Introduction
Hepatocellular carcinoma (HCC) is the fifth most
common cancer and the third leading cause of cancer-related
mortality worldwide. It has a five year natural mortality rate of
>95%, and it affects >500,000 people in the world each year;
>50% of the new HCC cases and deaths have occurred in China
(1). Despite considerable efforts
to improve the survival of patients with HCC, a satisfactory level
has not been achieved as only 15% of the patients are eligible for
optimal resection at diagnosis and the tumor cells exhibit an
inherent tumor chemo- and radioresistance.
DNA-repair systems, as the molecular basis of
defending against environmental damage to cell DNA, play an
important role in maintaining the genomic stabilization and
integrity. However, an elevated DNA repair capacity in tumor cells
leads to drug or radiation resistance and severely limits the
efficacy of chemotherapy and radiotherapy. Apurinic/apyrimidinic
endonuclease 1/redox factor-1 (APE1/Ref-1; APE1) is an essential
enzyme in the DNA base excision repair (BER) pathway, which plays a
critical role in the repair of DNA caused by oxidative and
alkylation damage (2). APE1
accounts for 95% of the abasic site cleavage activity in human
cells, and is essential for the protection of cells against the
toxic effects of endogenous and exogenous agents. In addition to
its DNA repair functions, APE1 participates in other crucial
cellular processes, including the response to oxidative stress and
regulation of some transcriptional factors, including p53 (3). Moreover, several studies demonstrated
that APE1 was high-expressed in several human tumors including
prostate, osteosarcomas, lung and cervical carcinoma, and the
elevated APE1 level was associated with chemo- and radioresistance
and poor clinical outcome (4,5).
Silencing of APE1 enhanced cell sensitization to chemotherapeutic
agents and radiation (6,7). In other types of cancer, the shift of
APE1 from nucleus to cytoplasm was observed compared with normal
tissues, and might play a pivotal role in carcinogenesis and
progression of several human tumors (8,9). Tumor
cells often show increased expression level and altered subcellular
localization of APE1, and are associated with chemo- and
radioresistance and tumor carcinogenesis and progression.
The tumor suppressor gene p53 is activated in
response to DNA damage and induces cell cycle arrest or apoptosis,
and thus helps to maintain genomic stability and prevent cancer
(10,11). Once p53 is activated, it induces
cell apoptosis by both transcription-dependent and -independent
mechanisms, or arrests cell cycle by transactivation of Waf-1/p21,
14-3-3σ. p53 is one of the most commonly mutated genes in human
cancer; more than half of all types of human cancer contain mutant
or inactive p53. The mutant p53 (mutp53) proteins not only lose
their tumor suppressive activities but often gain additional
oncogenic functions (12). p53
mutations are detected in human HCC and its inactivation is
correlated with chemo- and radioresistance, and the carcinogenesis
and progression of HCC (13,14).
APE1 as a redox regulator is responsible for
reducing wild-type p53 (wtp53), thus enhancing its DNA-binding
activity by redox-dependent and -independent mechanisms (15,16).
Moreover, the redox-independent activation of wtp53 is due to a
regulatory interaction of APE1 with the p53 C-terminal regulatory
domain (CRD) (15), which is intact
in most of the frequently encountered mutp53 proteins, containing
the most common R249S mutation in HCC. Although mutp53 proteins
have lost the sequence-specific DNA binding (SSDB) transcriptional
activity, they retained the potential to bind nonlinear DNA in a
DNA structure-dependent manner (17). Given the activation of wtp53 by APE1
and the intact CRD in mutp53, we propose that APE1 may also
regulate the DNA binding activity of mutp53. Elucidating the
combined expression of APE1 and p53 in human cells may be of major
clinical significance, and a clear understanding of the mechanisms
by which APE1 controls mutp53 expression may aid in the effective
use of chemotherapeutic agents or multigene therapy strategies in
the treatment of tumors.
In this study, we first investigated the expression
level and subcellular localization of APE1, and its correlation
with p53 expression and clinicopathological parameters in HCC.
