Introduction
Lynch syndrome (LS) is the most common hereditary
colorectal cancer syndrome (1), and
is an autosomal dominant disease that accounts for 1–5% of all
colorectal cancer (CRC) patients (2–4). LS is
associated with germline mutations in one of the DNA mismatch
repair (MMR) genes, such as MLH1, MSH2, MSH6
or PMS2 (MIM#s 120436, 609309, 600678, 600259,
respectively). Carriers of MMR gene mutations are at high risk of
developing LS associated cancer in the colorectum, endometrium,
small bowel, stomach, ovary, ureter, biliary tract, brain and skin.
Individuals at high-risk can be identified by germline genetic
testing.
To identify individuals at risk for LS, the
Amsterdam criteria and the Bethesda guidelines based on the family
cancer history and age at cancer diagnosis have been proposed
(5,6). However, MSH6 mutation carriers
do not necessarily satisfy these criteria, as they tend to develop
CRC at an older age than MLH1 or MSH2 mutation
carriers and have reduced penetrance (7–10).
Thus, there are some difficulties associated with using these
criteria in the clinical practice, and a molecular analysis of the
tumor may help to identify individuals who have LS. The tumors from
LS patients are characterized by the microsatellite instability
(MSI) and a loss of expression of MMR proteins detected by
immunohistochemistry (IHC). Routine MSI and IHC testing for CRC
patients is an effective screening tool to identify overlooked
cases of LS. Approximately 6–15% of all CRCs display high-level of
MSI (MSI-H) and in the majority of these cases, the MMR defects
result from inactivation of MLH1 by the methylation of its
promoter (11–13). The presence of MLH1 promoter
hypermethylation generally suggests sporadic CRC, although there
are some exceptions. In addition, the BRAF mutation,
p.V600E, has also been identified in colorectal tumors showing MMR
deficiency associated with the epigenetic silencing of the
MLH1 gene, and previous studies showed that tumors from
patients with germline mutations in the MMR genes did not show
somatic mutations in BRAF(14). Thus, the results of the analyses of
MSI, IHC, MLH1 methylation and BRAF mutation in
tumors may aid in the diagnosis of LS.
In the present study, we identified 15 novel
variants of MSH6 and evaluated the molecular and clinical
characteristics of these patients. This is the first report to
describe the detailed data of Japanese LS due to MSH6
mutation.
Materials and methods
Samples and DNA extraction
The CRC samples were collected from patients who
were treated at Saitama Cancer Center beginning in 1998, and 1720
paired surgical specimens of primary CRC and normal mucosa were
used for the present study. Informed consent was obtained from each
patient. Fresh tissue samples were immediately frozen at −80°C.
Genomic DNA was extracted using the standard method (15). This research protocol was approved
by the Institutional Review Board of Saitama Cancer Center.
Clinical database collection
The clinical features, such as the personal and
family cancer history and pathological findings of the tumors, were
collected from medical records or directly from patients who were
provided genetic counseling. The personal and family cancer history
was classified into five categories based on the clinical criteria
(Table I). The criteria were
defined based on both the Amsterdam criteria II (16) and revised Bethesda guidelines
(17).
 | Table IDefinition of the clinical
criteria. |
Table I
Definition of the clinical
criteria.
| Criteria |
|---|
| A2 | Amsterdam criteria
II: three or more family members affected with Lynch
syndrome-associated cancera, one
of whom was a first-degree relative of the other two, at least two
successive generations were affected and at least one cancer was
diagnosed at the age of <50. Familial adenomatous polyposis
(FAP) was excluded. |
| B1 | Revised Bethesda
guidelines 1: a patient who was diagnosed with colorectal cancer at
the age of <50. |
| B2 | Revised Bethesda
guidelines 2: a patient with synchronous or metachronous
colorectal, or other Lynch syndrome-related tumorb, regardless of the age at
diagnosis. |
| B4 | Revised Bethesda
guidelines 4: a patient with colorectal cancer who has one or more
first-degree relatives with a Lynch syndrome-related tumorb diagnosed at the age of <50. |
| B5 | Revised Bethesda
guidelines 5: a patient with colorectal cancer who has two or more
first- or second-degree relatives with Lynch syndrome-related
tumorb regardless of the age at
diagnosis. |
Analysis of microsatellite
instability
The MSI status of the colorectal cancers was
investigated according to a previously reported method (18). A reference panel of five MSI markers
recommended by the National Cancer Institute workshop: BAT-25,
BAT-26, D2S123, D5S346 and D17S250, were used. A variation in the
number and size of peaks of a marker in tumor DNA compared with
that of normal DNA was interpreted to indicate instability for the
markers. Tumors were classified as MSI-H if at least two of the
five markers showed instability compared with normal tissues, and
were classified as low-level of MSI (MSI-L) if one marker showed
instability. Tumors showing no instability were classified as
microsatellite stable (MSS).
