Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer worldwide and a major form of liver cancer
responsible for 90% of the primary malignant liver tumors in adults
(1). Most patients have a low
survival rate due to locally advanced or metastatic diseases and
surgery is feasible for only a low percentage of patients with HCC.
Although chemotherapy is ineffective due to toxicity and poor
response, recent trials with appropriate patient selection and
targeted molecular therapeutics are promising in the treatment for
advanced HCC (2,3). Sorafenib, a multiple tyrosine kinase
inhibitor, is one of the promising drugs clinically approved for
patients with advanced HCC. However, the response rate to sorafenib
in HCC is very low (2–3%) and it has been shown that activation of
the phosphoinositide 3-kinase/AKT (PI3K/AKT) signaling pathway
mediates acquired resistance to sorafenib in HCC (4). Therefore, development of novel
strategies for HCC treatment is required.
PI3K/AKT signaling is one of the major signaling
pathways activated in human cancer including HCC and is therefore
considered a suitable molecular target (5). Several PI3K inhibitors are in the
clinic and many more are in preclinical development. It has been
shown that novel inhibitors targeting the PI3K signaling cascade
cause decrease in cell proliferation and in some circumstances
promote cell death of HCC cells in vitro (6–8). There
are contradictory results regarding the effect of PI3K inhibition
on apoptosis and cell cycle in different cancer types including
HCC. Two PI3K inhibitors, LY294002 and ZSTK474, were found to
suppress cell growth without inducing apoptosis (9). Dan et al were demonstrated that
the inhibition of AKT suppressed proliferation by decreasing
expression of CycD1 and Ki-67, while not increasing apoptotic cell
numbers in six different cell lines from four different cancer
models and human cancer xenografts (9). In contrast, another study showed that
LY294002 induces apoptosis of human nasopharyngeal carcinoma in
vitro and in vivo (10).
Moreover, it has been reported that PI3K-mTOR inhibition does not
promote substantial apoptosis in the EGFR mutant lung cancer while
it induced apoptosis in HER2-amplified breast cancer (11). In EGFR mutant or KRAS mutant lung
cancer models, tumor regression associated with apoptosis was also
observed only when the PI3K/AKT pathway and MEK/MAPK pathway were
simultaneously blocked (12). Thus,
the literature suggests that the effect of inhibition of PI3K
signaling might cause different effects in a context-dependent
manner. Little is known about the effect of PI3K/AKT inhibition on
the cell cycle and apoptosis in HCC.
In the present study, we first analyzed the
activation status of AKT in normal liver, cirrhotic, HCC tissues
and HCC cell lines. Then, we functionally analyzed the effect of
AKT inhibition on cell proliferation and apoptosis by explaining
how the level of activated form of AKT induces apoptosis in HCC
cell lines.
Materials and methods
Cell culture
Human HCC cell lines (Mahlavu, SNU-449, SNU-475,
HepG2, PLC/PRF/5, SNU-398, HuH-7, Hep3B) were provided by Dr Mehmet
Öztürk (Bilkent University, Turkey). Cells were maintained in DMEM
with 10% FBS, 100 U/ml penicillin, 2 mM L-glutamine, and 100 mg/ml
streptomycin in 5% CO2 at 37°C (Biological Industries,
Israel). LY294002 (Calbiochem, Nottingham, UK) was used to inhibit
AKT signaling pathway, doxorubicin and cisplatin were used as an
apoptotic inducer.
Western blotting
Western blotting was performed as previously
described (13). For immunoblotting
p-AKT Ser 473(CS-4051), AKT (CS-7292), p-Rb Ser 608 (CS-2181), p-Rb
Ser 780 (CS-9307), p-Rb Ser 795 (CS-9301), p-Rb Ser 807/811
(CS-9308), Rb (CS-9309), p-MAPK p44/p42 (ERK1/2) Thr202 Tyr204
(CS-4377), p21/Cip1/waf1(CS-2946), p27 (sc-1641), p18 (sc-9965),
CycE (sc-247), CycA (sc-239), CycD1 (sc-718), CycH (sc-855), CycD3
(sc-6283), CDK2 (sc-6248), CDK4 (sc-601), CDK6 (sc-177) and CDK7
(sc-7344) and Calnexin (sc-11397) antibodies were used. Detection
was performed by Super Signal West Dura Extended Duration Substrate
(Pierce, IL, USA).
Cell proliferation analyses with BrdU
incorporation
DNA synthesis in LY294002-treated and -untreated
cells was determined by BrdU incorporation. Cells were seeded at a
density of 20×103 cells/well in 12-well plates. BrdU (30
μM) (Darmstadt, Germany) was added to media 4 h before ethanol
fixation. Following DNA denaturation, cells were incubated with
anti-BrdU monoclonal antibody (Dako, Denmark). Peroxidase labeled
IgG was used as secondary antibody and 3,3′-diaminobenzidine
tetrahydrochloride (DAB) substrate (Dako) was also used for
visualization. Cells were counterstained by hematoxylin. Positively
stained cells were counted with a light microscope and the cell
growth ratio (%) was calculated by dividing the number of BrdU
positive nuclei by the total number of nuclei.
Cell cycle analysis
Cell cycle distribution was quantified by flow
cytometry. Cells were trypsinized at 24 and 48 h after treatment
with LY294002. Pellets were resuspended and fixed in ethanol. After
washing, cells were incubated in 0.1% Triton X-100 and DNAse-free
RNAse (200 mg/ml), then stained with propidium iodide. Cells were
analyzed by BD FACSCanto version 5.03 Flow Cytometry and Cell cycle
distribution was analyzed by using BD FACS Diva version 5.03. and
Modfit LT 3.0 software (BD Biosciences, FACSCanto, San Jose, CA,
USA).
