Introduction
RU486 (mifepristone) is a derivative of the
progestin norethindrone, and acts as a progesterone receptor
antagonist (1,2) or glucocorticoid receptor antagonist
(3,4). RU486 has potential antitumor
antineoplastic effects, and thus has been used for the treatment of
several types of cancers. In breast cancer, RU486 was found to
inhibit cell growth in a progesterone receptor-dependent manner
(5). RU486 also induced cell growth
inhibition in human ovarian epithelial carcinoma (6). Furthermore, in vivo, RU486
reduced tumor growth in human meningioma-implanted athymic nude
mice (7). Although the effect of
RU486 on cancer cells has been studied, the apoptosis-induced
mechanism of RU486 remains unclear in human leukemia cells.
Proteins of the Bcl-2 family consist of
pro-apoptotic (Bax and Bak) and anti-apoptotic members (Bcl-2 and
Bcl-xL). The balance between the pro-apoptotic Bcl-2 family and the
anti-apoptotic Bcl-2 family regulates mitochondrial functions. When
the pro-apoptotic Bcl-2 family is enhanced or the anti-apoptotic
Bcl-2 family is reduced, the permeability of the mitochondrial
membrane is increased, and then apoptogenic factors, such as
cytochome c, second mitochondria-derived activator of
caspase (Smac)/direct inhibitor of apoptosis-binding protein with
low pI (DIABLO), Omi/Htra2, endonuclease G, and apoptosis-inducing
factor are released (8,9). Smac/DIABLO and Omi/Htra2 facilitate
caspase activation, and endonuclease G and AIF induce DNA
fragmentation. Released apoptotic proteins activate caspase
signaling and initiate caspase-mediated DNA fragmentation and cell
death signaling. Therefore, the Bcl-2 family is important for the
modulation of mitochondrial-mediated apoptosis.
In the present study, we examined the mechanism of
RU486-induced apoptosis in U937 human leukemia cells. We found that
reduction in mitochondrial membrane potential and activation of p38
MAPK are associated with RU486-induced apoptosis.
Materials and methods
Cells and materials
U937, A549, MDA231 and HCT116 cells were obtained
from the American Type Culture Collection (ATCC, Rockville, MD,
USA). The culture medium used throughout these experiments was
RPMI-1640 (U937, A549, and HCT116) or DMEM (MDA231), containing 10%
fetal bovine serum (FBS), 20 mM HEPES buffer and 100 μg/ml
gentamicin. Bcl-2-overexpressing U937 cells were generated using a
pMAX vector containing the human Bcl-2 gene (provided by Dr
Rakesh Srivastava, NIH/NIA). U937 cells (400 μl) in RPMI-1640
(20×106 cells/ml) were transfected by pre-incubation
with 15 μg of the Bcl-2 plasmid for 10 min at room temperature and
then electroporating at 500 V, 700 μF. The sample was immediately
placed on ice for 10 min and then 10 ml complete medium was added
and the cells were incubated at 37°C for 24 h. The cells were
selected in a medium containing 0.7 μg/ml geneticin (G418) for 4
weeks. Single-cell clones were obtained by limiting dilution and
were subsequently analyzed for an increase in Bcl-2 protein
expression relative to the identically cloned empty vector control.
Chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Anti-Bcl-2, anti-Bcl-xL, anti-Mcl-1, anti-XIAP, anti-cIAP1,
anti-cIAP2, anti-cytochrome c, and anti-PARP antibodies were
purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). Anti-c-FLIP(L) antibody was obtained from Alexis Corporation
(San Diego, CA, USA). Anti-phospho-ERK, anti-phospho-JNK, anti-JNK,
anti-phospho-p38 MAPK and anti-p38 antibodies were purchased from
Cell Signaling Technology (Beverly, MA, USA). Anti-ERK antibody was
obtained from Transduction Laboratories (Lexington, KY, USA).
Anti-Qps-2 antibody was purchased from Molecular Probes (Eugene,
OR, USA). Anti-actin antibody was obtained from Sigma-Aldrich.
Flow cytometric analysis
Cells were suspended in 100 μl of phosphate-buffered
saline (PBS), and 200 μl of 95% ethanol was added during vortexing.
Cells were incubated at 4°C for 1 h, washed with PBS and
resuspended in 250 μl of 1.12% sodium citrate buffer (pH 8.4)
together with 12.5 μg of RNase. Incubation was continued at 37°C
for 30 min. The cellular DNA was then stained by applying 250 μl of
propidium iodide (50 μg/ml) for 30 min at room temperature. The
stained cells were analyzed by fluorescence-activated cell sorting
on a FACScan flow cytometer for the relative DNA content based on
red fluorescence.
Cell death assessment by DNA
fragmentation assays
The cell death detection ELISAPLUS kit (Boehringer
Mannheim, Indianapolis, IN, USA) was used for assessing apoptotic
activity by detecting fragmented DNA within the nucleus in the
RU486-treated cells. Briefly, each culture plate was centrifuged
for 10 min at 200 × g, the supernatant was removed, and the pellet
was lysed for 30 min. After centrifuging the plate again at 200 × g
for 10 min, the collected supernatant containing cytoplasmic
histone-associated DNA fragments was incubated with an immobilized
anti-histone antibody, and the reaction products were determined by
spectrophotometry. Finally, the absorbance at 405 nm and 490 nm
(reference wavelength), upon incubating with a peroxidase substrate
for 5 min, was determined with a microplate reader. Signals in the
wells containing the substrate only were subtracted as
background.
