Introduction
Hereditary multiple exostoses (HME) is an autosomal
dominant disease characterized by multiple cartilage-capped
outgrowths at the growth plates of long bones. HME usually presents
early in life with 80% of patients diagnosed before the age of 10
years (1). The prevalence of HME is
estimated to be ~1 in 100,000 in the European population (2) and at least 1 in 50,000 in the state of
Washington (3). HME leads to
serious complications, such as compression of peripheral nerves and
vessels, bone deformity, short stature and interference with joint
movement (3). Each of these
complications may necessitate surgical removal of the exostosis.
However, the possibility of the recurrence of exostosis is high,
and patients usually have multiple surgeries in the same area.
Genetic studies have identified the link between HME
and 3 loci (4–7): EXT1, which maps to 8q24.1,
EXT2, which maps to 11p13 and EXT3, which is located
on the short arm of chromosome 19, while its exact position has not
been successfully mapped. It has been estimated that 60–70% of HME
patients have mutations in EXT1, as well as 30–40% in
EXT2. To date, over 650 mutations have been found in these 2
genes, most of which are nonsense, frame shift, or splice-site
mutations (8,9), which result in truncation, premature
termination, premature degradation and nearly complete loss of
function of EXT proteins (10). EXT family members have been
identified as tumor-suppressor genes, and share a homologous
C-terminus glycosyltransferase domain which plays a key role in
heparan sulfate (HS) biosynthesis.
Here, we identified a three-generation Chinese
kindred with HME and discovered a novel nonsense mutation
c.1902C>A (p.Tyr634X) in the EXT1 gene exclusively in the
patients resulting in a truncated glycosyltransferase domain at the
C-terminus of the EXT1 protein.
Materials and methods
Patients and clinical data
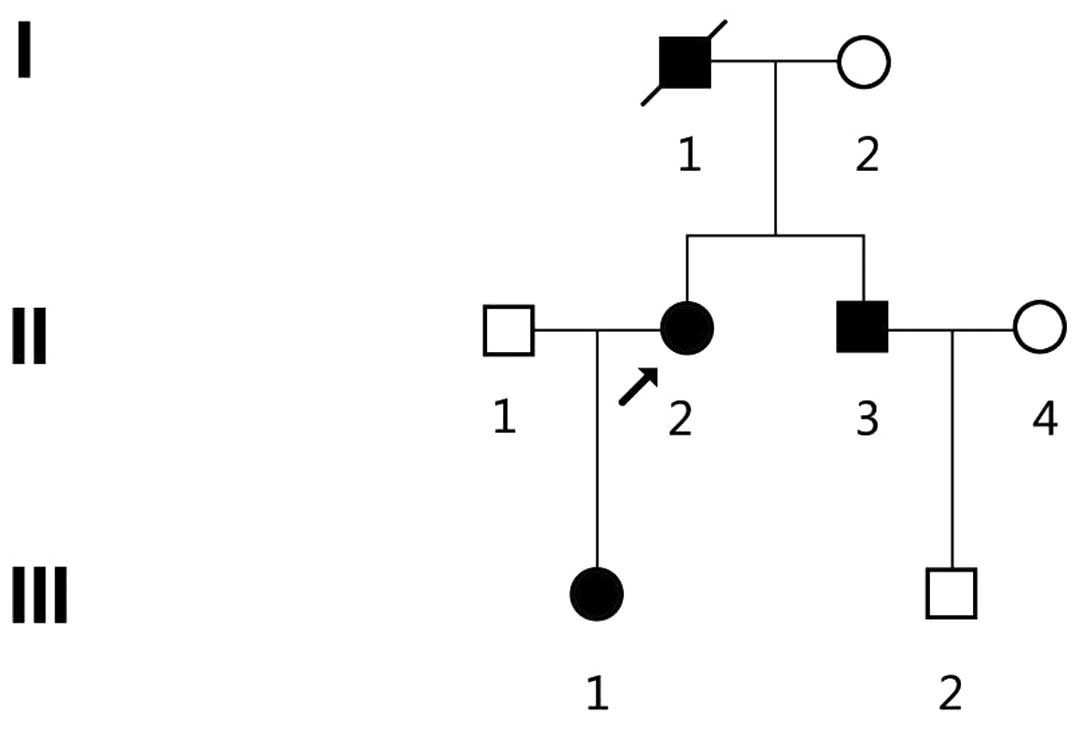
A three-generation Chinese kindred ZJ-H001 from
Zhejiang Province, China was identified (Fig. 1). Diagnosis of HME was based on
radiological examinations which indicated at least 2
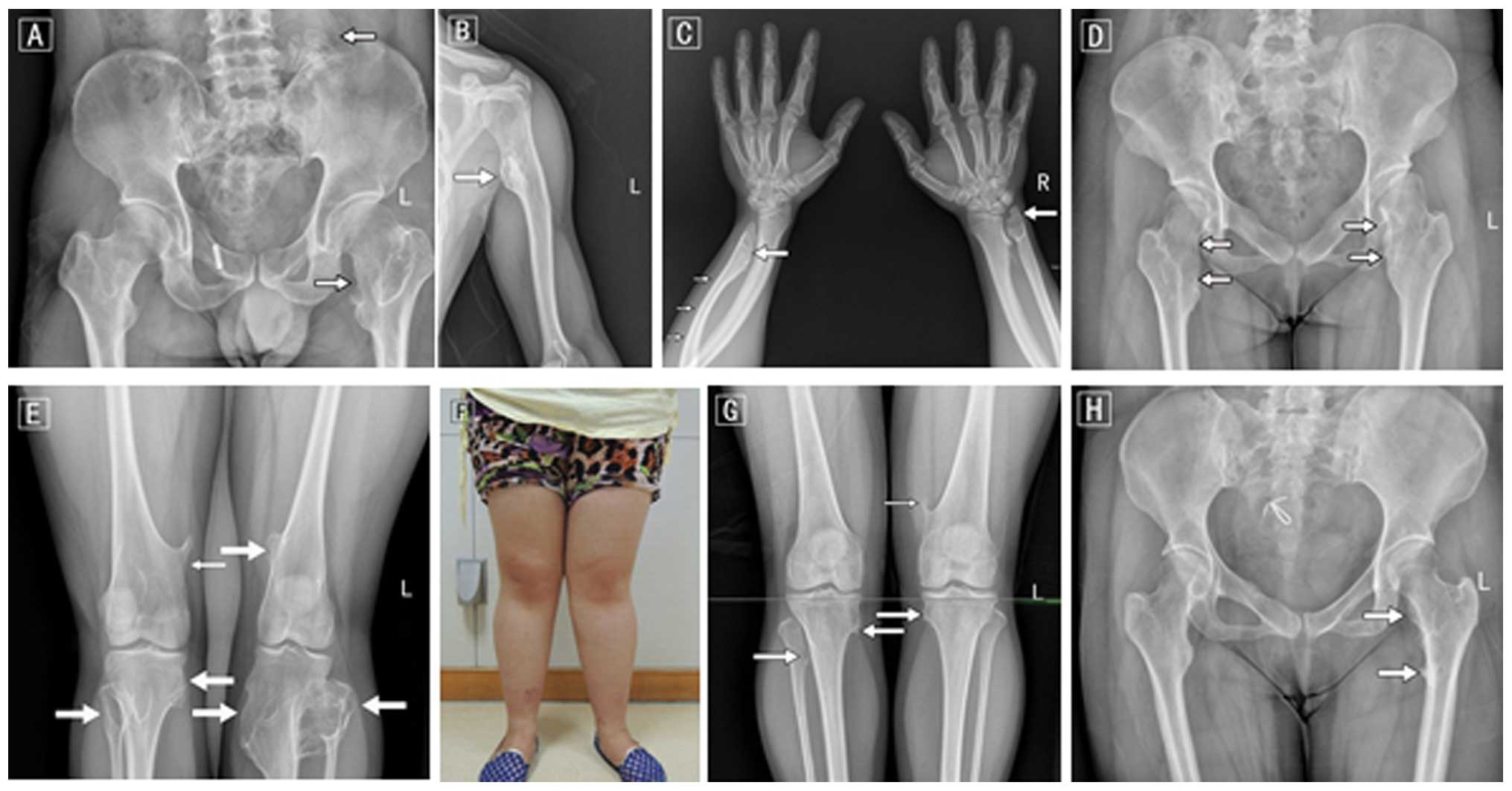
osteochondromas in the juxta-epiphyseal region of long bones, and
was further confirmed by joint and long bone palpitations (Fig. 2). There were 3 patients with HME (1
male and 2 females) in the kindred, inherited from generation to