Luciferase reporter assays
Cells were transiently transfected with both the
E2F1 luciferase reporter construct [pGL33 + TAP73 (−833) E2F1
responsive vector, kindly provided by Dr Emre Sayan] and the pRL-TK
plasmid vector (kindly provided by Mehmet Ozturk) by using FuGENE
HD transfection reagent (Roche, Mannheim, Germany). Cells were
harvested and luciferase activity was measured using the Dual
Luciferase Reporter Assay kit (Promega, Madison, WI, USA) according
to the manufacturer’s instructions. Levels of reporter gene
induction in transfected cells were calculated by normalizing the
Firefly luciferase levels to total Renilla luciferase levels.
Detection of apoptosis
The Annexin V-FITC Apoptosis Detection kit (BD
Biosciences) was used to analyze apoptosis by flow cytometry
according to the manufacturer’s recommendations. Cells were
transiently transfected with pBabe puroL Akt K179M T308A S473A
(ΔN-AKT) plasmid (kindly provided by Aykut Uren) for 48 h. They
were then treated with 10 μM cisplatin, 5 and 10 μM doxorubicin
and/or 25 μM LY294002 for both 24 and 48 h before staining with
Annexin V. Cells were analyzed by flow cytometry (FACSCalibur; BD
Biosciences) and results were analyzed by BD CellQuestPro.
Immunohistochemistry
Formalin-fixed, paraffin-embedded liver tissue
samples were obtained from 73 patients with HCC in cirrhotic
background and 17 patients with cirrhosis who underwent complete
resection without any preoperative treatment in Dokuz Eylül and Ege
University Hospitals in Izmir, Turkey. Normal liver biopsies were
used as controls (n=22). The study was approved by the Ethics
Committees of both Dokuz Eylül and Ege University Medical Schools.
Written informed consent was obtained from all patients prior to
liver transplantation or liver biopsy sampling. Tissue sections
were stained with hematoxylin and eosin (H&E) and then examined
by light microscopy. The histopathological diagnosis of all
patients was carried out according to the WHO histopathological
classification of liver disease. Immunostaining was performed using
an automated immunohistochemical stainer according to the
manufacturer’s guidelines (Biogen, Lab Vision Autostainer 360, CA,
USA). The antigen retrieval step was performed with proteinase K.
Endogenous peroxidase and avidin activities were blocked by 3%
H2O2 and the avidin-biotin block solution,
respectively. Sections were then incubated with anti-p-AKT antibody
(CS-4051) at 1:100 dilutions. The reaction product was developed
using 3,3′-diaminobenzidine tetrahydrochloride (DAB) and sections
were counterstained with hematoxylin. Sections were then evaluated
by two pathologists (O.S., F.Y., D.N.). The extent of p-AKT
expression was semi quantitatively graded as 0 (no stain), 1+
(<20% of the cells), 2+ (positivity between 20–60% of cells),
and 3+ (>60% of cells).
Statistical analysis
All graphic data are expressed as the mean ± SE.
Statistical analysis of data was performed using the GraphPad Prism
and Statistical Package for Social Sciences 15.0 (SPSS Inc.,
Chicago, IL, USA). Analysis of variance (ANOVA) was used to
determine the significance of the differences between means of
multiple groups. The association of p-AKT expression of tissue
samples and clinicopathological characteristics of patients was
examined with the χ2 test. p<0.05 was considered to
indicate a statistically significant difference.
Results
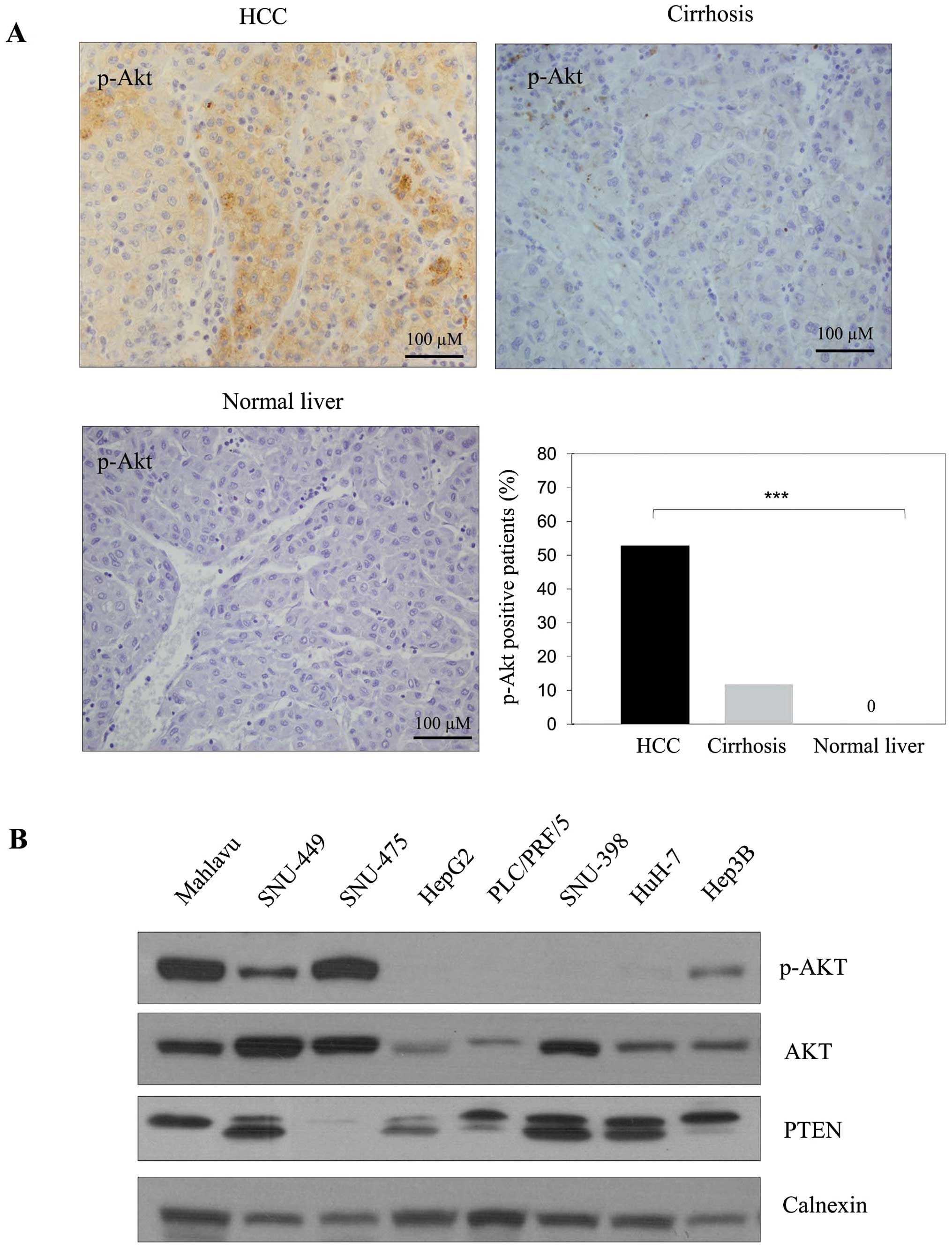
p-AKT expression markedly decreases from
HCC tissues to normal liver tissues
The p-AKT expression was observed in 53% of HCC
tissues and in 12% of cirrhotic tissues (Fig. 1A). Although the p-AKT staining was
significantly strong in HCC tumor tissues, little or no staining
was observed in cirrhotic and normal liver tissues, respectively
(p<0.001). p-AKT signaling was higher in HCC patients with more
than three nodules than in patients with less than three.