DEVDase activity assay
After treatment, cells were lysed, and 20 μg of cell
lysates was incubated with 100 μl reaction buffer (1% NP-40, 20
mmol/l Tris-HCl (pH 7.5), 137 mmol/l NaCl, 10% glycerol) containing
the caspase substrate (DEVD-chromophore p-nitroanilide) at 5
μmol/l. Lysates were incubated at 37°C for 2 h. The absorbance at
405 nm was measured with a spectrophotometer.
Western blot analysis
Cellular lysates were prepared by suspending
1.2×106 cells in 100 μl of lysis buffer (137 mM NaCl, 15
mM EGTA, 0.1 mM sodium orthovanadate, 15 mM MgCl2, 0.1%
Triton X-100, 25 mM MOPS, 100 μM phenylmethylsulfonyl fluoride, and
20 μM leupeptin, adjusted to pH 7.2). The cells were disrupted by
sonication and extracted at 4°C for 30 min. The proteins were
electrotransferred to Immobilon-P membranes (Millipore Corp., USA).
Detection of specific proteins was carried out with an ECL western
blotting kit according to the manufacturer’s instructions.
Analysis of cytochrome c release
U937 human leukemia cells (1.2×106
cells/ml) were harvested, washed once with ice-cold PBS and gently
lysed for 2 min in 80 μl ice-cold lysis buffer [250 mM sucrose, 1
mM EDTA, 20 mM Tris-HCl (pH 7.2), 1 mM DTT, 10 mM KCl, 1.5 mM
MgCl2, 5 μg/ml pepstatin A, 10 μg/ml leupeptin and 2
μg/ml aprotinin]. Lysates were centrifuged at 12,000 × g at 4°C for
10 min to obtain the supernatants (cytosolic extracts free of
mitochondria) and the pellets (fraction that contains
mitochondria). The resulting cytosolic fractions were used for
western blot analysis using an anti-cytochrome c
antibody.
Determination for the mitochondrial
membrane potential by rhodamine 123
Rhodamine 123 (Molecular Probes, Inc., Eugene, OR,