generation (Fig. 1 and Table I), and all 3 patients and 3
unaffected family members (I2, II4 and III2) were enrolled in the
present study. All 3 patients showed multiple exostoses, arising
from the humerus, ulna, hands, hips, femurs, tibia, fibula and knee
joints. Shorter limb lesions were observed in all 3 patients, limb
deformity was noted in 2 patients and hip and knee valgus or joint
activity was only found in 1 patient. None of the 3 patients had
serious complications. The only clinical manifestation was a
painless lump on the bone and occasionally pain (mean VAS score
5.7) with functional limitations near knees or hips. The medical
history was obtained by using a questionnaire regarding the
subjective degree of HME, age at onset, evolution and other
relevant clinical manifestations (Table
I). The median age of the HME patients was 29.5 years. All 3
patients presented with osteochondromas before 10 years of age
(median age of onset, 5), and none of the patients had any surgical
removal of osteochondromas prior to the present study. The average
height (160.2 cm for males and 153.5 cm for females) was much lower
than the average height of the same age group reported by the 2010
National Physique Monitoring Bulletin Administration of Sports of
China. In addition to the kindred ZJ-H001, 200 ethnically unrelated
healthy individuals (107 males and 93 females, aged from 22 to 40
years) were included in the present study. All study procedures
were approved by the Ethics Review Committee of the Zhejiang
Provincial People’s Hospital and were carried out following written
informed consent obtained from each individual and/or parents of
the children.
 | Table IClinical data of the 3 patients with
HME. |
Table I
Clinical data of the 3 patients with
HME.
| ID | Gender | Age of onset
(years) | Age at present study
(years) | Height (cm) | Localization | Classification |
|---|
| II2 | Female | 5 | 40 | 156.4 | Ulna, hips, femurs,
tibia, fibula | IB |
| II3 | Male | 7 | 39 | 160.2 | Humerus, hips,
femurs, tibia, fibula | IIB |
| III1 | Female | 3 | 19 | 150.5 | Ulna, hands, hips,
femurs, tibia, fibula | IIIB |
Molecular analysis
Genomic DNA was purified from peripheral blood
leukocytes using an AxyPrep Nucleic Acid Purification kit (Axygen,
Union City, CA, USA) according to the manufacturer’s instructions.