Furthermore p-AKT level was significantly higher in patients with
moderate/poorly differentiated HCC than in patients with well
differentiated tumors (Table I).
There was no significant correlation between activated AKT level
and tumor size or gender.
 | Table IClinicopathological characteristics of
primary hepatocellular carcinoma patients. |
Table I
Clinicopathological characteristics of
primary hepatocellular carcinoma patients.
| Characteristics | No. of patients | p-AKT | P-value |
|---|
|
|---|
| (+) | (%) |
|---|
| Gender |
| Female | 10 | 5 | 50 | 1.000 |
| Male | 43 | 24 | 55 | |
| Tumor size |
| <5 cm | 32 | 15 | 46 | 0.360 |
| ≥5 cm | 10 | 7 | 70 | |
| No. of tumor
nodules |
| <3 | 22 | 8 | 36 | 0.038a |
| ≥3 | 23 | 16 | 70 | |
| Tumor grade |
| Well
differentiated | 9 | 3 | 33 | 0.01a |
| Moderately
differentiated | 27 | 15 | 55 | |
| Poorly
differentiated | 7 | 4 | 57 | |
PI3K/AKT signaling is often active in HCC
cell lines
The p-AKT expression was high in Mahlavu and
SNU-475, moderate in Hep3B and SNU-449 and almost absent in HepG2,
PLC/PRF/5, SNU-398 and HuH-7 cells. The expression of total AKT was
observed at different levels in these cell lines. When PTEN level
was tested as a negative upstream regulator of PI3K/AKT signaling,
a positive correlation was found between dephosphorylated
functional PTEN and p-AKT levels (Fig.
1B). This suggests functional PTEN might block Akt
phosphorylation as expected.
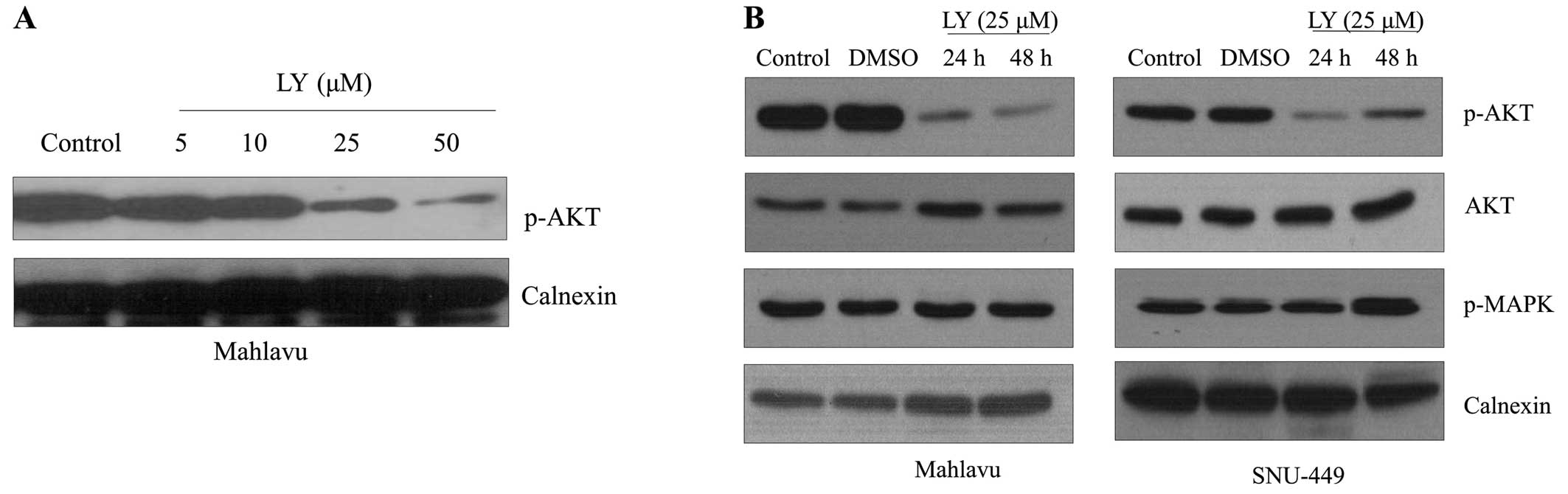
LY294002 treatment inhibits AKT
activation in a dose-dependent manner in both Mahlavu and SNU-449
cell lines
To define the influence of AKT activation on the
proliferation of HCC cell lines, we first analyzed the
dose-dependent effect of LY294002 on the active phosphorylated form
of AKT in Mahlavu cells. LY294002 treatment for 48 h resulted in a
progressive decrease in the p-AKT level in Mahlavu cells in a
dose-dependent manner (Fig. 2A).