Then, we performed electromobility shift assay (EMSA) and
quantitative chromatin immunoprecipitation assay (ChIP) to
determine whether APE1 can regulate mutp53 DNA binding activity. We
showed that the increased APE1 expression level was significantly
correlated with p53 expression, and the high APE1
expression/p53+ status indicated a higher tumor grade.
We also present evidence that APE1 enhanced reduced mutp53 binding
to the nonlinear DNA in a redox-dependent manner. In addition, APE1
could not increase p21 mRNA and protein levels in mutp53 cells. To
our knowledge, this is the first direct evidence to show that APE1
stimulates mutp53 DNA binding activity, and these findings provide
new insights into the functional linkage between APE1 and p53 in
cancer therapy.
Materials and methods
Patients and tissues
Tumor tissues were obtained from 103 patients with
hepatoma at the Department of Cancer Center, Daping Hospital, Third
Military Medical University, China, from 1991 to 2004. The local
ethics committee approved this study. No chemotherapy or
radiotherapy was administered to patients prior to surgery.
Histological grading according to Edmondson and Steiner’s standard:
grade I, 5 cases; grade II, 27 cases; grade III, 56 cases and grade
IV, 15 cases. In HCC cases, 40 liver samples were sufficiently
large to include both the tumor and the surrounding cirrhosis. Ten
patients who underwent resection of hepatic angioma were used as
controls.
Immunohistochemistry and APE1
scoring
The expression of APE1 protein was analyzed using
immunohistochemistry. Sections from paraffin-embedded tumors were
incubated overnight with mouse anti-human APE1 monoclonal antibody
(Novus Biologicals, Littleton, CO, USA) at a 1:2,000 dilution and
anti-p53 antibody (DO-1) (Santa Cruz Biotechnologies, Inc., Santa
Cruz, CA, USA) at a 1:500 dilution, and then incubated with goat
anti-mouse secondary antibody from Pierce (Rockford, IL, USA).
Antigen-antibody complexes were visualized by incubation with
3,3′-diaminobenzidine (DAB) substrate and counterstained with
diluted Harris hematoxylin. Tissues were scored for: i) percentage
of cell staining and ii) intensity of staining (low, moderate, or
high). To be defined as low expression, the tissue needed to meet
weak staining and positive cell percentage <50% or moderate
staining and positive percentage <25%.
Cell culture
Human hepatoma cell lines HepG2 (harboring wtp53),
Hep3B (P53 null) and MHCC97L (carrying mutp53) were obtained from
the Cell Institute of Shanghai (Academia Sinica, Shanghai, China).
Cells were maintained as monolayer cultures in Dulbecco’s modified
Eagle’s medium (DMEM) (Invitrogen, Grand Island, NY, USA)
supplemented with 10% fetal bovine serum, 50 mg/ml
penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Cells
were grown at 37°C in a humidified incubator under 5%
CO2.
Infections
Adenovirus vector Ad5/F35-siAPE1 carrying human APE1
siRNA sequence was constructed by Xiang et al (7). The control adenovirus, Ad5/F35-EGFP,
was purchased from Vector Gene Technology Co., Ltd. (Beijing,
China). HepG2 and MHCC97L cell lines were infected with
Ad5/F35-EGFP or Ad5/F35-siAPE1 for 2 h and then washed to remove
the adenoviruses. Cells were cultured for another 48 h and then
analyzed by western blotting or prepared for subsequent
experiments. Cells were transfected with p3XFLAG-CMV/APE1, the
wild-type (WT) APE1, using Lipofectamine 2000 transfection reagent
(Invitrogen) according to the manufacturer’s protocol. As controls,
cells were transfected with the p3XFLAG-CMV-14 empty vector
(Sigma-Aldrich). At 24-h post-transfection, the transfected cells
were transferred into normal growth medium. After a further 24 h,
cells were prepared for subsequent experiments.