Sanger direct sequencing DNA sequence analysis of the EXT1 and EXT2
coding region including the 100-bp flanking intron-exon junctions
by polymerase chain reaction (PCR) of genomic DNA followed by
sequencing reactions with Sanger sequencing chemistry using the
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems Inc., Foster City, CA, USA) on an ABI 3730XL automated
sequencer. Primer sequences are listed in Table II. Sequence data were analyzed by
Sequencer Demo 3.0 and Mutation Surveyor Demo V4.0 using the
reference sequences from NCBI (NM_000127.2 for EXT1, NM_001178083.1
for EXT2).
| Table IIPrimer sequences used for sequencing
the EXT1 and EXT2 genes. |
Table II
Primer sequences used for sequencing
the EXT1 and EXT2 genes.
| Primers | Reverse | Forward |
|---|
| EXT1 gene |
| Exon 1.1 |
5′-CCCTTCGGTCTTTCATCTTT-3′ |
5′-GACGTGACGCTCGGCCAAT-3′ |
| Exon 1.2 |
5′-GGAGTTGGCATCTCGCTTCT-3′ |
5′-GCAAGGCAATCACTTCGTC-3′ |
| Exon 1.3 |
5′-AAGGGGAAAGAGGACTGAGG-3′ |
5′-GGAGAGAAGAACACAGCGGTAG-3′ |
| Exon 2 |
5′-CCTCCACCCCTCACTTGTCA-3′ |
5′-GCAACCCAACCTCCTTCCTC-3′ |
| Exon 3 |
5′-TCTGGTTATTGAAAGGGGTGGA-3′ |
5′-GCAGTGTCAAAAATGCCAGTCA-3′ |
| Exon 4 |
5′-TTTGTGGAGTTTGTCAGGAATG-3′ |
5′-GAAGCCAAATGCTATGAAGAAT-3′ |
| Exon 5 |
5′-CAATGCAGGGTGTTAGATGGA-3′ |
5′-TAAAGTGGGAGGGAGGGTAGA-3′ |
| Exon 6 |
5′-GTAACGAGGCAGGATGAATGA-3′ |
5′-AAATCATCCAGGAGGGAACAT-3′ |
| Exon 7 |
5′-CTGATTTGAAAGCCTATTGTGG-3′ |
5′-AATGTTCTGAGGTTGTGTGGGA-3′ |
| Exon 8 |
5′-GTGCTAACAGGAATCGGGCT-3′ |
5′-TGAGATTCCTTCGGTGTTGAG-3′ |
| Exon 9 |
5′-CCAACTGAAAATGTTACTCTACC-3′ |
5′-ATTATGAATTAGTGGGGAGAAGG-3′ |
| Exon 10 |
5′-GCACCAATCATACACTCTTTTCTA-3′ |
5′-ATGGGTATGTGTTTTCTGTCTCA-3′ |
| Exon 11 |
5′-TTTCCACGAAGTTTGAGCTTTT-3′ |
5′-GCTCATTTGCCTGACTCCATT-3′ |
| EXT2 gene |
| Exon 1 |
5′-GCAGGAGTGGAAATCGGAG-3′ |
5′-ATTGCCCTCCAGGAATGTTA-3′ |
| Exon 2 |
5′-ACCAACTCAAGAGCAGAAGCA-3′ |
5′-GGCGTGGTGGTCACAGTTAC-3′ |
| Exon 3 |
5′-TGCCAGGACATAAGCCCTAACT-3′ |
5′-CTGTTGGGATTTCCAGGAGTTT-3′ |
| Exon 4 |
5′-AAACAAAGGAGAGAACGGAGT-3′ |
5′-TGATTCAAGGATAGAACGCAG-3′ |
| Exon 5 |
5′-CACAAGACACCAGACATCCAAG-3′ |
5′-GTGGAGGTGAAGACTGGTAAGG-3′ |
| Exon 6 |
5′-CCTTGGTTTGTGAACTGCTCT-3′ |
5′-GGCGTCAACCCTTGTAGAAAC-3′ |
| Exon 7 |
5′-AAGTAAACCCCACTCAGGCATT-3′ |
5′-GAAGGAGGTTTGGGATGTTGTT-3′ |
| Exon 8 |
5′-ACTGCTGAAACCCTGCTGTG-3′ |
5′-AAGTGTGCCTGGTTGGAGTG-3′ |
| Exon 9 |
5′-CCCAAGTATAAAAAGCACACTCTC-3′ |
5′-TAAAGGAATTAGCCTAACCTGGAG-3′ |
| Exon 10 |
5′-GTATGCCAGGGCTTGGAGTT-3′ |
5′-GTAAAAGCCACCAAGCCTGC-3′ |
| Exon 11 |
5′-CCCACACTCTTACGCACACCT-3′ |
5′-CTTTGGATTTGATGAGAGCCG-3′ |
| Exon 12 |
5′-ATGGTTATCTCGAAGTGACAGGA-3′ |
5′-TCTCCAGAATCCCATTATGACCT-3′ |
| Exon 13 |
5′-GACCGCATCAATCATAGAACCT-3′ |
5′-GCCTCCTTTTACCCTTCCTATT-3′ |
| Exon 14 |
5′-GACCCTTCCAGCCATTACAAA-3′ |
5′-CTTGTGAGTTCTGCCGTTGG-3′ |
| Exon 15.1 |
5′-AGGAATTGGTGTTTAAGACACAG-3′ |
5′-TTTGCTGTTATCTCTCAACCTCT-3′ |
| Exon 15.2 |
5′-ACGCTGACTGGCAAAACAACTA-3′ |
5′-TACATCAATGAGTTCTTTCAGGGA-3′ |
In silico analysis
In the present study, we used Mutation Taster
software (11) to determine
possible changes in protein structure that may affect
phenotype.