Since the MAPK signaling pathway is one of the best-characterized
signaling pathways that regulate cell proliferation and there is a
cross-talk between Ras/MAPK and PI3K/AKT signaling at different
levels in a context-dependent manner (14), we further analyzed the effect of
LY294002 on the expression of Phospho-p44/42 MAPK (Erk1/2)
(Thr202/Tyr204). The inhibition of AKT signaling did not affect
activation of MAPK signaling in HCC cell lines (Fig. 2B).
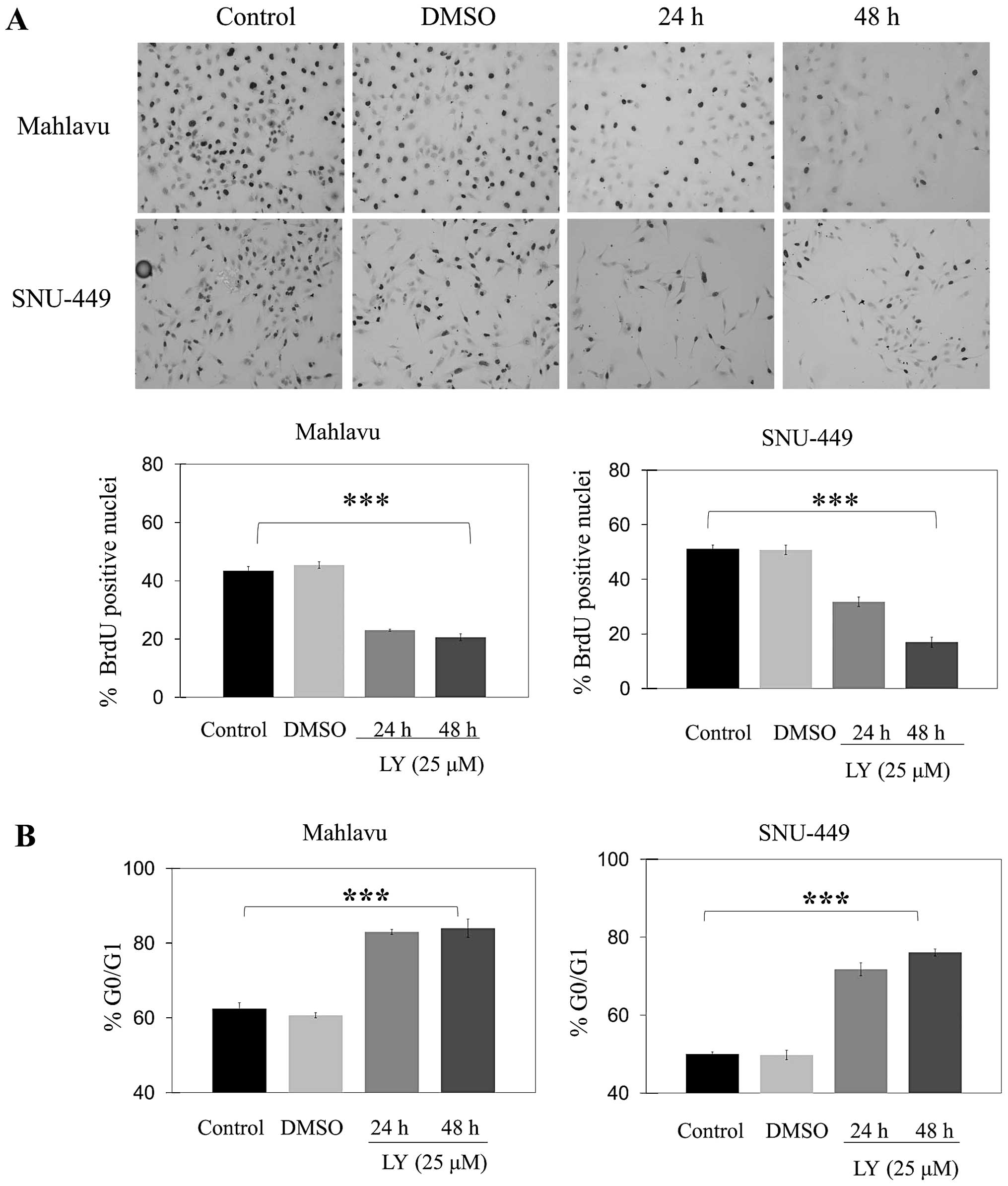
Inhibition of AKT induces cell cycle
arrest at G0/G1 and increases sensitivity to chemical
induced-apoptosis in HCC cell lines
Treatment with LY294002 caused a significant
decrease in the percentage of cells in S phase at 24 and 48 h as
compared to controls in both Mahlavu and SNU-449 cell lines
(Fig. 3A). LY294002 caused a
significant (p<0.001) increase in the percentage of cells at
G0/G1 phase at 24 and 48 h in both Mahlavu and SNU-449 (Fig. 3B). Furthermore, there was no
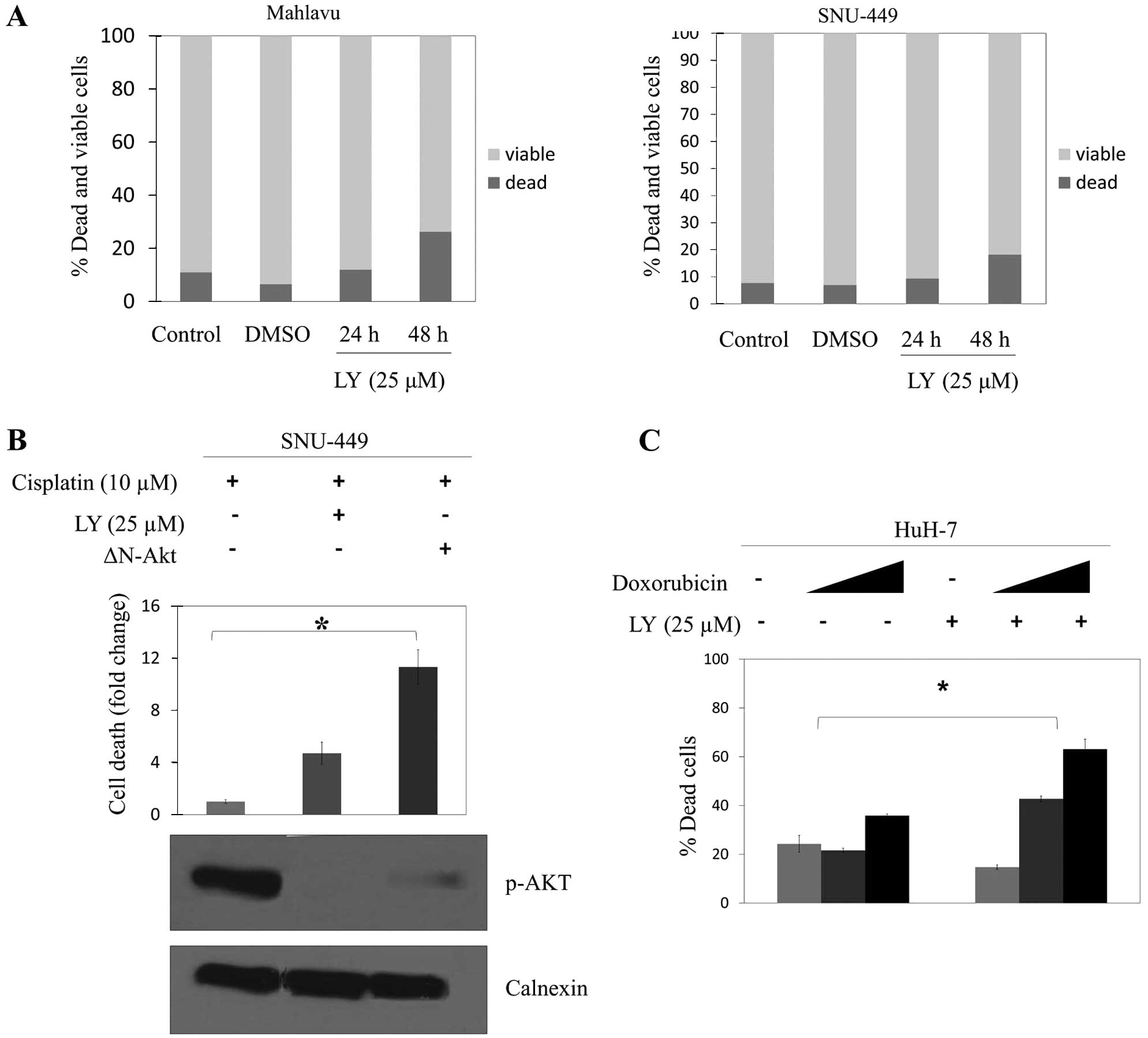
statistically significant difference in the apoptotic death in
either Mahlavu or SNU-449 cells after treatment of LY294002 for 24
h (Fig. 4A). However, 48 h LY294002
treatment amplified cisplatin-induced apoptosis in SNU-449 cells.
When the p-AKT level was decreased specifically by transient
transfection of ΔN-AKT plasmid into SNU-449 cells, they became even
more sensitive to apoptosis induced by cisplatin (Fig. 4B). HuH-7 cells which do not have
basal phosphorylated AKT were markedly affected and killed by the
treatment of doxorubicin in a concentration-dependent manner
(Fig. 4C).
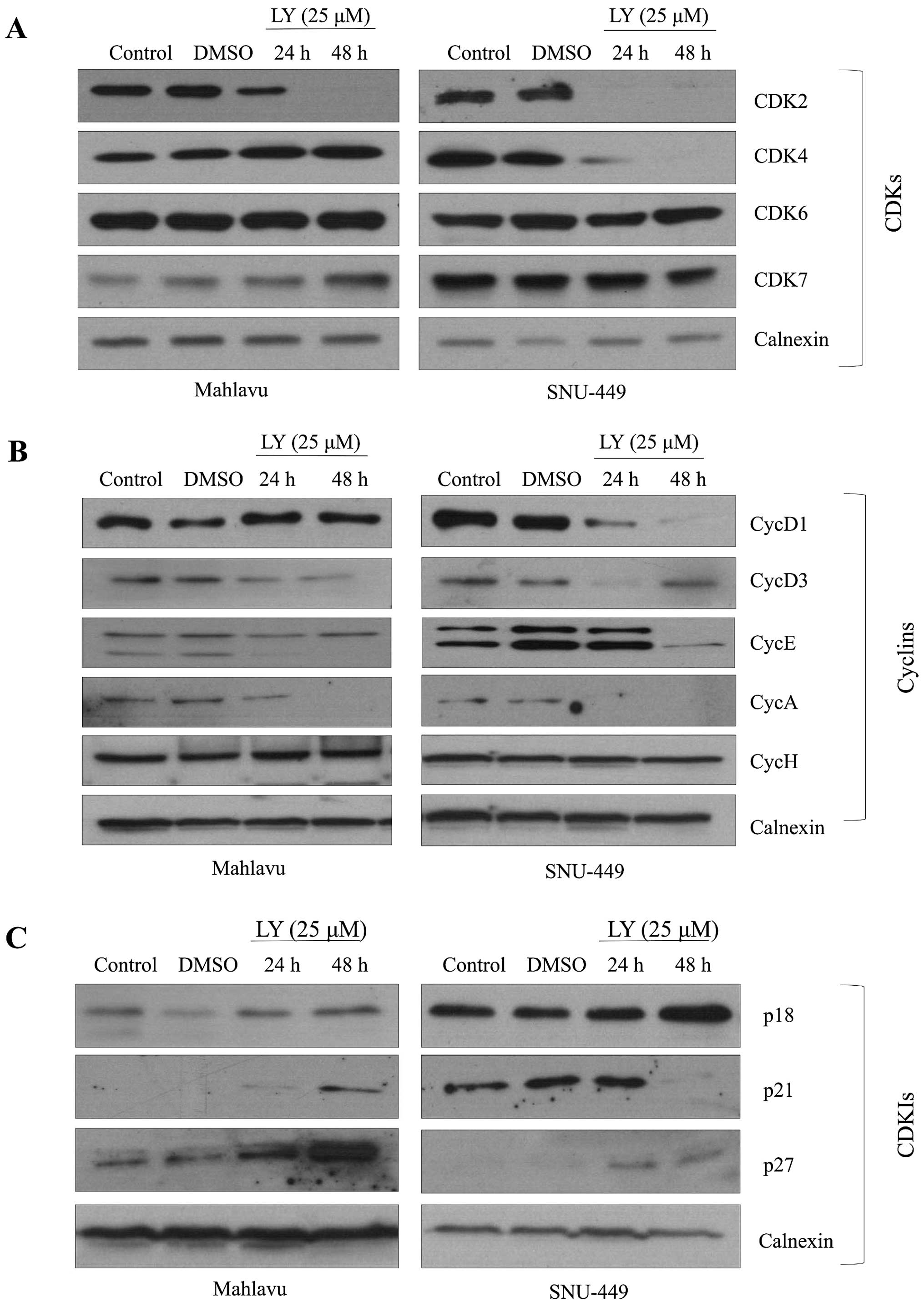
LY294002-induced G0/G1 arrest is
correlated with differential expression of cell cycle
effectors
To understand the mechanisms behind G1 arrest under
the effect of PI3K/AKT inhibition, G1 cell cycle arrest-related
molecules were analyzed. We showed that LY294002 decreased CDK2
expression strongly while it did not affect the expression of CDK6
in both Mahlavu and SNU-449 cells. Meanwhile, inhibition of AKT
caused a decrease in the expression of CDK4 in the SNU-449 cell