Western blot analysis
Equal amounts of nuclear or cytosolic extract or
whole-cell lysate, obtained from HepG2, Hep3B and MHCC97L cells,
were electrophoresed by 10% SDS- polyacrylamide gels. The proteins
were then transferred to polyvinylidene difluoride membranes
(Bio-Rad, Hercules, CA, USA) and blocked in Tris-Buffered Saline
and Tween-20 (TBST) [50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 0.1%
(volume/volume) Tween-20] containing 5% (weight/volume)defatted
milk for 1 h at room temperature. Membranes were incubated with the
specific primary antibody. After three washes with TBST, the
membranes were incubated for 1 h at room temperature with a
horseradish peroxidase-conjugated secondary antibody (1:2,000)
(Pierce). Then, the membranes were washed three times with TBST and
the blots were reacted with chemiluminescence reagents and revealed
with Biomax-Light films (Kodak, Rochester, NY, USA). Band
intensities were analyzed using the Gel Doc 2000 apparatus and
software (Quantity One; Bio-Rad). Suppliers of incubation
conditions for antibodies used for western blotting were: anti-APE1
monoclonal (Novus Biologicals), 1 h at 37°C, dilution 1:5,000;
anti-p21 monoclonal (Santa Cruz Biotechnologies, Inc.), overnight
at 4°C, dilution 1:500; anti-p53 monoclonal (DO-1), overnight at
4°C, dilution 1:500; anti-β-actin monoclonal (Santa Cruz
Biotechnologies, Inc.), 1 h at 37°C, dilution 1:2,000.
Electrophoretic mobility shift
assays
Nuclear extracts were prepared using the NE-PER
Nuclear and Cytoplasmic Extraction Reagents (Thermo Scientific)
according to the manufacturer’s instructions. Electrophoretic
mobility shift assays (EMSAs) were performed following the
manufacturer’s instruction manual of LightShift Chemiluminescent
EMSA kit (Pierce). Briefly, 5 μg of nuclear extracts was incubated
with 3′-biotin labeled and purified double-stranded oligonucleotide
probe. The p53 response element of the
p21Waf1-promoter (p53-RE) was prepared
either in its linear form (P53RE-linF,
5′-gctctgccGAACATGTCCCAACATGTTGccgctctg-3′ and P53RE-linR,
5′-cagagcggCAACATGTTGGGACATGTTCggcagagc-3′) or in a stem loop
conformation (P53RE-strF, 5′-ccgcggtaccattacctaaggcgtc-3′ and
P53RE-strR
5′-gacgccttaggtacctgccGAACATGTCCCAACATGTTgggcctgatggtaccgcgg-3′) as
previously described (18)
(Invitrogen, Shanghai, China). Upper case letters indicate the
p53-binding sites. Following incubation, samples were separated on
a prerun 5% polyacrylamide gel at 100 V for 90 min and then
transferred to a Zeta-Probe GT nylon membrane (Bio-Rad). The probes
were detected by HRP-conjugated streptavidin (1:300) and the bands
visualized by ECL reagents provided with the kit. The resultant
bands were quantified using Quantity One imaging software
(Bio-Rad).
Chromatin immunoprecipitation assays
Chromatin immunoprecipitation (ChIP) assays were
performed to analyze the DNA binding affinity of p53 to the p21
promoter region of its downstream genes using a ChIP kit
(Millipore) according to the manufacturer’s instructions. Cells
incubated in 10-cm petri dishes and subjected to various infection
treatments were harvested and crosslinked with 1% formaldehyde.
Pellets were collected and resuspended in ice-cold lysis buffer,
and then the chromatin was broken down through ultrasonication to
the DNA fragments at an average size of 200–500 bp, as empirically
estimated by agarose gel electrophoresis. Immunoprecipitations were
performed with p53 antibody (DO-1) to fractionate p53 protein-DNA
complexes and IgG (as a negative control). Quantitative PCR was
then performed to amplify the p21 gene promoter region (containing
potential specific p53 binding sites) using primers:
CCCTTCCTCACCTGAAAACA and GTGGCTCTGTTGGCTTTCTG. The predicted sizes
of the products were 116 bp. Preimmunoprecipitation lysates were
also included as input controls.
mRNA analysis by quantitative RT-PCR
Infected and control cells (1×106) were
harvested and washed using cold PBS once. Total RNA was extracted
by using TRIzol (Invitrogen) and chloroform/isoamyl alcohol
precipitation following the manufacturer’s instructions. RNA
concentrations were determined by spectro-photometer (Eppendorf AG,
Hamburg, Germany). Then, 1 μg of total RNA was reverse transcribed
into single-stranded DNA by using SuperScript II (Invitrogen).