Evolutionary conservation and structural
prediction
Clustal ×1.83 was used to compare human wild-type
EXT1 protein sequence (NP_000118) with the orthologs from mouse,
Drosophila, Danio, Caenorhabditis and
Xenopus (sequences obtained from http://www.ensembl.org/). We used PredictProtein
(https://www.predictprotein.org/) to
predict the possible structural changes caused by the nonsense
mutation.
Results
DNA sequencing identifies a novel EXT1
mutation in all of the HME patients
We sequenced each exon of EXT1 and
EXT2 genes in all 3 patients (II2, II3 and III1) and the 3
unaffected family members (I2, II4 and III2) in the kindred
ZJ-H001. As previously reported by others, the present study also
identified 3 SNPs, c.28C>A, c.1065C>T and c.1761G>A, which
are located in exons 1, 3 and 9 of the EXT1 gene,
respectively (Table III). All
patients were carriers for genetic lesions of EXT1. Moreover, we
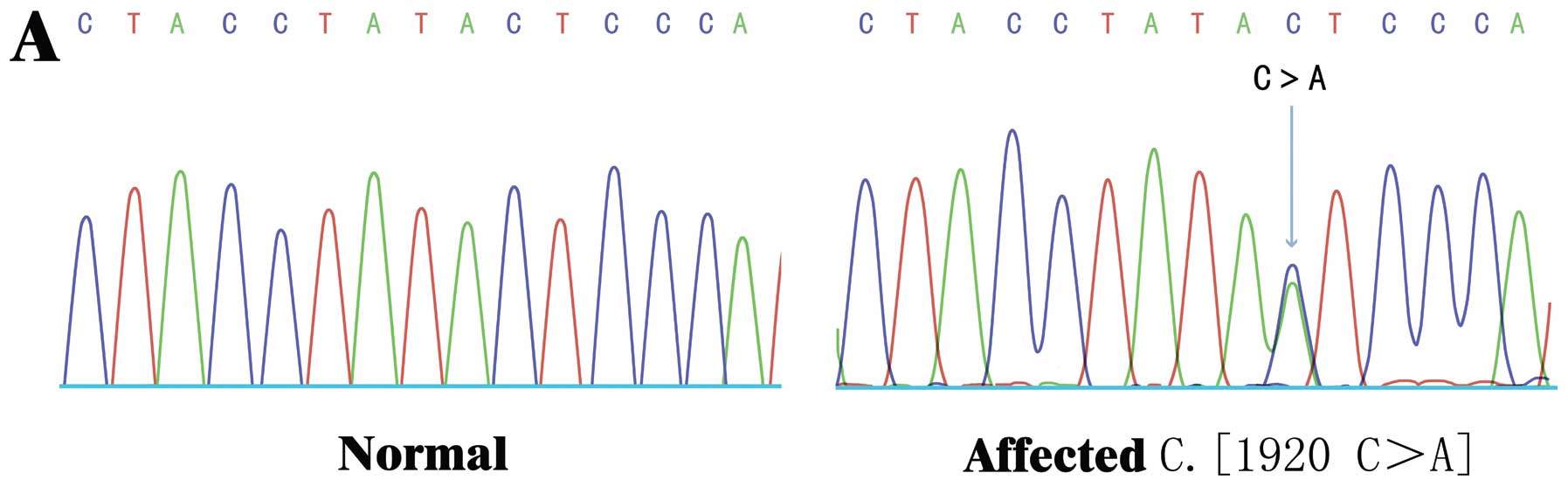
identified a novel nonsense mutation in the EXT1 gene c.1902
C>C/A (amino acid 634Y>X) in all 3 HME patients (Fig. 3), and 3 novel synonymous mutations
in the unaffected family members c.1005C>C/T, c.1761G>A and
c.1839G>A/G (amino acid 335C>C, 587E>E and 613T>T,
respectively) (Table IV). Of note,
this novel 634Y>X mutation was exclusively identified in all 3
affected patients, but not in the unaffected family members. To
assess the possibility that this novel EXT1 mutation is a
disease-causing mutation, we further sequenced 200 ethnically
unrelated healthy individuals and confirmed that none of these 200
healthy donors carried the 634Y>X mutation. Thus, our data
suggest that the novel 634Y>X nonsense mutation in the
EXT1 gene was a disease-causing mutation in the Chinese
pedigree (ZJ-H001) with HME.