Methylation of the MLH1 promoter and BRAF
mutation
The methylation status of the MLH1 promoter
in MSI-H CRC was investigated by methylation-specific PCR. In
addition, a mutational analysis of BRAF was performed by
PCR-RFLP and sequencing and the analysis focused specifically on
the V600E mutation. These analyses were carried out by the methods
previously reported (19).
Mutation analysis of the MMR genes
Germline mutations in MLH1, MSH2 and
MSH6 were investigated in patients with MSI-H CRC. The
mutation analyses were performed by direct sequencing of the entire
coding region and large deletions/duplications in the MLH1
and MSH2 genes were also investigated by multiplex
ligation-dependent probe amplification (MLPA). The mutation
description is according to the reference sequence: NM_000249.2,
NM_000251.1 and NM_000179.2. The G39E variant in MSH6
(rs1042821) was considered to be non-pathogenic due to the high
frequency of the allele (20) and
the variant was excluded from this report. If a pathogenic mutation
was detected in the CRC patients, their relatives were referred to
for genetic testing. Informed consent was obtained from each
relative before blood samples were collected. The mutation analyses
were performed by limiting it to the probands' mutation site.
Immunohistochemical analysis for the MMR
protein
Immunohistochemical staining was performed for the
MLH1, MSH2, MSH6 and PMS2 proteins. A total of 70 tumor specimens
showing MSI-H were tested. The tumor and normal frozen samples were
dissected in 4-μm serial sections. Frozen sections were fixed with
4% paraformaldehyde containing 0.5% triton. Mouse anti-human
monoclonal antibodies against MLH1 (G168–728; Cell Marque, Rocklin,
CA, USA, at 1:500 dilution), MSH2 (NA27; Calbiochem, Billerica, MA,
USA, at 1:500 dilution) and PMS2 (PDM171; Diagnostic BioSystems,
Pleasanton, CA, USA, at no dilution) and a rabbit anti-human
monoclonal antibody against MSH6 (ab92471; Cambridge, UK, at 1:100
dilution) were used for the present study. The incubation periods
were 45 min at room temperature for MLH1 and MSH2, 2 h at room
temperature for MSH6 and overnight at 4°C for PMS2. The reviewers
of the immunostaining samples were blinded to the results of the
genetic testing. Stained samples were classified into positive
(normal expression of nuclear staining) or negative (loss of
nuclear staining) groups. It is known that loss of only the MSH6 or
PMS2 protein expression is associated with the respective genetic
mutations, but that loss of the MSH2 protein expression is
accompanied by loss of MSH6, and loss of MLH1 is accompanied by
loss of PMS2 (21).
In silico analysis
In order to classify the MSH6 missense
variants into pathogenic or non-pathogenic, the clinical and
molecular data and in silico predictions were integrated.
The in silico tools, CoDP (22), SIFT (23), PolyPhen-2 (24) and PON-MMR (25) were used in the present study.
PolyPhen-2 calculates the values of both HumDiv and HumVar. The
HumDiv is used for the diagnosis of Mendelian diseases, and HumVar
is used for the evaluation of rare alleles potentially involved in
complex phenotypes (24). Both
values were used in the present study.
Statistical analysis
The data were calculated as totals, means, medians
or percentages. Student's t-test, the Mann-Whitney U-test, one-way
analysis of variance (ANOVA) or the Kruskal-Wallis test was used
for statistical analyses as appropriate. The analyses were carried
out using the PASW Statistics 18.0.0 software program (SPSS, Inc.,
Chicago, IL, USA), with a value of P<0.05 considered to indicate
a statistically significant result.
Results
Variant analysis
We identified nine pathogenic variants which were
truncating mutations and eight missense variants in MSH6.