USA) uptake by mitochondria is directly proportional to its
membrane potential. U937 cells subjected to a 4-h treatment were
incubated with rhodamine 123 (5 μM) for 30 min in the dark at 37°C.
The cells were harvested and suspended in PBS. The mitochondrial
membrane potential was subsequently analyzed using a flow cytometer
(Becton-Dickinson, Franklin Lakes, NJ, USA).
Statistical analysis
Data were analyzed with one-way ANOVA, followed by
post hoc comparisons (Student-Newman-Keuls) using the Statistical
Package for Social Sciences 8.0 (SPSS Inc., Chicago, IL, USA).
Results
Cellular features characteristic of
apoptosis in U937 cells exposed to RU486
In order to investigate whether RU486 induces
apoptosis, U937 cells were treated with various concentrations of
RU486 for 12 h. We first determined the apoptosis in U937 cells
using flow cytometric analysis to detect hypodiploid cell
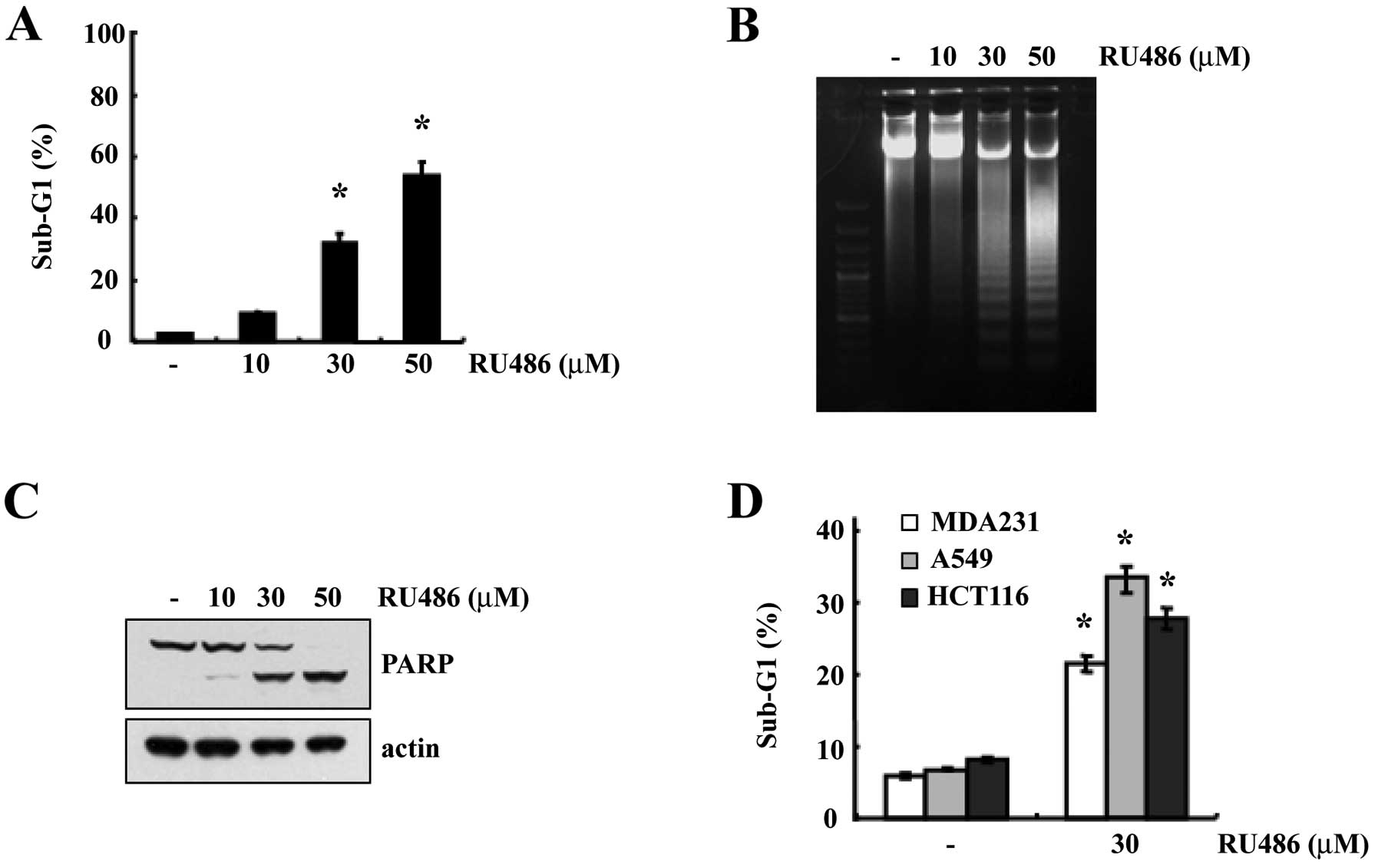
populations. As shown in Fig. 1A,
treatment of U937 cells with RU486 resulted in a markedly increased
accumulation of sub-G1 phase cells in a dose-dependent manner.
Next, we also investigated whether RU486 induces the apoptotic DNA
fraction in U937 cells. As shown in Fig. 1B, increasing concentrations of RU486
induced the progressive accumulation of apoptotic DNA. Since cells
undergoing apoptosis execute the death program by activating
caspases and cleaving PARP (10),
we next analyzed PARP cleavage. Exposure to RU486 induced a
dose-dependent cleavage of PARP (Fig.
1C). These results suggest that RU486 induced apoptosis in the
U937 human leukemia cells. In addition, RU486-induced apoptosis was
also observed in a variety of tumor cell types [breast carcinoma