line, but not in Mahlavu cells (Fig.
5A). LY294002 reduced expression of CycD1 and CycE in both
SNU-449 and Mahlavu. Since CycD3, an isoform of CycD, plays an
important role especially in the proliferation of liver cells, we
further analyzed expression of CycD3 after PI3K inhibition. It
caused a significant decrease in the expression of CycD3 in both
HCC cell lines. Moreover, we found that there was no expression of
CycA as S phase regulator under the PI3K/AKT inhibition in both
cell lines. CycH functions as a CDK activating kinase by forming
complex with CDK7. Both CycH and its kinase partner participate in
a transcriptional regulation process during cell cycle. PI3K/AKT
inhibition did not affect CycH and CDK7 expression levels in
SNU-449 and Mahlavu cells (Fig.
5B). We also analyzed the expression of CDKIs and observed that
LY294002 treatment increased the levels of p27 and p21 expression
in both cell lines, as it caused a moderate increase in the level
of p18 expression in the SNU-449 cell line (Fig. 5C).
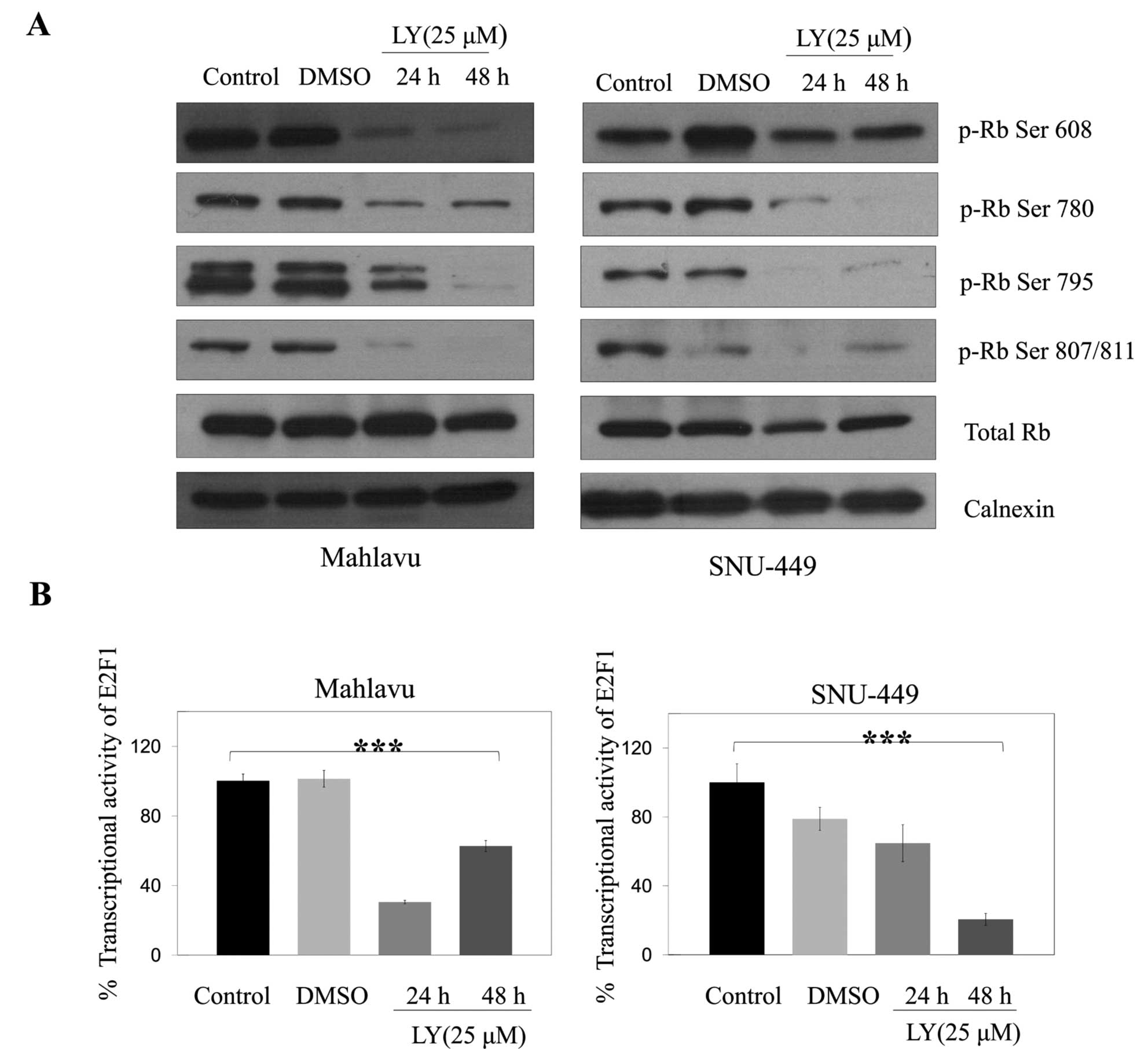
Retinoblastoma protein (Rb) is inactivated by
CDK-dependent phosphorylation or by proteolytic degradation, and
downregulation of CDK4/6 has also been associated with low level of
Rb (15). Since LY294002 reduced
the amount and potential activity of CDK4 and CDK6 in SNU-449 and
Mahlavu cells, we examined the levels and phosphorylation status of
Rb in these cells. LY294002 treatment strongly decreased levels of
different phosphorylated forms of Rb (Ser 608, Ser 780, Ser 795,
Ser 807/811) and no difference in the level of total Rb was
observed in either Mahlavu or SNU-449 cell lines (Fig. 6A) consistent with the inhibition of
cell cycle arrest. When we examined transcriptional activity of
E2F1 controlled by Rb phosphorylation, consistent with the decrease
in hyperphosphorylated form of Rb, inhibitor treatment caused a
significant (p<0.001) decrease in the level of E2F1 activity
over 24 and 48 h in Mahlavu and only over 48 h in SNU-449 cells
(Fig. 6B).
Discussion
The PI3K/AKT signaling pathway plays a pivotal role