Quantitative RT-PCR was performed using SYBR Premix Ex Taq (Takara)
in a LightCycler 480 real-time PCR system (Roche, Indianapolis, IN,
USA). Primers for p21 were: CTGGAGACTCTCAGGGTCGAAA and
TTCTCCAAGAGGAAGCCCTAATC; and for control β-actin:
GATCATTGCTCCTCCTGAGC and TGTGGACTTGGGAGAGGACT. Gene expression was
determined by normalization against β-actin expression.
Statistical analysis
Associations between categorical groups (i.e., APE1
expression and clinicopathologic data, APE1 and p53 expression,
APE1/p53 expression and tumor histologic grade) were examined using
Chi-square analysis. All p-values were two sided, and p-values
<0.05 were considered to indicate statistically significant
differences. For ChIP assays, data were obtained from three
independent experiments and expressed as mean ± standard deviation
values, and then analyzed using the one-way ANOVA test with
computer SPSS software SPSS 10.0 (SPSS, Chicago, IL, USA).
Results
APE1 immunohistochemistry and
clinicopathologic parameters
We investigated the expression of APE1 in 10 normal
liver tissues, 40 liver cirrhosis tissues and 103 cases of HCC
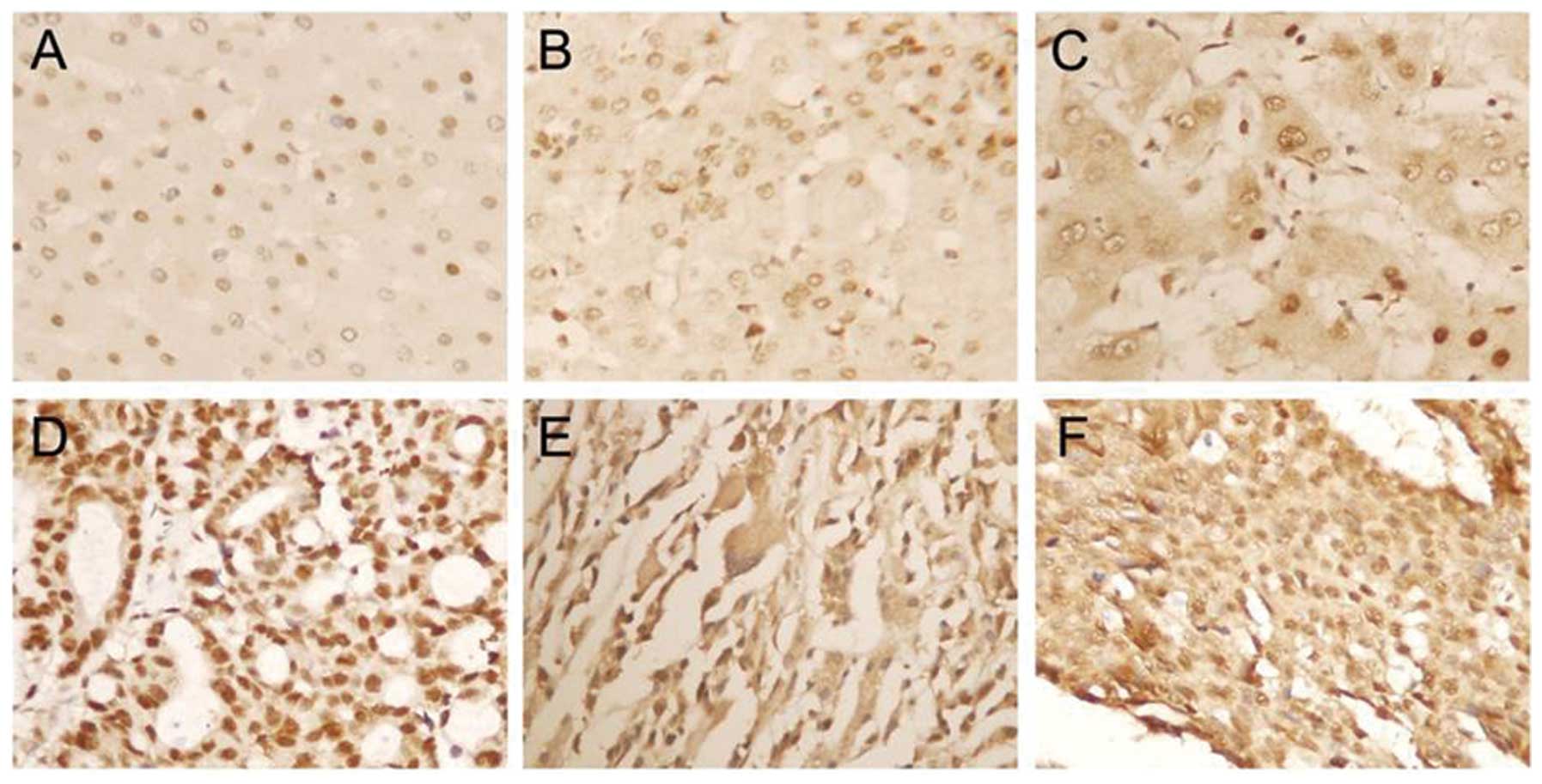
tissues using immunohistochemistry. As shown in Fig. 1, APE1 staining was mainly located in
the nucleus in normal liver tissues, and was located not only in
the nucleus, but also in the cytoplasm in liver cirrhosis and HCC
tissues. Twenty-five of 40 liver cirrhosis tissues (62.5%) were
nucleus staining and 8 tissues (20%) showed both the nucleus and
cytoplasm staining. Three subcellular locations of APE1 protein
were observed, and were nucleus staining in 48 cases (46.6%),
cytoplasm staining in 4 cases (3.9%) and staining in both the
nucleus and cytoplasm in 51 cases (49.5%) in HCC tissues.
Additionally, there was a significant difference in the cytoplasmic
and nuclear staining intensity of APE1 among normal liver tissue,
liver cirrhosis and HCC tissues. As shown in Table I, APE1 expression was not related to