cells (MDA231), lung carcinoma cells (A549) and colon cancer cells
(HCT116)], demonstrating that RU486-induced apoptosis is a common
response in various types of cancer cells (Fig. 1D).
Inhibition of RU486-induced apoptosis by
a caspase-3 inhibitor
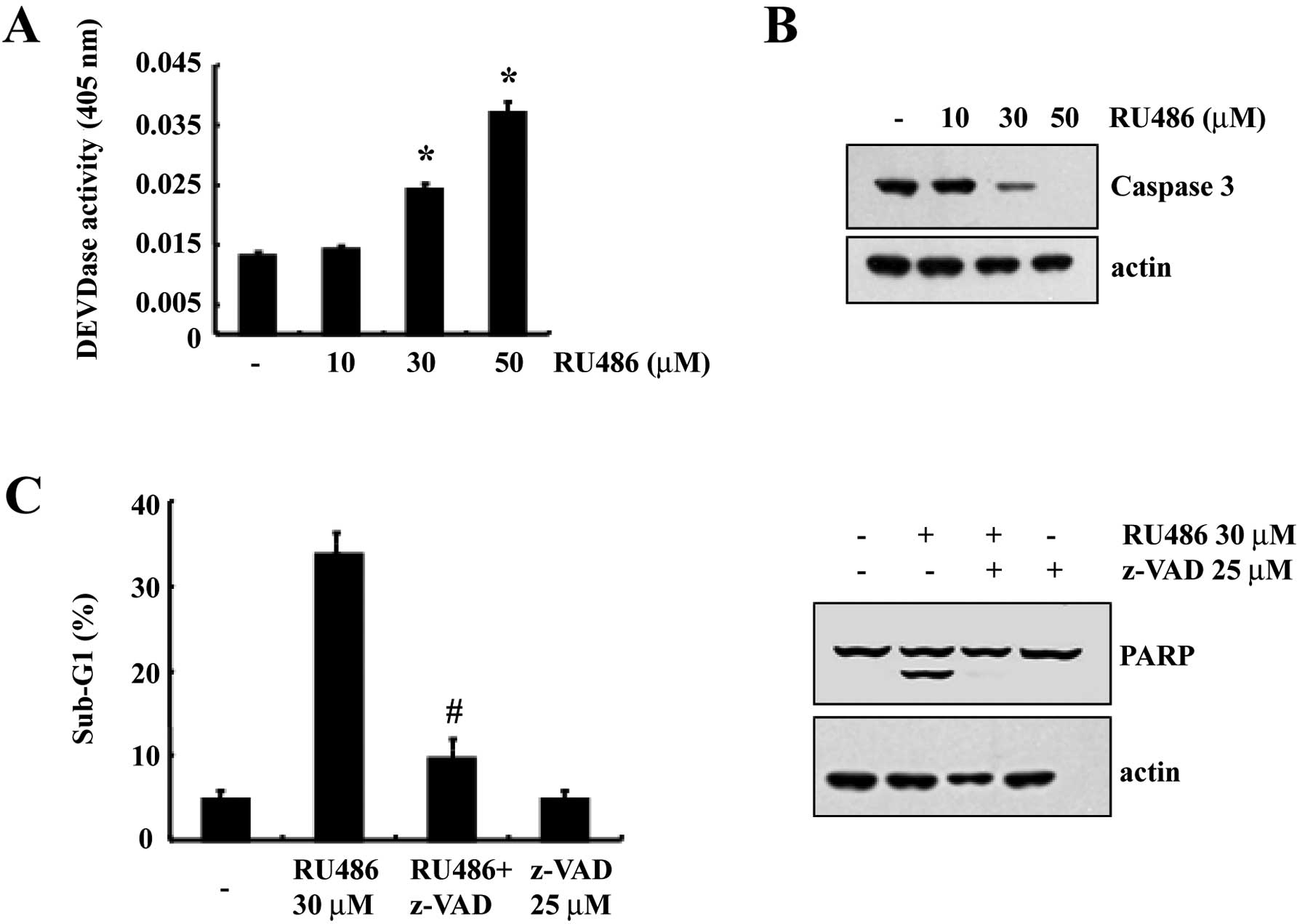
We next analyzed whether treatment with RU486
results in the activation of caspases, a key executioner of
apoptosis. Exposure of U937 cells to RU486 strongly stimulated
caspase-3 activity in a dose-dependent manner (Fig. 2A). As shown in Fig. 2B, western blot analysis showed that
RU486 concentration-dependently induced a marked change in the
cleavage of caspase-3. In order to confirm that the activation of
caspase-3 is a key step in the RU486-induced apoptotic pathway,
U937 cells were pretreated with z-VAD-fmk (25 μM), a cell-permeable
pan-caspase inhibitor, followed by treatment with 30 μM RU486 for
12 h. As shown in Fig. 2C,
RU486-induced apoptosis was significantly prevented by pretreatment
with the potent inhibitor of caspases, z-VAD-fmk, as determined by
FACS analysis. We also found that z-VAD-fmk prevented
caspase-related events such as cleavage of PARP (Fig. 2C). These results suggest that
RU486-induced cell death is associated with caspase-3
activation.
RU486 induces the downregulation of
anti-apoptotic proteins and cytochrome c release
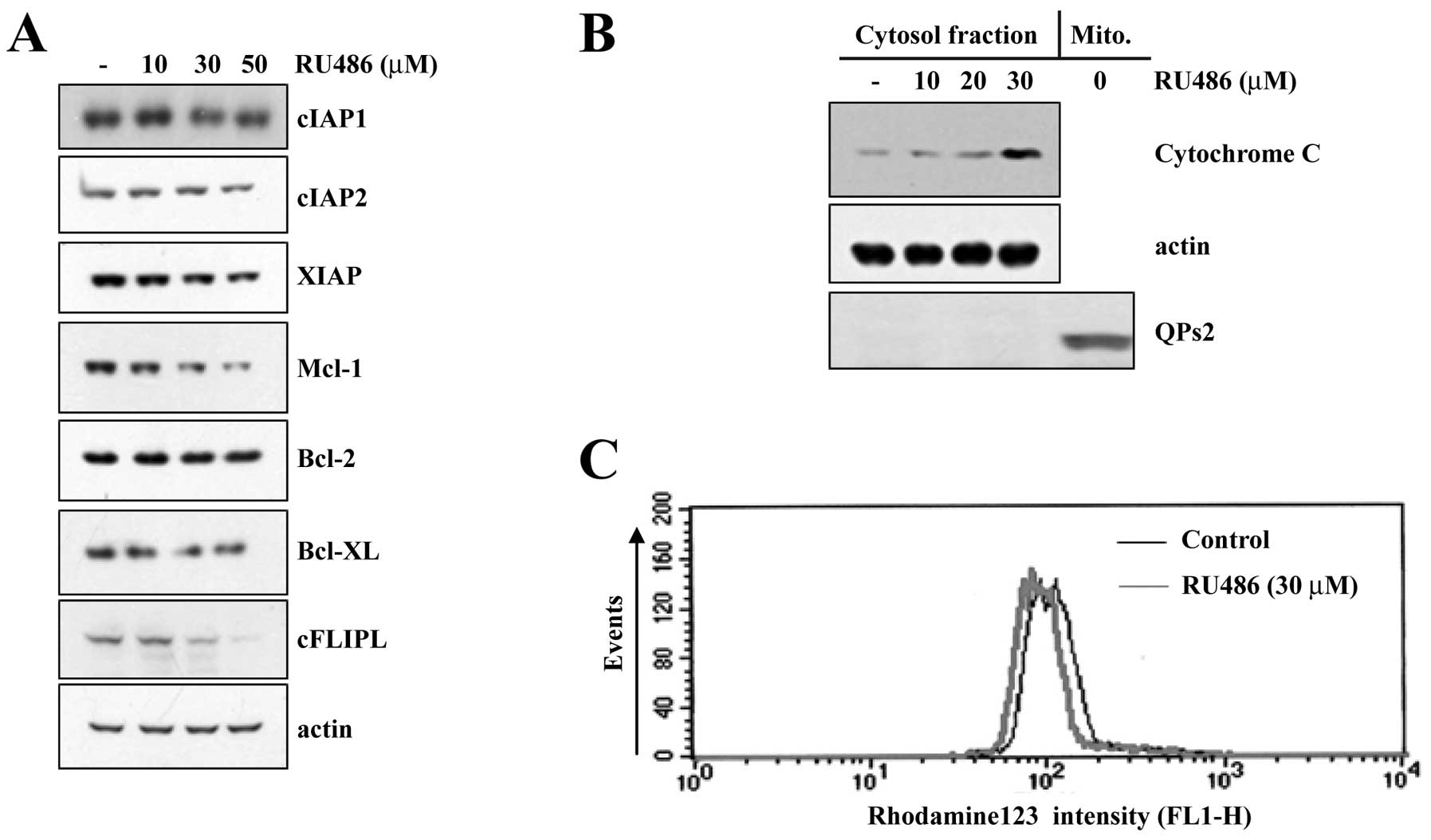
To investigate the underlying mechanisms involved in
RU486-induced apoptosis, we analyzed the changes in the expression
levels of various anti-apoptotic proteins. As shown in Fig. 3A, protein levels of Bcl-2, Bcl-xL,
cIAP1 and cIAP2 were not altered in response to RU486 treatment.
