitors in the treatment of multiple myeloma. Leukemia.
23:1964–1979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ye L, Dinkova-Kostova AT, Wade KL, Zhang
Y, Shapiro TA and Talalay P: Quantitative determination of
dithiocarbamates in human plasma, serum, erythrocytes and urine:
pharmacokinetics of broccoli sprout isothiocyanates in humans. Clin
Chim Acta. 316:43–53. 2002. View Article : Google Scholar
|
|
48
|
Miller WH Jr, Schipper HM, Lee JS, Singer
J and Waxman S: Mechanisms of action of arsenic trioxide. Cancer
Res. 62:3893–3903. 2002.PubMed/NCBI
|