age, gender, tumor size, serum HBsAg and TNM stage, whereas there
was a significant difference between the APE1 protein expression
among patients with different histologic classification (Grade I–II
vs. Grade III–IV).
 | Table IThe relationship between
clinicopathologic factors and APE1 protein expression in 103
hepatoma cases. |
Table I
The relationship between
clinicopathologic factors and APE1 protein expression in 103
hepatoma cases.
| | Nucleus
expression | Cytoplasm
expression |
|---|
| |
|
|
|---|
| Clinicopathologic
data | No. of cases | − | + | ++ | − | + | ++ |
|---|
| Age (years) |
| <45.5 | 37 | 2 | 6 | 29 | 15 | 15 | 7 |
| ≥45.5 | 66 | 2 | 13 | 51 | 33 | 21 | 12 |
| Gender |
| Male | 93 | 3 | 17 | 73 | 46 | 30 | 17 |
| Female | 10 | 1 | 2 | 7 | 3 | 6 | 1 |
| Tumor size
(cm) |
| Single tumor
≤3 | 24 | 1 | 7 | 16 | 11 | 7 | 6 |
| Single tumor
3–5 | 15 | 0 | 4 | 11 | 5 | 3 | 7 |
| Single tumor ≥5,
or ≥ two tumors | 64 | 3 | 8 | 53 | 32 | 26 | 6 |
| Serum HBsAg |
| Positive | 81 | 2 | 11 | 68 | 43 | 26 | 12 |
| Negative | 22 | 2 | 8 | 12 | 5 | 10 | 7 |
| TNM stage |
| II | 38 | 2 | 5 | 31 | 10 | 20 | 8 |
| IIIA | 16 | 0 | 3 | 13 | 8 | 8 | 0 |
| IIIB | 2 | 0 | 0 | 2 | 0 | 2 | 0 |
| IVA | 36 | 1 | 8 | 27 | 21 | 6 | 9 |
| IVB | 11 | 1 | 3 | 7 | 9 | 0 | 2 |
| Histologic
grade |
| Grade I–II | 32 | 3 | 9 | 20 | 22 | 10 | 0 |
| Grade III–IV | 71 | 1 | 10 | 60a | 26 | 26 | 19b |
Relationship between APE1/mutp53
expression and tumor grade malignancy
We also investigated the expression of p53 in 10
normal liver tissues, 40 liver cirrhosis tissues and 103 cases of
HCC tissues using immunohistochemical assay. Immunohistochemical
staining showed that p53 staining was mainly located in the nucleus
in 60.2% (62/103) HCC tissues, and no p53 staining was detected in
both normal liver tissues and liver cirrhosis tissues. Also, 44.66%
(46/103) HCC tissues showed lower APE1 expression, and the
remaining 55.34% (57/103) of tissues showed high APE1 expression.