However, protein levels of Mcl-1, cFLIP(L) and XIAP were markedly
reduced in U937 cells following treatment at the indicated
concentrations of RU486 in a dose-dependent manner. Accumulating
evidence suggests that mitochondria play an essential role in
apoptosis by releasing apoptogenic effectors such as cytochrome
c. When we performed western blot analysis using cytosolic
fractions to examine the release of mitochondrial cytochrome
c in RU486-treated U937 cells, RU486 treatment markedly
induced a dose-dependent release of cytochrome c into the
cytoplasm (Fig. 3B). In addition,
the role of mitochondria in RU486-induced apoptosis of U937 cells
was further investigated by examining the effect of RU486 on
mitochondrial membrane potential (MMP, Δψm). Exposure of U937 cells
to 30 μM RU486 for 4 h led to a significant reduction in the MMP
level (Fig. 3C).
Effect of ectopic expression of Bcl-2 and
antioxidant on RU486-induced apoptosis
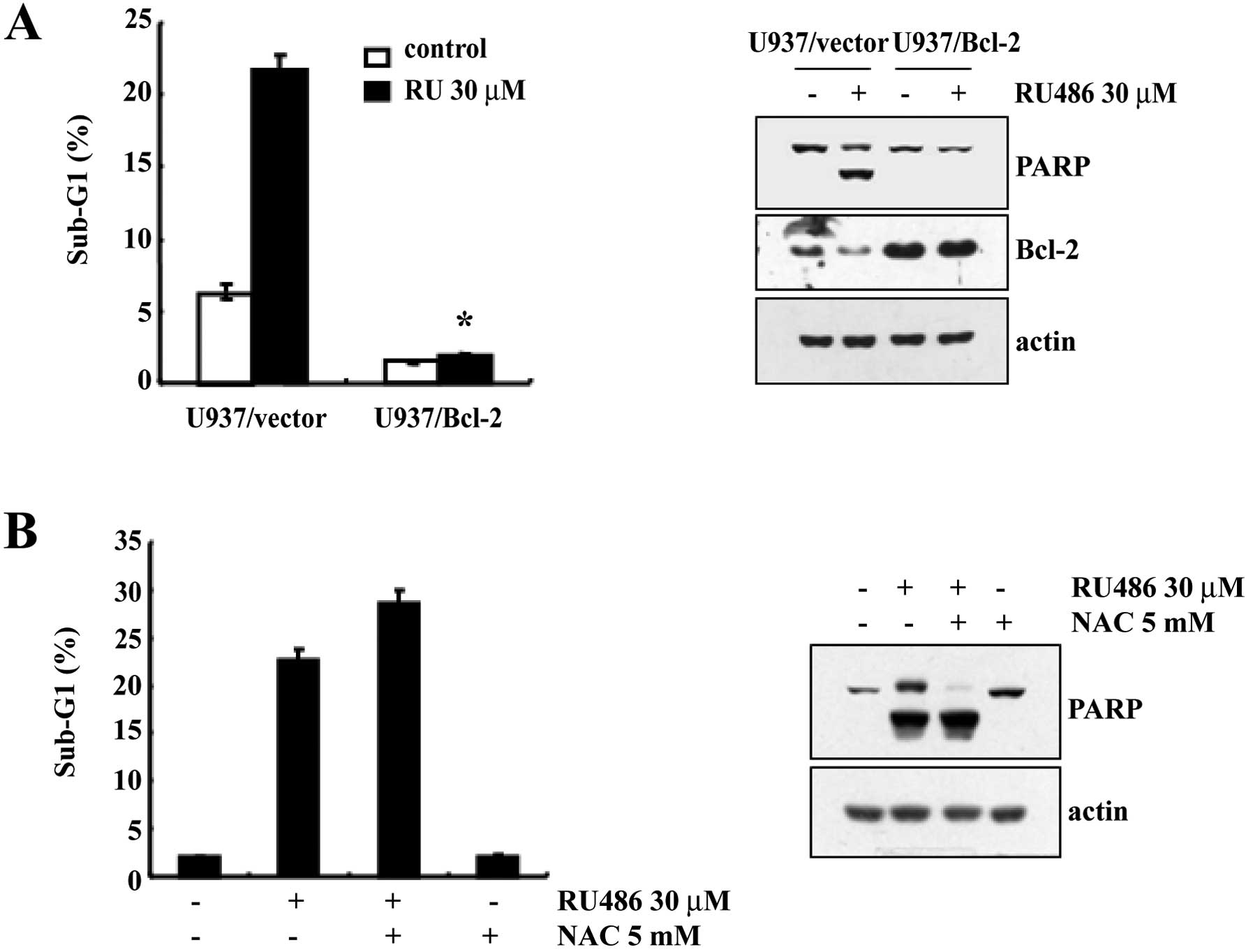
In order to evaluate the functional role played by
Bcl-2 in preventing RU486-induced apoptosis, cells stably
overexpressing Bcl-2 were established. As shown in Fig. 4A, treatment of U937/Vector cells
with 30 μM RU486 for 12 h resulted in a markedly increased
accumulation of cells in the sub-G1 phase. In contrast, the
accumulation of cells in the sub-G1 phase induced by RU486 was
inhibited by Bcl-2 overexpression. Subsequent western blot analysis
demonstrated that the proteolytic cleavage of PARP in U937/Vector
cells was more prominent than that in the U937/Bcl-2 cells when
exposed to RU486 (Fig. 4A). Taken
together, these results indicate that ectopic expression of Bcl-2
inhibits RU486-induced apoptosis.