| Table IIISNPs identified in the EXT1
gene. |
Table III
SNPs identified in the EXT1
gene.
| ID | Nucleotide
change | Trivial name | Location | Reported |
|---|
| II3 | c.28C>A
(het) | p.10R>R | Exon 1 | Y |
| III1 | c.28C>A
(het) | p.10R>R | Exon 1 | Y |
| III2 | c.28C>A
(het) | p.10R>R | Exon 1 | Y |
| I2 | c.1065C>T
(het) | p.C355C | Exon 3 | Y |
| II2 | c.1065C>T
(het) | p.C355C | Exon 3 | Y |
| II3 | c.1065C>T
(hom) | p.C355C | Exon 3 | Y |
| III1 | c.1065C>T
(het) | p.C355C | Exon 3 | Y |
| I2 | c.1761G>A
(het) | 587E>E | Exon 9 | Y |
| II3 | c.1761G>A
(het) | 587E>E | Exon 9 | Y |
| III1 | c.1761G>A
(het) | 587E>E | Exon 9 | Y |
| III2 | c.1761G>A
(het) | 587E>E | Exon 9 | Y |
| Table IVChanges in the EXT1 gene
detected in the patients by direct sequencing. |
Table IV
Changes in the EXT1 gene
detected in the patients by direct sequencing.
| ID | Location | Nucleotide
change | Amino acid
change | Mutation type | Category |
|---|
| I2 | Exon 3 | 1005C>CT | 335C>C | Synonymous | SNV |
| I2 | Exon 9 | 1761G>A | 587E>E | Synonymous | SNV |