in hepatocarcinogenesis; therefore, it is an attractive candidate
as an anticancer drug target for HCC treatment (2,3,5,16).
Aberrant activation of the PI3K/AKT pathway is often found in
different types of human cancer, including HCC, which might be
explained by the hotspot PI3KCA mutations and AKT gene
amplifications as well as the increase in phosphorylation of AKT
(17).
In the present study, we analyzed the activated form
of AKT (p-Ser 473) to define the activation status of PI3K/AKT
signaling pathway in HCC patients. Immunostaining showed a very
high degree of positive staining for the expression of p-Ser 473
AKT as 38 of 73 (53%) among HCC tissues describing high activation
level of PI3K/AKT signaling pathway. There was also little and/or
no staining for the activated form of AKT in cirrhotic and normal
liver tissues. Consistent with our data, Li et al (18) reported that immunoreactivity score
of the activated AKT was very low (0–3) in >80% of cirrhotic and
normal tissues, while it was high (4–6) in 65%
of HCC patient tissues. Furthermore, Schmitz et al (19) found that there was no staining for
p-AKT in normal liver and increased p-AKT expression was
significantly associated with a lower, overall disease-specific
survival. As a result, our data, in agreement with other studies,
suggested that the p-AKT level can be used as a prognostic and
early disease recurrence risk factor in different studies based on
HCC patient data (18–20).
In accordance with our immunohistochemistry data,
increased levels of activated AKT were also observed in 5 of the 8
(62.5%) HCC cell lines that we tested and were correlated with the
level of PTEN expression. Moreover, p-AKT expression was positively
correlated with the number of nodules in HCC patients and was also
significantly higher in poorly differentiated HCC patient samples
when compared with well-differentiated ones.
To test the effect of AKT inhibition on the cell
cycle progression of HCC cells, we then performed cell cycle
analyses by flow cytometry and BrdU incorporation assay. Results
clearly indicated that G1 arrest occurred and the number of cells