The pathogenic germline mutations were c.619G>T; p.E207X,
c.1088delC, c.1149delG, c.1571dupA, c.1909_1913delCTCCT,
c.2773G>T; p.Q925X, c.2932C>T; p.Q978X, c.3261dupC and
c.3961A>T; p.R1321X. The missense variants were c.532C>T;
p.R178C, c.1190A>G (rs63750065); p.Y397C, c.1505T>C; p.I502T,
c.2780T>C; p.I927T, c.3244C>T (rs186240214); p.P1082S,
c.3464A>G; p.Q1155R, c.3947G>A; p.G1316E and c.4071T>G;
p.I1357M (Table II) and these are
all variants of uncertain significance (VUS). These variant
carriers did not have other mutations in MLH1 and
MSH2 except p.R178C variant carrier. None of the nine
pathogenic mutations are listed in the InSiGH (17), MMRUV (26) or HGMD (27) databases, and hence, these are novel
mutations in MSH6 associated with LS. Other than p.Y397C and
p.P1082S, none of the missense variants were listed in these
databases. The Allele frequencies of the missense variants were
searched from the dbSNP (28) and
1000 genomes (http://www.1000genomes.org/) databases. None of the
variants were reported in either of these databases, nor were they
polymorphism with a population-based allele frequency >0.01
(Table II), except for
c.3244C>T, which had an allele frequency of 0.0014.
| Table IIDetected variants and molecular
features of the colorectal tumors. |
Table II
Detected variants and molecular
features of the colorectal tumors.
| Patient no. | Genetic
testing | MSI | IHC | MLH1
methylation | BRAF
mutation |
|---|
|
|
|
|---|
| Exon | Mutation | Consequence | MAF | BAT-25 | BAT-26 | D5S346 | D17S250 | D2S123 | Missing
protein |
|---|
| 1 | 3 | c.619G>T;
p.E207X | Nonsense | ND | + | + | − | − | − | MSH6 | − | − |
| 2 | 4 | c.1088delC | Frameshift | ND | + | + | − | − | − | MSH6 | − | − |
| 3 | 4 | c.1149delG | Frameshift | ND | + | + | + | − | + | MSH6 | − | − |
| 4 | 4 | c.1571dupA | Frameshift | ND | + | + | + | + | + | MSH6 | − | − |
| 5 | 4 |
c.1909_1913delCTCCT | Frameshift | ND | + | + | + | − | + | MSH6 | − | − |
| 6 | 4 | c.2773G>T;
p.Q925X | Nonsense | ND | + | + | − | − | + | MSH6 | − | − |
| 7 | 4 | c.2932C>T;
p.Q978X | Nonsense | ND | + | + | − | − | − | MSH6 | − | − |
| 8 | 5 | c.3261dupC | Frameshift | ND | + | + | − | + | LOH | MSH2, MSH6 | − | − |
| 9 | 9 | c.3961A>T;
p.R1321X | Nonsense | ND | + | + | − | − | − | None | − | − |
| 10 | 3 | c.532C>T;
p.R178C | Missense | ND | + | + | + | + | + | MSH2, MSH6 | − | − |
| 11 | 4 | c.1190A>G;
p.Y397C | Missense | ND | + | + | + | + | + | MLH1, PMS2 | − | − |
| 12 | 4 | c.1505T>C;
p.I502T | Missense | ND | + | + | + | + | + | MLH1, PMS2 | + | + |
| 13 | 4 | c.2780T>C;
p.I927T | Missense | ND | + | + | + | LOH | + | MLH1, PMS2 | + | + |
| 14 | 5 | c.3244C>T;
p.P1082S | Missense | 0.0014 | + | + | − | + | + | MLH1, PMS2 | + | − |
| 15 | 6 | c.3464A>G;
p.Q1155R | Missense | ND | + | + | + | + | + | MSH2, MSH6 | − | − |
| 16 | 9 | c.3947G>A;
p.G1316E | Missense | ND | + | + | + | + | − | MSH6 | − | − |
| 17 | 10 | c.4071T>G;
p.I1357M | Missense | ND | + | + | + | + | + | MSH2, MSH6 | − | − |
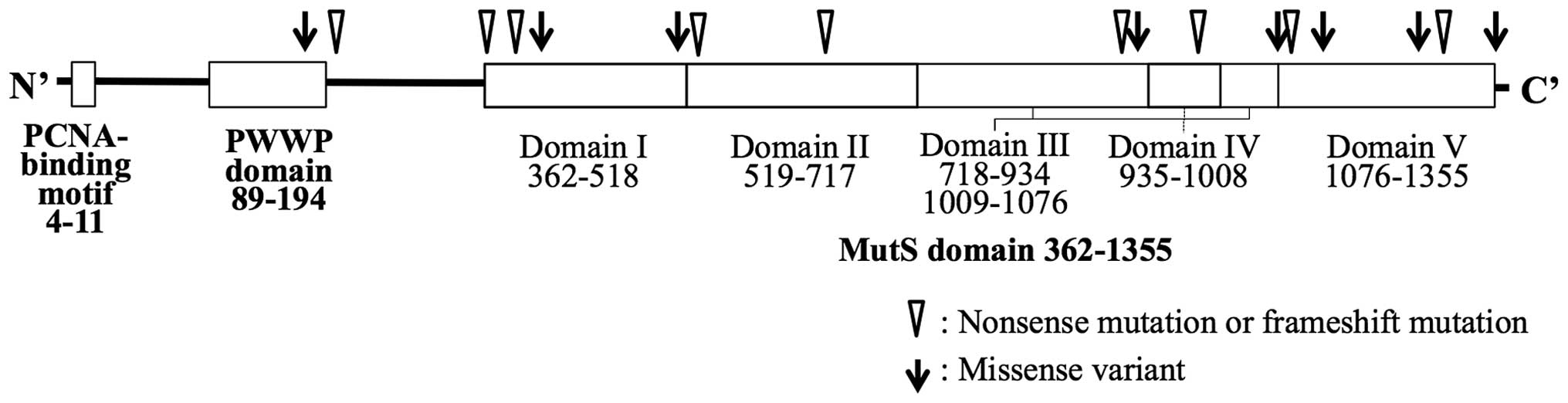
The distribution of the truncating mutations and
missense variants are shown in Fig.