Using the Chi-square test, we found that the APE1 high expression
was associated with mutp53 status.
As shown in Table
II, we separated these HCC patients into Grade I–II (32 cases)
and Grade III–IV (71 cases). According to APE1 expression level and
p53 status, 103 HCC patients were separated into four groups:
APE1+/p53−, APE1+/p53+,
APE1++/p53− and
APE1++/p53+. The level of APE1 combination
with the mutp53 status was significantly correlated with tumor
grade, and APE1++/p53+ status indicated a
higher tumor grade.
| Table IIRelationship between APE1/P53
expression and tumor grade malignancy. |
Table II
Relationship between APE1/P53
expression and tumor grade malignancy.
| Histologic
grade | Total | APE1/P53
expression | P-value |
|---|
|
|---|
| +/− | +/+ | ++/− | ++/+ |
|---|
| Grade I–II | 32 | 15 | 7 | 4 | 6 | |
| Grade III–IV | 71 | 16 | 8 | 6 | 41 | |
| Total | 103 | 31 | 15 | 10 | 47 | <0.01 |
Mutant p53 binds strongly and
specifically to nonlinear DNA
We selected the p53 response element of the
p21waf1-promoter (p53-RE) as target DNA to analyze the
binding of human p53 protein. The p53-RE was prepared either in its