entering S phase significantly decreased after LY294002 treatment.
G1 arrest in these cells was correlated with the downregulation of
the expression of CycD1, CycD3, CycE, CycA, CDK2 and CDK4 as well
as decreased retinoblastoma protein activity. However, the
inhibition of AKT by LY294002 did not induce basal apoptotic
machinery in either the SNU-449 or the Mahlavu cells. There are
contradictory results on the effect of AKT inhibition on apoptosis
in different human cancer types. For instance, the PI3K inhibitor
LY294002 triggers apoptosis in colorectal cancer (21), pancreatic cancer (22) and androgen-sensitive prostate cancer
(23). However, Dan et al
(9), demonstrated that both PI3K
inhibitors, LY294002 and ZSTK474, suppress proliferation by
decreasing expression of CycD1 and Ki-67, while they do not
increase apoptosis in prostate cancer, lung cancer, glioblastoma
and colorectal cancer cell lines and in human cancer xenografts.
Additionally, Faber et al (11), showed that the PI3K-mTOR inhibition
does not promote substantial apoptosis in EGFR mutant lung cancer
while it induces apoptosis in HER2-amplified breast cancer.
Moreover, in EGFR mutant or KRAS mutant lung cancer
models, tumor regression associated with apoptosis was observed
only when the PI3K/AKT pathway and MEK/MAPK pathway were
simultaneously blocked (9,24). In our study, the inhibition of the
PI3K/AKT pathway by LY294002 treatment did not affect the MEK/MAPK
pathway and caused growth inhibition without apoptosis. When we
decreased the level of phosphorylated form of AKT by transient
transfection of ΔN-AKT and treated with LY294002 in SNU-449 cells,
the level of apoptosis was increased independent of cisplatin
treatment. In light of these results, the level of activated AKT
may regulate apoptotic response in basal or induced conditions in
HCC cells. Collectively, our data suggest that overexpression of
the activated AKT may be a biomarker for the prognosis of HCC. The
increased p-AKT level may, on the other hand, be a biomarker for
predicting treatment response when conventional chemotherapeutic
agents are used as apoptotic inducers.