1. The majority of the mutations were located in the MutS
domain, but no spatial preference of the mutation site in the
domain was found. The molecular features of the tumors are shown in
Table II and the clinical features
of the carriers are shown in Table
III.
| Table IIIClinical features of the
carriers. |
Table III
Clinical features of the
carriers.
| | Colorectal
cancer | Clinical
criteriac | | |
|---|
| |
|
| | |
|---|
| Patient no. | Gender | Age at
diagnosis | Location of
tumora | Pathological
typeb | Dukes' stage | A2 | B1–5 | Multiple
cancerd (LS-related cancer) | Family cancer
historye (LS-related cancer) |
|---|
| 1 | Male | 51 | S | tub2+muc | B | NE | NE | - | Mo, endometrial or
cervical (55), brain (70); At1–3, endometrial or cervical (NA) |
| 2 | Female | 70 | D | tub1 | A | + | 2,4,5 | S, colon, M,
endometrial (74) | Br, colon (80); Ne,
colon (46); DtE+, endometrial (43) |
| 3 | Female | 46 | S | tub2 | B | − | 1,2 | S, endometrial,
ovary | GF, stomach
(81) |
| 4 | Female | 53 | R | tub2+muc | C | − | 2 | S, endometrial | - |
| 5 | Male | 50 | T | tub2 | B | − | - | - | Fa, bladder (45),
stomach (64) |
| 6 | Male | 62 | S | tub2+muc | B | − | 4 | - | DtE+,
endometrial (30) |
| 7 | Male | 56 | R | tub2 | B | − | 2 | S, stomach,
colon | GF, stomach
(73) |
| 8 | Male | 53 | A | por | D | − | 5 | - | Mo, gallbladder
(68); Un, rectal (55) |
| 9 | Male | 41 | S | por+muc | D | + | 1,2,5 | S, colon | Mo, colon (65); Un
1, colon (60); Un 2, colon (70) |
| 10 | Female | 36 | S | por+muc | B | − | 1,2 | M, endometrial
(38) | GF, stomach
(70) |
| 11 | Female | 57 | A | tub2 | B | − | - | - | Mo, colon (72) |
| 12 | Female | 70 | C | por | C | − | - | - | Br, stomach
(63) |
| 13 | Female | 86 | T | tub2+muc | D | − | 2 | S, colon | - |
| 14 | Male | 54 | A | tub2+muc | B | − | - | - | Fa, stomach
(59) |
| 15 | Male | 51 | R | tub2+muc | B | − | 2 | M, colon (52) | - |
| 16 | Female | 49 | T | tub2 | A | − | 1 | - | Br, colon (53),
pancreas (53) |
| 17 | Male | 68 | R | tub2 | B | − | - | - | - |
Molecular features of the variant
carriers
As shown in Table
II, the dinucleotide markers were more stable than the
mononucleotide markers in tumors from the pathogenic variant
carriers, but not in the missense variant carriers. Of the nine
tumors from the carriers of the pathogenic variants, eight showed
the absence of staining for MSH6 and retained staining for both
MLH1 and PMS2 in the IHC analysis. Only one tumor from pR1321X
mutation carrier showed expression of all proteins. In short, in
seven of the nine tumors from the pathogenic variant carriers, IHC
showed a specific pattern with negative staining for MSH6 and
positive staining for MSH2, MLH1 and PMS2. On the other hand, four
of the eight tumors from the missense variant carriers showed a
loss of MSH6 expression and the other four showed a loss of the
expression of both MLH1 and PMS2. Of the four tumors showing
negative staining for MSH6, three had also lost the expression of
the MSH2 protein. Most tumors without MLH1 expression had the
BRAF mutation and/or hypermethylation of MLH1
promoter (Table II).