Since oxidative stress-mediated cellular changes are
induced in cells exposed to cytotoxic drugs and ROS is known to be
a mediator of caspase-dependent cell death (11–13),
we investigated whether ROS generation induced by RU486 is directly
associated with the induction of apoptosis. However, pretreatment
with N-acetylcysteine (NAC) did not prevent RU486-induced increase
in the sub-G1 cell population and cleavage of PARP (Fig. 4B). These data indicate that ROS
generation is not critical for the induction of apoptosis by
RU486.
p38 MAPK pathways play important roles in
RU486-induced apoptosis
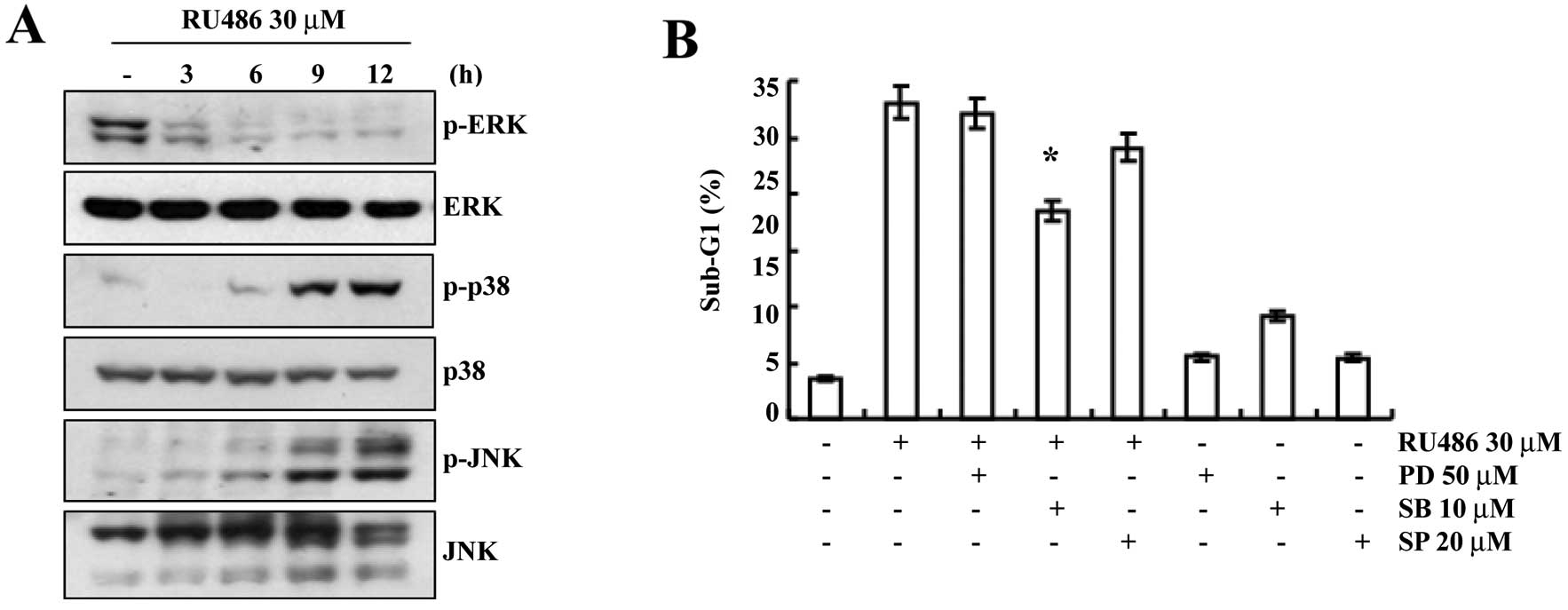
We investigated the effect of RU486 treatment on the
expression and activity of MAPKs in order to determine whether this
signaling pathway is involved in mediating the observed apoptotic
response. As shown in Fig. 5A,
RU486 treatment induced a decrease in the phosphorylated ERK levels
in a time-dependent manner. However, the phosphorylation of p38
MAPK and JNK gradually increased after RU486 treatment. We then
evaluated the possible roles of MAPKs in RU486 treatment-induced
apoptosis. As shown in Fig. 5B,
pretreatment with SB203580 (a specific inhibitor of p38 MAPK)
decreased the sub-G1 phase cell population (17.3%) when compared
with the sub-G1 phase cell population following RU486 treatment
(28.8%), while treatment of SP600125 (a potent inhibitor of JNK)
and PD98059 (a potent inhibitor of ERK) did not significantly
decrease the number of cells with sub-G1 DNA content. These results
indicate that the activation of the p38 MAPK pathway plays an
important role in regulating RU486-induced apoptosis in U937
cells.