| II2 | Exon 10 | 1902C>CA | 634Y>X | Nonsense | SNV |
| II3 | Exon 10 | 1902C>CA | 634Y>X | Nonsense | SNV |
| III1 | Exon 10 | 1902C>CA | 634Y>X | Nonsense | SNV |
| III2 | Exon 9 | 1839G>AG | 613T>T | Synonymous | SNV |
In silico analyses indicate the critical
impact of the Y634X mutation on EXT1 function
To understand the potential impact of the 634Y>X
nonsense mutation on EXT1 function, we further performed in
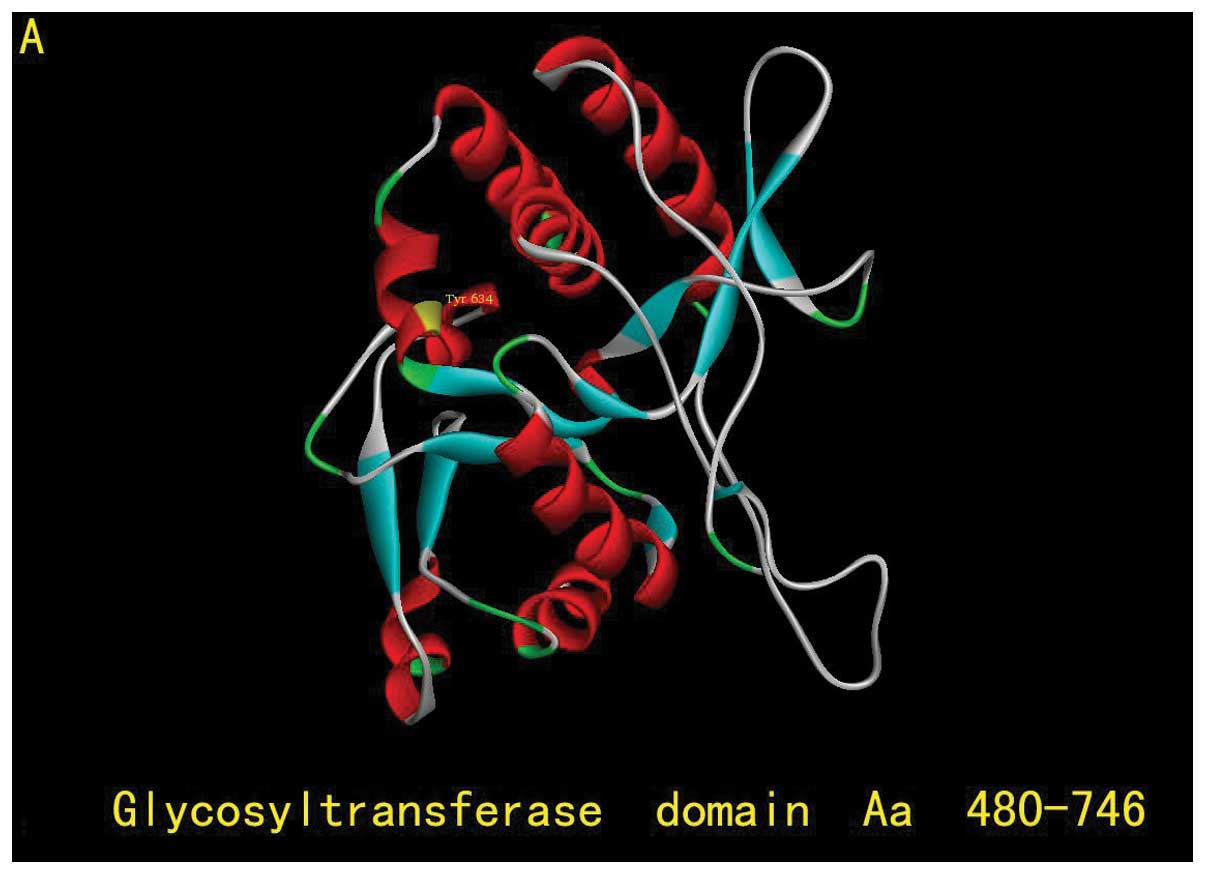
silico analyses. EXT1 protein is composed of 746 amino acids
with 2 critical domains: the exostosin domain (aa110–396) and the
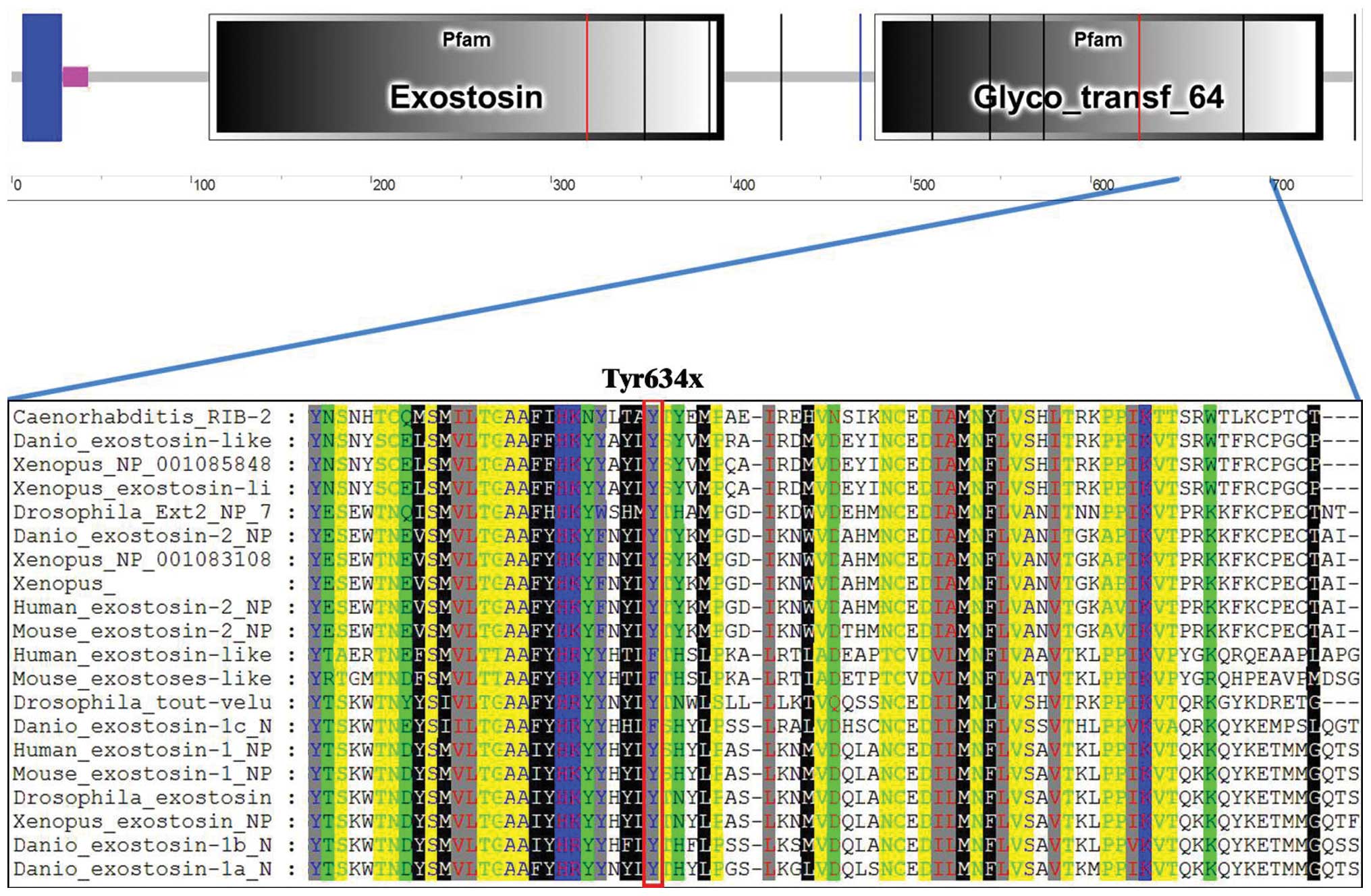
glycosyltransferase domain (aa480–729) (Fig. 3B). Try634 is located in the
glycosyltransferase domain, which is highly conserved among many
species (Fig. 4), indicating the
potential conserved role of Try634 in EXT1 function. The 634Y>X
nonsense mutation results in the premature termination of EXT1
protein translation and generates a truncated EXT1 protein lacking
112 amino acids at the C-terminus (Fig.