Clinical features of the variant
carriers
The mean and median age of the patients carrying
pathogenic variants at the diagnosis of CRC was 53.6 and 53.0 years
(range, 41–70, patient nos. 1–9), respectively. The mean and median
age of the patients with hypermethylation in the MLH1
promoter (patient nos. 12–14) were 70.0 and 70.0 years (range,
54–86), while those of the patients without hypermethylation was
53.0 and 54.0 years (range, 36–68) in the missense variant carriers
(patient nos. 10, 11, 15–17), respectively (Table II). The patients with
hypermethylation in the MLH1 promoter tended to develop CRC
at a more advanced age than did the carriers of the pathogenic
variants (Student's t-test, P=0.040; Mann-Whitney U-test, P=0.064).
Meanwhile, in the pathogenic variant carriers, including affected
relatives, the mean and median ages at the diagnosis of endometrial
cancer was 49.2 and 46.0 years (range, 30–74) respectively,
indicating that endometrial cancer developed earlier than CRC.
As shown in Table
III, of the nine pathogenic variant carriers, seven had tumors
in the left side colon or rectum, and five had multiple cancers. In
the histological findings of the CRCs, four pathogenic variant
cases contained mucinous adenocarcinoma and poorly differentiated
adenocarcinoma was observed in two cases. Tumor infiltrating
lymphocytes were observed in most cases (data not shown).
Concerning the clinical criteria, of the nine pathogenic variant
carriers, two fulfilled the Amsterdam criteria II and seven
fulfilled one of the criteria included in the revised Bethesda
guidelines. Four cases had a family cancer history that more than
two relatives affected LS-related cancer. On the other hand, in the
missense variant carriers, none of the patients fulfilled the
Amsterdam criteria II. Although four fulfilled at least one of the
criteria in the revised Bethesda guidelines, the B4 and B5 criteria
related to a family history of LS-related cancer were not fulfilled
in any of the cases.
Prediction by in silico tools
We also investigated the impact of the missense
variants of MSH6 to classify them as either pathogenic or
non-pathogenic by an in silico prediction using the CoDP,
SIFT, PolyPhen-2 and PON-MMR programs (Table IV). PON-MMR classified six variants
as unclassified variants. Excluding the results obtained from the
PON-MMR program, five missense variants gave consistent results;
the Y397C, Q1155R and G1316E variants were predicted to be
pathogenic, and the I502T and P1082S variants were predicted to be
non-pathogenic.
| Table IVPrediction results of in
silico tools in missense variants. |
Table IV
Prediction results of in
silico tools in missense variants.
| | | PolyPhen-2c | |
|---|
| | |
| |
|---|
| Missense
variant | CoDPa | SIFTb | HumDiv | HumVar | PON-MMRd |
|---|
| R178C | 0 (0.009) | 0 | 1 | 0 | U |
| Y397C | 2 (0.976) | 1 | 2 | 1 | U |
| I502T | 0 (0.001) | 0 | 0 | 0 | 0 |
| I927T | 0 (0.113) | 1 | 2 | 1 | U |
| P1082S | 0 (0.018) | 0 | 0 | 0 | U |
| Q1155R | 2 (0.980) | 1 | 2 | 2 | U |
| G1316E | 2 (0.999) | 1 | 2 | 2 | U |
| I1357M | 0 (0.002) | 1 | 0 | 0 | 0 |
Discussion
Accumulating evidence indicates that difference of
the mutated MMR genes, geographic region and gender affects the
clinical phenotype of Lynch syndrome (29). However, the majority of previous
studies have been conducted on Caucasian European populations and
there have been few reports on Japanese LS patients carrying
MSH6 mutations. In the present study, we screened colorectal
cancer by MSI testing, we performed a germline mutation analysis
and identified nine pathogenic and eight unclassified missense
variants in MSH6, 15 of which were novel variants. A
comprehensive investigation of the molecular and clinical
characteristics of the carriers of those variants was performed,
and several unique features in Japanese LS patients carrying
MSH6 mutations were found.
The mean age at the diagnosis of colorectal
carcinoma in the MSH6 pathogenic variant carriers was 53.6
years, and the earliest onset was at age 41 in the present study.
It was previously reported that the mean age at diagnosis of
colorectal carcinoma in MLH1 or MSH2 mutation
carriers was 43–46 years but was 51–57 years in MSH6
mutation carriers in Western countries (30). Thus, the development of colorectal
carcinomas in Japanese MSH6 mutation carriers was similar to
the previous reports from Western countries. On the other hand,
mean age at the diagnosis of endometrial carcinoma was 49.2 years,
and the earliest onset was at age 30 (the daughter of patient no.