linear form (p53-RElin) or in a stem loop conformation
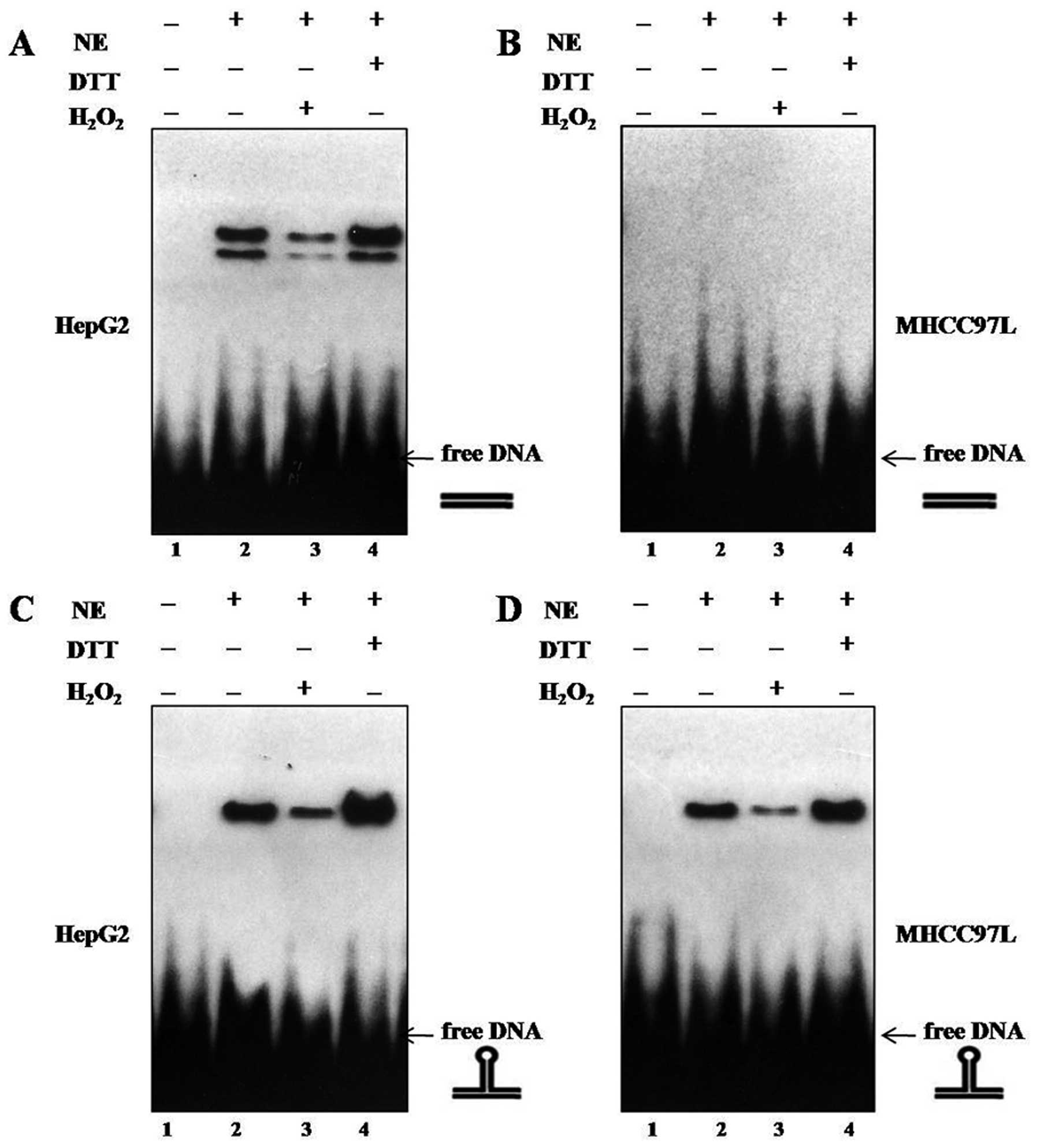
(p53-REstruct) as previously described (18). As shown in Fig. 2A and C, we demonstrated that reduced
wtp53 proteins bound efficiently to linear and nonlinear DNA,
indicating that binding of wtp53 to either DNA conformation
requires a reduced status of the p53-DBD, in accordance with
previous reports (16,19,20).
Compared to the high-affinity binding of wtp53, mutp53 bound
efficiently to nonlinear DNA (Fig.
2D), but not to linear DNA (Fig.
2B), in line with the findings of Gohler et al, and the
specific and selective binding of mutp53 to nonlinear DNA provide
mutp53 proteins as DNA structure-specific DNA-binding (DSSB)
proteins (17). DTT application
enhanced the mutp53 DNA binding to nonlinear DNA, whereas addition
of H2O2 attenuated the binding of mutp53 to
the nonlinear DNA substrate p53-REstruct, indicating
that binding of mutp53 to nonlinear DNA requires a reduced p53
status as well as in wtp53 DNA binding (16) (Fig.
2D). Thus, enhanced DNA binding to nonlinear DNA by addition of
DTT corresponds to DNA binding by reduced mutp53.