Acknowledgements
This study was supported by a grant from the
Scientific and Technological Research Council of Turkey (TUBITAK)
(SBAG-105S092) and Dokuz Eylul University, DEU-2005.KB.SAG.08.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Shen YC, Hsu C and Cheng AL: Molecular
targeted therapy for advanced hepatocellular carcinoma: current
status and future perspectives. J Gastroenterol. 45:794–807. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Asghar U and Meyer T: Are there
opportunities for chemotherapy in the treatment of hepatocellular
cancer? J Hepatol. 56:686–695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen KF, Chen HL, Tai WT, et al:
Activation of phosphatidylinositol 3-kinase/Akt signaling pathway
mediates acquired resistance to sorafenib in hepatocellular
carcinoma cells. J Pharmacol Exp Ther. 337:155–161. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Courtney KD, Corcoran RB and Engelman JA:
The PI3K pathway as drug target in human cancer. J Clin Oncol.
28:1075–1083. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen TA, Wang JL, Hung SW, Chu CL, Cheng
YC and Liang SM: Recombinant VP1, an Akt inhibitor, suppresses
progression of hepatocellular carcinoma by inducing apoptosis and
modulation of CCL2 production. PLoS One. 6:e233172011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Masuda M, Shimomura M, Kobayashi K, Kojima
S and Nakatsura T: Growth inhibition by NVP-BEZ235, a dual
PI3K/mTOR inhibitor, in hepatocellular carcinoma cell lines. Oncol
Rep. 26:1273–1279. 2011.PubMed/NCBI
|
|
8
|
Jung KH, Choi MJ, Hong S, et al: HS-116, a
novel phosphatidylinositol 3-kinase inhibitor induces apoptosis and
suppresses angiogenesis of hepatocellular carcinoma through
inhibition of the PI3K/AKT/mTOR pathway. Cancer Lett. 316:187–195.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dan S, Yoshimi H, Okamura M, Mukai Y and
Yamori T: Inhibition of PI3K by ZSTK474 suppressed tumor growth not
via apoptosis but G0/G1 arrest. Biochem Biophys Res Commun.
379:104–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jiang H, Fan D, Zhou G, Li X and Deng H:
Phosphatidylinositol 3-kinase inhibitor (LY294002) induces
apoptosis of human nasopharyngeal carcinoma in vitro and in vivo. J
Exp Clin Cancer Res. 29:342010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Faber AC, Li D, Song Y, et al:
Differential induction of apoptosis in HER2 and EGFR addicted
cancers following PI3K inhibition. Proc Natl Acad Sci USA.
106:19503–19508. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Donev IS, Wang W, Yamada T, Li Q, Takeuchi
S, Matsumoto K, Yamori T, Nishioka Y, Sone S and Yano S: Transient
PI3K inhibition induces apoptosis and overcomes HGF-mediated
resistance to EGFR-TKIs in EGFR mutant lung cancer. Clin Cancer
Res. 17:2260–2269. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cokakli M, Erdal E, Nart D, et al:
Differential expression of Caveolin-1 in hepatocellular carcinoma:
correlation with differentiation state, motility and invasion. BMC
Cancer. 9:652009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Aksamitiene E, Kiyatkin A and Kholodenko
BN: Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt
pathways: a fine balance. Biochem Soc Trans. 40:139–146. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hallstrom TC and Nevins JR: Balancing the
decision of cell proliferation and cell fate. Cell Cycle.
8:532–535. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mínguez B, Tovar V, Chiang D, Villanueva A
and Llovet JM: Pathogenesis of hepatocellular carcinoma and
molecular therapies. Curr Opin Gastroenterol. 25:186–194.
2009.PubMed/NCBI
|
|
17
|
Karakas B, Bachman KE and Park BH:
Mutation of the PIK3CA oncogene in human cancers. Br J Cancer.
94:455–459. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li W, Tan D, Zhang Z, Liang JJ and Brown
RE: Activation of Akt-mTOR-p70S6K pathway in angiogenesis in
hepatocellular carcinoma. Oncol Rep. 20:713–719. 2008.PubMed/NCBI
|
|
19
|
Schmitz KJ, Wohlschlaeger J, Lang H, et
al: Activation of the ERK and AKT signalling pathway predicts poor
prognosis in hepatocellular carcinoma and ERK activation in cancer
tissue is associated with hepatitis C virus infection. J Hepatol.
48:83–90. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakanishi K, Sakamoto M, Yamasaki S, Todo
S and Hirohashi S: Akt phosphorylation is a risk factor for early
disease recurrence and poor prognosis in hepatocellular carcinoma.
Cancer. 103:307–312. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Semba S, Itoh N, Ito M, Harada M and
Yamakawa M: The in vitro and in vivo effects of
2-(4-morpholinyl)-8-phenyl-chromone (LY294002), a specific
inhibitor of phosphatidylinositol 3′-kinase, in human colon cancer
cells. Clin Cancer Res. 8:1957–1963. 2002.
|
|
22
|
Bondar VM, Sweeney-Gotsch B, Andreeff M,
Mills GB and McConkey DJ: Inhibition of the phosphatidylinositol
3′-kinase-AKT pathway induces apoptosis in pancreatic carcinoma
cells in vitro and in vivo. Mol Cancer Ther.
1:989–997. 2002.
|
|
23
|
Lin J, Adam RM, Santiestevan E and Freeman
MR: The phosphatidylinositol 3′-kinase pathway is a dominant growth
factor-activated cell survival pathway in LNCaP human prostate
carcinoma cells. Cancer Res. 59:2891–2897. 1999.
|
|
24
|
Engelman JA, Chen L, Tan X, et al:
Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D
and PIK3CA H1047R murine lung cancers. Nat Med. 14:1351–1356. 2008.
View Article : Google Scholar : PubMed/NCBI
|