6) in the present study. Previous studies from Western countries
reported that mean age at the diagnosis of endometrial cancer was
~48 years in MLH1 or MSH2 mutation carriers (9,30,31)
and 56.5 years in MSH6 mutation carriers (32). Thus, the endometrial carcinoma in
MSH6 mutation carriers was generally diagnosed at an older
age (~5 years later) than colorectal carcinoma in the previous
reports. However, Japanese MSH6 mutation carriers tended to
develop endometrial carcinoma earlier than colorectal carcinoma.
Although the small number of cancer cases limited statistical
power, this result may provide evidence to support the development
of effective surveillance programs for Japanese LS due to
MSH6 mutations. In other words, the finding of the
early-onset of endometrial carcinoma in MSH6 mutation
carriers in our study suggests that the surveillance program for
endometrial carcinoma may need to be started from an earlier age in
Japan.
Another marked finding of the present study was the
location of colorectal carcinomas in MSH6 mutation carriers.
The CRCs from MMR mutation carriers are mostly located in the
right-sided colon, in contrast to sporadic colorectal carcinomas,
which are generally detected in the distal region (17). However, the present study noted that
the majority (77.8%) of colorectal carcinomas were located distally
in Japanese MSH6 mutation carriers, and these were all
MSI-H. Berends et al(8)
reported that two-thirds of the colorectal carcinomas from
MSH6 mutation carriers were located on the left side, but
more than half of these were MSS or MSI-low. Furthermore,
approximately one-third of the tumors were detected to express the
MSH6 protein in their study, but only one tumor (11%) was detected
with MSH6 protein expression by IHC in our study. Thus, the MSI and
IHC status differed in the two studies.
In general, the Amsterdam criteria and Bethesda
guidelines are used to screen patients for Lynch syndrome. Of the
nine mutation carriers in the present study, two fulfilled the
Amsterdam criteria II and seven fulfilled the revised Bethesda
guidelines but these criteria missed one LS case. Therefore, MSI
testing for all CRC cases would be a useful screening method for
high-risk individuals with an MSH6 mutation. However, some
MSH6 mutation carriers developed MSI-L or MSS tumors
(32). While, an IHC analysis is
another potential screening strategy for LS, a recent study
reported that ~30% of colon tumors in carriers of pathogenic
MSH6 mutations still showed MSH6 protein expression
(33). Thus, neither of these
methods is a perfect screening tool, and a combination of MSI and
IHC testing to screen MSH6 mutation carriers would be a more
appropriate method.
To classify the missense variants detected in the
present study, we integrated the molecular and clinical data and
the results of an in silico prediction. A classification
system for VUS was previously proposed, based on the probability of
pathogenicity (34). The system
classifies genetic variants into five categories: class-5,
definitely pathogenic; class-4; likely pathogenic; class-3,
uncertain; class-2, likely not pathogenic or of little clinical
significance; and class-1, definitely neutral. Based on this
system, the eight missense variants were classified into five
categories.
Hypermethylation of the MLH1 promoter and
loss of MLH1 and PMS2 expression detected by IHC was observed in
the tumors from the I502T, I927T and P1082S variant carriers. Two
of these patients were over 70 years old at the time of the
diagnosis of CRC, and two did not fulfill the clinical criteria.
Therefore, these tumors were considered to be sporadic and these
variants were categorized as class-2 or −3.
The R178C variant carrier also harbored an H639Y
variant in MSH2. The variant in MSH2 was classified
as pathogenic by integrative consideration of a clinical report
(35), functional assay results
(35–37) and in silico prediction. The
loss of MSH2 and MSH6 expression was also observed by IHC and R178C
was predicted to be non-pathogenic by the CoDP, SIFT and PolyPhen-2
programs. However, since the R178C (c.532C>T) variant has thus
far not been reported, it is considered to be class-3.
The remaining four variants, i.e. Y397C, Q1155R,
G1316E and I1357T, did not show either hypermethylation of the
MLH1 promoter or the BRAF mutation. The Q1155R
carrier had multiple CRCs and the G1316E carrier was diagnosed with
CRC before age 50. In addition, the tumors of these variant
carriers had lost the MSH6 protein, as determined by IHC and
demonstrated by MSI-H. We also performed an in silico
prediction, and most tools predicted that Q1155R and G1316E were
pathogenic. After taking all these findings into account, these
variants were considered to be class-4.