3B). Since the glycosyltransferase domain plays an essential
role in HS biosynthesis, loosing the C-terminus of the
glycosyltransferase domain may have a significant impact on EXT1
function, particularly on HS biosynthesis. Consistently, by online
prediction tool, Mutation Taster, it was predicted that
substitution of the Y634 to the terminator codon affects protein
function and causes diseases (probability score, 1). Finally, we
used the PredictProtein tool (12)
to predict the possible structural changes caused by the 634Y>X
mutation, and found that the 634Y>X mutation may affect the
secondary structure of the EXT1 protein by changing the numbers of
α-helices and β-pleated sheets in addition to truncating the 112
amino acids at the C-terminus of EXT1 (Fig. 5).
Discussion
The EXT family of genes consists of tumor-suppressor
genes whose loss of function or dysregulation causes hereditary
multiple exostoses. The EXT proteins are endoplasmic
reticulum-localized type II transmembrane proteins comprising an
N-terminal cytoplasmic tail, a transmembrane domain, a stalk, and a
large globular domain (13). EXT1
and EXT2 form heteroligomeric glycosyltransferases in Golgi
apparatus, and are tightly associated with glycosyltransferase
activities involved in the polymerization of heparan sulfate (HS)
via alternating addition of GlcAc and GlcNAc residues to lengthen
HS chains. The EXT1/EXT2 complex possesses substantially higher
glycosyltransferase activity than either EXT1 or EXT2 alone
(14,15). HS proteoglycans are known to be
necessary for signaling of fibroblast growth factors (FGFs),
vascular endothelial growth factor (VEGF), and transforming growth
factor-β (TGF-β) and are involved in the gradient formation of
morphogens such as hedgehog or bone morphogenetic proteins (BMPs)
(16). Mutations in the
glycosyltransferase genes, usually by causing a frame-shift in
protein elongation or missense in amino acid code, creates
truncated forms of the enzymes that these genes encode, leading to
lower enzyme activity and less HS chain synthesis (10,17)
Critical loss of full length HS chains results in deranged
ligand-receptor interactions as well as abnormal ligand diffusion
gradients for a number of signaling pathways (Ihh, BMPs, FGF, Wnt),
resulting in the development of osteochondroma (18–20).
Mice lacking EXT1 or EXT2 were found to be embryonic
lethal, and the embryos failed to undergo gastrulation, possibly as
a result of a disruption of several signaling pathways critical to
mesoderm development and formation of extra-embryonic structures.
Heterozygous null EXT2+/− mice showed growth plate
disturbances similar to those in their EXT1+/−
counterparts, with a disorganized proliferative zone and changes in
Ihh expression domains (21).
Generally, EXT1 mutations are correlated to more
severe presentations of the disease; male patients usually exhibit
more severe symptoms when compared with female patients which is
hypothesized to be caused by a later growth plate closure, allowing
more time for exostosis formation. Accordingly, patients with a
greater number of exostoses (>20) usually have more disabilities
and deformities (22–24). In our HME family, all patients
carred the heterozygous mutation of EXT1 while no EXT2 mutation was
noted. Compared with the second generation, patient III1 showed an
increasing severity of the disease with larger tumor volume, larger
tumor quantity, more onset positions and deformity, which may be
due to the fact that the growth plate of patients II2 and II3 had
been closed for a long time or due to the degeneration of
osteochondromas. This finding supports the variation in the
expressivity of HME that is in favor of genetic anticipation in
this disease (22). Future studies
using a larger patient population and applying the whole exome
sequencing technology to identify more disease-causing mutations or
investigating the epigenetic mechanisms contributing to disease
progression should enhance insights regarding the link between
genotype and disease phenotype as well as the molecular and
pathophysiological mechanisms of HME.
Multiple osteochondromas usually increase in size
and number during both childhood and adolescence. Although they
have a predilection for the juxta-epiphyseal region of the long
bones, in fact, they can be present on any bone of the body
including short bones, flat bones and irregular bones. It has been
shown that many HME patients do not present any clinical symptoms
in the early onset or even in later stages. There is strong
evidence to suggest that rare mutations in a set of key genes are
responsible for a substantial portion of complex HME disease
(25). HME is also known to be
characterized by a wide clinical heterogeneity. Evolutionary forces
generate vast genetic heterogeneity by introducing many new
variants in each generation. Current sequencing technologies offer
the possibility of finding rare disease-causing mutations and
mutation-associated genes. The present study identified a novel
disease-causing EXT1 mutation exclusively in all patients in
a Chinese pedigree with HME, which not only highlights the critical
pathophysiological role of the EXT1 gene in HME, but also
supports the high value of developing rapid and accurate genetic
tools for HME diagnosis.
Our study of a Chinese HME kindred identified one
novel disease-causing mutation c.1902C>A (p.Tyr634X) in the
EXT1 gene, and in silico analysis revealed the
significant impact of this variation on the glycosyltransferase
activity of EXT1. This offers new clinical and genetic data for
better understanding the pathogenesis of this disease. Moreover,
our findings also provide important information for genetic
counseling and designing optimal strategies for the quick and
accurate molecular diagnosis of HME.