Taken together, our results demonstrated that RU486
induced apoptosis through reduction in mitochondrial membrane
potential and activation of p38 MAPK in U937 human leukemia
cells.
Discussion
RU486 is an anti-progestin, which was primarily used
as an emergency contraceptive or abortion-inducing drug (14). RU486 was also found to demonstrate
promising results in cancer therapy. However, it was unclear
whether the mechanism of RU486 involves apoptosis of cancer cells.
In this study, we tested whether RU486 has potential for the
treatment of human leukemia and examined the mechanisms of
RU486-induced apoptosis in human leukemia cells. We observed the
following. i) RU486 activated the caspase-dependent apoptotic
pathway in a dose-dependent manner in U937 cells, which was
significantly prevented by pretreatment with a pan-caspase
inhibitor z-VAD-fmk (Fig. 2). ii)
RU486-induced apoptosis was closely correlated with reduction in
mitochondrial membrane potential (Fig.
3). iii) p38 MAPK inhibition by SB203580 (a specific inhibitor
of p38 MAPK) effectively inhibited the cell death induced by RU486,
demonstrating its critical role in this event (Fig. 5).
A previous report demonstrated that RU486 activity
was partially due to promotion of cellular apoptosis through
increased NF-κB binding resulting in overexpression of Bax and
downregulation of Bcl-2 in a human endometrial epithelial cell line
EM42 (15). Furthermore, in our
previous study, RU486 sensitized TRAIL-mediated apoptosis through
downregulation of Bcl-2 in human renal carcinoma Caki cells
(16). As shown in Fig. 4, ectopic expression of Bcl-2
significantly attenuated RU486-induced apoptosis and PARP cleavage
in U937 cells. Since Bcl-2 inhibits members of the caspase family
(17), ectopic expression of Bcl-2
inhibited RU486-mediated apoptosis in leukemia U937 cells (Fig. 4A).
Mitogen-activated protein kinases (MAPKs) are
serine/threonine kinases and induce the phosphorylation of multiple
substrates, which results in modulation of cellular proliferation,
motility and cell death (18–21).
MAPKs mainly consist of three family members (ERK, p38 MAPK and
JNK). Although the function of each MAPK is dependent on cell type,
stimuli, and duration of stimuli, ERK commonly has been known as an
anti-apoptotic signal (22,23). Activation of the ERK signaling
pathway is involved in induction of cellular proliferation,
differentiation and migration (24–26).
In contrast, p38 MAPK and JNK are associated with apoptotic
signaling (27). For examples,
multiple stresses such as DNA damage, UV irradiation, and oxidative
stress activate p38 MAPK and/or JNK, thus induce apoptosis
(28–31). While RU486 activated p38 MAPK and
JNK in a time-dependent manner (Fig.
5A), phosphorylation of ERK decreased in the RU486-treated
cells. Among MAPKs, p38 MAPK was solely involved in RU486-induced
apoptosis (Fig. 5B).
In conclusion, the results of our studies, for the
first time, provide mechanistic evidence that RU486 induces
apoptosis by loss of MMP, p38 MAPK activation and caspase
activation. The apoptosis-inducing ability of RU486 makes it a
potentially effective, preventive and/or therapeutic agent against
leukemia. However, additional in vivo studies are needed to
establish the role of RU486 as a chemopreventive and/or therapeutic
agent for cancer.
References
|
1
|
Herrmann W, Wyss R, Riondel A, et al: The
effects of an antiprogesterone steroid in women: interruption of
the menstrual cycle and of early pregnancy. CR Seances Acad Sci
III. 294:933–938. 1982.(in French).
|
|
2
|
Robbins A and Spitz IM: Mifepristone:
clinical pharmacology. Clin Obstet Gynecol. 39:436–450. 1996.
View Article : Google Scholar
|
|
3
|
Gagne D, Pons M and Philibert D: RU 38486:
a potent antiglucocorticoid in vitro and in vivo. J Steroid
Biochem. 23:247–251. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jung-Testas I and Baulieu EE: Inhibition
of glucocorticosteroid action in cultured L-929 mouse fibroblasts
by RU 486, a new anti-glucocorticosteroid of high affinity for the
glucocorticosteroid receptor. Exp Cell Res. 147:177–182. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bardon S, Vignon F, Chalbos D and
Rochefort H: RU486, a progestin and glucocorticoid antagonist,
inhibits the growth of breast cancer cells via the progesterone
receptor. J Clin Endocrinol Metab. 60:692–697. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rose FV and Barnea ER: Response of human
ovarian carcinoma cell lines to antiprogestin mifepristone.