In a previous report, the Y397C mutation was found
in a patient with ureter cancer, which is one of the LS-associated
cancers (38), but the tumor did
not have MSI. The in silico analysis predicted that it was
pathogenic. Since the information was limited, this variant is
presently considered to be class-3.
The carrier of I1357T did not fulfill any of the
clinical criteria, and this variant was predicted to be
non-pathogenic by the in silico tools. However, the rectal
carcinoma of this carrier showed MSI-H and loss of the MSH6
protein. Therefore, I1357T is considered to be class-3.
The category of all variants can be altered by the
inclusion of additional data, such as the results of a segregation
analysis and functional assays of the MMR activity. More
MSH6 variant carriers will need to be identified and
analyzed and the classification of the variants will be
continuously refined based on the accumulation of clinical and
molecular data.
Acknowledgements
We thank Akemi Takahashi for the technical
assistance. The present study was supported in part by a
grant-in-aid for Cancer Research from the Ministry of Health,
Labour and Welfare.
Abbreviations:
|
CRC
|
colorectal cancer
|
|
IHC
|
immunohistochemistry
|
|
LS
|
Lynch syndrome
|
|
MLPA
|
multiplex ligation-dependent probe
amplification
|
|
MMR
|
mismatch repair
|
|
MSI
|
microsatellite instability
|
|
MSI-H
|
high-level of microsatellite
instability
|
|
MSI-L
|
low-level of microsatellite
instability
|
|
MSS
|
microsatellite stable
|
|
VUS
|
variants of uncertain significance
|
References
|
1
|
Lynch HT and de la Chapelle A: Hereditary
colorectal cancer. N Engl J Med. 348:919–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Grover S and Syngal S: Genetic testing in
gastroenterology: Lynch syndrome. Best Pract Res Clin
Gastroenterol. 23:185–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hampel H, Frankel WL, Martin E, et al:
Feasibility of screening for Lynch syndrome among patients with
colorectal cancer. J Clin Oncol. 26:5783–5788. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aaltonen LA, Salovaara R, Kristo P, et al:
Incidence of hereditary nonpolyposis colorectal cancer and the
feasibility of molecular screening for the disease. N Engl J Med.
338:1481–1487. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wijnen J, de Leeuw W, Vasen H, et al:
Familial endometrial cancer in female carriers of MSH6 germline
mutations. Nat Genet. 23:142–144. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vasen HF, Mecklin JP, Khan PM and Lynch
HT: The International Collaborative Group on Hereditary
Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum.
34:424–425. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Baglietto L, Lindor NM, Dowty JG, et al:
Risks of Lynch syndrome cancers for MSH6 mutation carriers.
J Natl Cancer Inst. 102:193–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berends MJW, Wu Y, Sijmons RH, et al:
Molecular and clinical characteristics of MSH6 variants: an
analysis of 25 index carriers of a germline variant. Am J Hum
Genet. 70:26–37. 2002.
|
|
9
|
Hendriks YMC, Wagner A, Morreau H, et al:
Cancer risk in hereditary nonpolyposis colorectal cancer due to
MSH6 mutations: impact on counseling and surveillance.
Gastroenterology. 127:17–25. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wijnen JT, Vasen HF, Khan PM, et al:
Clinical findings with implications for genetic testing in families
with clustering of colorectal cancer. N Engl J Med. 339:511–518.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cunningham JM, Christensen ER, Tester DJ,
et al: Hypermethylation of the hMLH1 promoter in colon
cancer with microsatellite instability. Cancer Res. 58:3455–3460.
1998.PubMed/NCBI
|
|
12
|
Herman JG, Umar A, Polyak K, et al:
Incidence and functional consequences of hMLH1 promoter
hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA.
95:6870–6875. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kane MF, Loda M, Gaida GM, et al:
Methylation of the hMLH1 promoter correlates with lack of
expression of hMLH1 in sporadic colon tumors and mismatch
repair-defective human tumor cell lines. Cancer Res. 57:808–811.
1997.PubMed/NCBI
|
|
14
|
Bessa X, Ballesté B, Andreu M, et al: A
prospective, multicenter, population-based study of BRAF
mutational analysis for Lynch syndrome screening. Clin
Gastroenterol Hepatol. 6:206–214. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akagi K, Uchibori R, Yamaguchi K, Kurosawa
K, Tanaka Y and Kozu T: Characterization of a novel oncogenic
K-ras mutation in colon cancer. Biochem Biophys Res Commun.
352:728–732. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vasen HF, Watson P, Mecklin JP and Lynch
HT: New clinical criteria for hereditary nonpolyposis colorectal
cancer (HNPCC, Lynch syndrome) proposed by the International
Collaborative group on HNPCC. Gastroenterology. 116:1453–1456.