Acknowledgements
We thank all the family members for their interest
and cooperation, and Dr Yufei Xu (Novartis Institutes for
BioMedical Research) for his critical reading of the manuscript.
The present study was supported by the Natural Science Foundation
of Zhejiang Province (Y2100731) to Q.B.
References
|
1
|
Solomon L: Hereditary multiple exostosis.
Am J Hum Genet. 16:351–363. 1964.PubMed/NCBI
|
|
2
|
Hennekam RC: Hereditary multiple
exostoses. J Med Genet. 28:262–266. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmale GA, Conrad EU III and Raskind WH:
The natural history of hereditary multiple exostoses. J Bone Joint
Surg Am. 76:986–992. 1994.PubMed/NCBI
|
|
4
|
Ahn J, Lüdecke HJ, Lindow S, et al:
Cloning of the putative tumour suppressor gene for hereditary
multiple exostoses (EXT1). Nat Genet. 11:137–143. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cook A, Raskind W, Blanton SH, et al:
Genetic heterogeneity in families with hereditary multiple
exostoses. Am J Hum Genet. 53:71–79. 1993.PubMed/NCBI
|
|
6
|
Le Merrer M, Legeai-Mallet L, Jeannin PM,
et al: A gene for hereditary multiple exostoses maps to chromosome
19p. Hum Mol Genet. 3:717–722. 1994.PubMed/NCBI
|
|
7
|
Wu YQ, Heutink P, de Vries BB, et al:
Assignment of a second locus for multiple exostoses to the
pericentromeric region of chromosome 11. Hum Mol Genet. 3:167–171.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jennes I, Pedrini E, Zuntini M, et al:
Multiple osteochondromas: mutation update and description of the
multiple osteochondromas mutation database (MOdb). Hum Mutat.
30:1620–1627. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ciavarella M, Coco M, Baorda F, et al: 20
novel point mutations and one large deletion in EXT1 and
EXT2 genes: report of diagnostic screening in a large
Italian cohort of patients affected by hereditary multiple
exostosis. Gene. 515:339–348. 2013.PubMed/NCBI
|
|
10
|
Wuyts W and Van Hul W: Molecular basis of
multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum
Mutat. 15:220–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schwarz JM, Rödelsperger C, Schuelke M and
Seelow D: MutationTaster evaluates disease-causing potential of
sequence alterations. Nat Methods. 7:575–576. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Roy A, Kucukural A and Zhang Y: I-TASSER:
a unified platform for automated protein structure and function
prediction. Nat Protoc. 5:725–738. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Varki A, Esko JD and Colley KJ: Cellular
organization of glycosylation. Essentials of Glycobiology. Varki A,
Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW
and Etzler ME: Chapter 3. 2nd edition. Cold Spring Harbor
Laboratory Press; New York: 2009
|
|
14
|
Busse M, Feta A, Presto J, et al:
Contribution of EXT1, EXT2, and EXTL3 to heparan sulfate chain
elongation. J Biol Chem. 282:32802–32810. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McCormick C, Duncan G, Goutsos KT and
Tufaro F: The putative tumor-suppressors EXT1 and EXT2 form a
stable complex that accumulates in the Golgi apparatus and
catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA.
97:668–673. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nadanaka S and Kitagawa H: Heparan
sulphate biosynthesis and disease. J Biochem. 144:7–14. 2008.
View Article : Google Scholar
|
|
17
|
Stickens D, Clines G, Burbee D, et al: The
EXT2 multiple exostoses gene defines a family of putative
tumour suppressor genes. Nat Genet. 14:25–32. 1996.
|
|
18
|
Bornemann DJ, Park S, Phin S and Warrior
R: A translational block to HSPG synthesis permits BMP signaling in
the early Drosophila embryo. Development. 135:1039–1047.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bornemann DJ, Duncan JE, Staatz W, et al:
Abrogation of heparan sulfate synthesis in Drosophila
disrupts the Wingless, Hedgehog and Decapentaplegic signaling
pathways. Development. 131:1927–1938. 2004.PubMed/NCBI
|
|
20
|
Bishop JR, Schuksz M and Esko JD: Heparan
sulphate proteoglycans fine-tune mammalian physiology. Nature.
446:1030–1037. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stickens D, Zak BM, Rougier N, et al: Mice
deficient in Ext2 lack heparan sulfate and develop exostoses.
Development. 132:5055–5068. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Francannet C, Cohen-Tanugi A, Le Merrer M,
et al: Genotype-phenotype correlation in hereditary multiple
exostoses. J Med Genet. 38:430–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pedrini E, Jennes I, Tremosini M, et al:
Genotype-phenotype correlation study in 529 patients with multiple
hereditary exostoses: identification of ‘protective’ and ‘risk’
factors. J Bone Joint Surg Am. 93:2294–2302. 2011.PubMed/NCBI
|
|
24
|
Alvarez C, Tredwell S, De Vera M and
Hayden M: The genotype-phenotype correlation of hereditary multiple
exostoses. Clin Genet. 70:122–130. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Stieber JR and Dormans JP: Manifestations
of hereditary multiple exostoses. J Am Acad Orthop Surg.
13:110–120. 2005.PubMed/NCBI
|