Oncogene. 12:999–1003. 1996.PubMed/NCBI
|
|
7
|
Olson JJ, Beck DW, Schlechte JA and Loh
PM: Effect of the antiprogesterone RU-38486 on meningioma implanted
into nude mice. J Neurosurg. 66:584–587. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cheng EH, Wei MC, Weiler S, et al: BCL-2,
BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and
BAK-mediated mitochondrial apoptosis. Mol Cell. 8:705–711. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ruffolo SC and Shore GC: BCL-2 selectively
interacts with the BID-induced open conformer of BAK, inhibiting
BAK auto-oligomerization. J Biol Chem. 278:25039–25045. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O’Brien MA, Moravec RA and Riss TL: Poly
(ADP-ribose) polymerase cleavage monitored in situ in apoptotic
cells. Biotechniques. 30:886–891. 2001.PubMed/NCBI
|
|
11
|
Wen J, You KR, Lee SY, Song CH and Kim DG:
Oxidative stress-mediated apoptosis. The anticancer effect of the
sesquiterpene lactone parthenolide. J Biol Chem. 277:38954–38964.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sheng-Tanner X, Bump EA and Hedley DW: An
oxidative stress-mediated death pathway in irradiated human
leukemia cells mapped using multilaser flow cytometry. Radiat Res.
150:636–647. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fleury C, Mignotte B and Vayssière JL:
Mitochondrial reactive oxygen species in cell death signaling.
Biochimie. 84:131–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mahajan DK and London SN: Mifepristone
(RU486): a review. Fertil Steril. 68:967–976. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Han S and Sidell N: RU486-induced growth
inhibition of human endometrial cells involves the nuclear
factor-kappa B signaling pathway. J Clin Endocrinol Metab.
88:713–719. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Min KJ, Jang JH, Lee JT, Choi KS and Kwon
TK: Glucocorticoid receptor antagonist sensitizes TRAIL-induced
apoptosis in renal carcinoma cells through up-regulation of DR5 and
down-regulation of c-FLIP(L) and Bcl-2. J Mol Med (Berl).
90:309–319. 2012. View Article : Google Scholar
|
|
17
|
Cory S and Adams JM: The Bcl-2 family:
regulators of the cellular life-or-death switch. Nat Rev Cancer.
2:647–656. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choudhury GG, Karamitsos C, Hernandez J,
Gentilini A, Bardgette J and Abboud HE: PI-3-kinase and MAPK
regulate mesangial cell proliferation and migration in response to
PDGF. Am J Physiol. 273:F931–F938. 1997.PubMed/NCBI
|
|
19
|
Seger R and Krebs EG: The MAPK signaling
cascade. FASEB J. 9:726–735. 1995.PubMed/NCBI
|
|
20
|
Xi XP, Graf K, Goetze S, Fleck E, Hsueh WA
and Law RE: Central role of the MAPK pathway in ang II-mediated DNA
synthesis and migration in rat vascular smooth muscle cells.
Arterioscler Thromb Vasc Biol. 19:73–82. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bonni A, Brunet A, West AE, Datta SR,
Takasu MA and Greenberg ME: Cell survival promoted by the Ras-MAPK
signaling pathway by transcription-dependent and -independent
mechanisms. Science. 286:1358–1362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kitagawa D, Tanemura S, Ohata S, et al:
Activation of extracellular signal-regulated kinase by ultraviolet
is mediated through Src-dependent epidermal growth factor receptor
phosphorylation. Its implication in an anti-apoptotic function. J
Biol Chem. 277:366–371. 2002. View Article : Google Scholar
|
|
23
|
Klein JB, Buridi A, Coxon PY, et al: Role
of extracellular signal-regulated kinase and phosphatidylinositol-3
kinase in chemoattractant and LPS delay of constitutive neutrophil
apoptosis. Cell Signal. 13:335–343. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Whelchel A, Evans J and Posada J:
Inhibition of ERK activation attenuates endothelin-stimulated
airway smooth muscle cell proliferation. Am J Respir Cell Mol Biol.
16:589–596. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Qui MS and Green SH: PC12 cell neuronal
differentiation is associated with prolonged p21ras activity and
consequent prolonged ERK activity. Neuron. 9:705–717. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Graf K, Xi XP, Yang D, Fleck E, Hsueh WA
and Law RE: Mitogen-activated protein kinase activation is involved
in platelet-derived growth factor-directed migration by vascular
smooth muscle cells. Hypertension. 29:334–339. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen YR, Wang X, Templeton D, Davis RJ and
Tan TH: The role of c-Jun N-terminal kinase (JNK) in apoptosis
induced by ultraviolet C and gamma radiation. Duration of JNK
activation may determine cell death and proliferation. J Biol Chem.
271:31929–31936. 1996. View Article : Google Scholar
|
|
29
|
Turner NA, Xia F, Azhar G, Zhang X, Liu L
and Wei JY: Oxidative stress induces DNA fragmentation and caspase
activation via the c-Jun NH2-terminal kinase pathway in H9c2
cardiac muscle cells. J Mol Cell Cardiol. 30:1789–1801. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Naderi J, Hung M and Pandey S: Oxidative
stress-induced apoptosis in dividing fibroblasts involves
activation of p38 MAP kinase and over-expression of Bax: resistance
of quiescent cells to oxidative stress. Apoptosis. 8:91–100. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Luo Y, Umegaki H, Wang X, Abe R and Roth
GS: Dopamine induces apoptosis through an oxidation-involved
SAPK/JNK activation pathway. J Biol Chem. 273:3756–3764. 1998.
View Article : Google Scholar : PubMed/NCBI
|