1999. View Article : Google Scholar
|
|
17
|
Umar A, Boland CR, Terdiman JP, et al:
Revised Bethesda Guidelines for hereditary nonpolyposis colorectal
cancer (Lynch syndrome) and microsatellite instability. J Natl
Cancer Inst. 96:261–268. 2004. View Article : Google Scholar
|
|
18
|
Ohsawa T, Sahara T, Muramatsu S, et al:
Colorectal cancer susceptibility associated with the hMLH1
V384D variant. Mol Med Rep. 2:887–891. 2009.
|
|
19
|
Asaka S, Arai Y, Nishimura Y, et al:
Microsatellite instability-low colorectal cancer acquires a
KRAS mutation during the progression from Dukes' A to Dukes'
B. Carcinogenesis. 30:494–499. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Campbell PT, Curtin K, Ulrich CM, et al:
Mismatch repair polymorphisms and risk of colon cancer, tumour
microsatellite instability and interactions with lifestyle factors.
Gut. 58:661–667. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jass JR: Classification of colorectal
cancer based on correlation of clinical, morphological and
molecular features. Histopathology. 50:113–130. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Terui H, Akagi K, Kawame H and Yura K:
CoDP: predicting the impact of unclassified genetic variants in
MSH6 by the combination of different properties of the
protein. J Biomed Sci. 20:252013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ng PC and Henikoff S: Predicting
deleterious amino acid substitutions. Genome Res. 11:863–874. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Adzhubei IA, Schmidt S, Peshkin L, et al:
A method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ali H, Olatubosun A and Vihinen M:
Classification of mismatch repair gene missense variants with
PON-MMR. Hum Mutat. 33:642–650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Woods MO, Williams P, Careen A, Edwards L,
Bartlett S, McLaughlin JR and Younghusband HB: A new variant
database for mismatch repair genes associated with Lynch syndrome.
Hum Mutat. 28:669–673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Stenson PD, Ball EV, Mort M, Phillips AD,
Shaw K and Cooper DN: The Human Gene Mutation Database (HGMD) and
its exploitation in the fields of personalized genomics and
molecular evolution. Curr Protoc Bioinformatics.
39:1.132012.PubMed/NCBI
|
|
28
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: the NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Engel C, Loeffler M, Steinke V, et al:
Risks of less common cancers in proven mutation carriers with lynch
syndrome. J Clin Oncol. 30:4409–4415. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peltomäki P: Lynch syndrome genes. Fam
Cancer. 4:227–232. 2005.
|
|
31
|
Vasen HFA: Review article: The Lynch
syndrome (hereditary nonpolyposis colorectal cancer). Aliment
Pharmacol Ther. 26:113–126. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao Y, Hu F, Wang F, Han B, Li D, Li X
and Zhu S: Meta-analysis of MSH6 gene mutation frequency in
colorectal and endometrial cancers. J Toxicol Environ Health A.
72:690–697. 2009.
|
|
33
|
Okkels H, Lindorff-Larsen K,
Thorlasius-Ussing O, et al: MSH6 mutations are frequent in
hereditary nonpolyposis colorectal cancer families with normal
pMSH6 expression as detected by immunohistochemistry. Appl
Immunohistochem Mol Morphol. 20:470–477. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Plon SE, Eccles DM, Easton D, et al:
Sequence variant classification and reporting: recommendations for
improving the interpretation of cancer susceptibility genetic test
results. Hum Mutat. 29:1282–1291. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ellison AR, Lofing J and Bitter GA:
Functional analysis of human MLH1 and MSH2 missense variants and
hybrid human-yeast MLH1 proteins in Saccharomyces
cerevisiae. Hum Mol Genet. 10:1889–1900. 2001.PubMed/NCBI
|
|
36
|
Gammie AE, Erdeniz N, Beaver J, Devlin B,
Nanji A and Rose MD: Functional characterization of pathogenic
human MSH2 missense mutations in Saccharomyces cerevisiae.
Genetics. 177:707–721. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Polaczek P, Putzke AP, Leong K and Bitter
GA: Functional genetic tests of DNA mismatch repair protein
activity in Saccharomyces cerevisiae. Gene. 213:159–167.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Furihata M, Shuin T, Takeuchi T, Sonobe H,
Ohtsuki Y, Akiyama Y and Yuasa Y: Missense mutation of the hMSH6
and p53 genes in sporadic urothelial transitional cell carcinoma.
Int J Oncol. 16:491–496. 2000.PubMed/NCBI
|