APE1 stimulates mutant p53 DNA binding to
nonlinear DNA
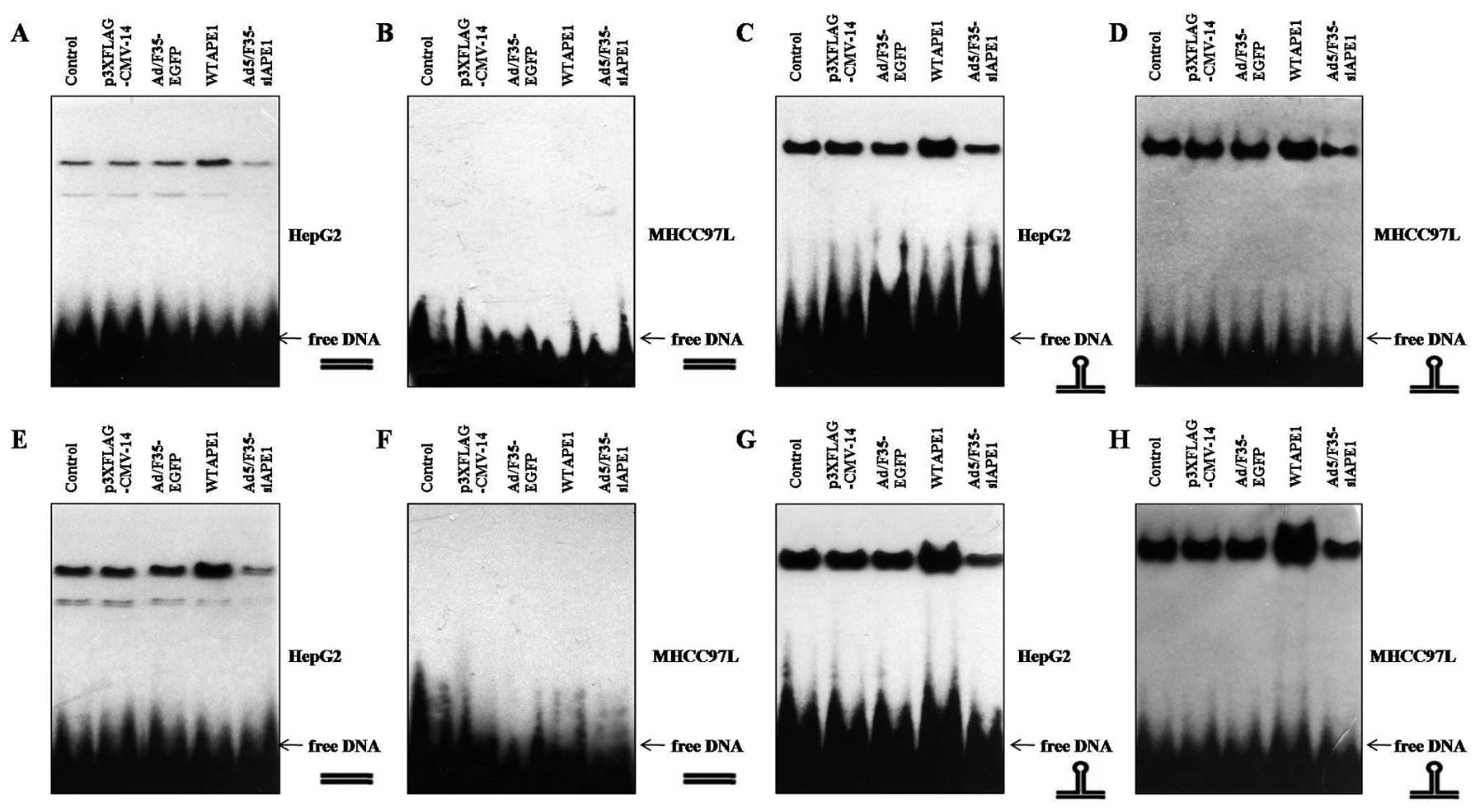
To test whether APE1 influences p53 DNA binding,
cells were infected with p3XFLAG-CMV/APE1 (WTAPE1) or
p3XFLAG-CMV-14 empty vector, Ad5/F35-APE1 siRNA or Ad5/F35-EGFP. We
found that addition of the WTAPE1 further and strongly enhanced
binding of wtp53 to p53-RElin DNA (Fig. 3A) and p53-REstruct DNA
(Fig. 3C) compared with
p3XFLAG-CMV-14, as previously described by Jayaraman et al
(15). Moreover, Ad5/F35-APE1 siRNA
attenuated wtp53 DNA binding to either DNA conformation compared
with Ad5/F35-EGFP (Fig. 3A and C).
Of note, WTAPE1 enhanced binding of mutp53 to
p53-REstruct DNA (Fig.
3D), but not to p53-RElin DNA (3B). Additionally,
Ad5/F35-siAPE1 attenuated p53 DNA binding to
p53-REstruct compared with Ad5/F35-EGFP (Fig. 3D).
To further examine whether APE1 also influences p53
DNA binding independently of its reducing activities, we examined
the effects of APE1 in the presence of DTT. As shown in Fig. 3E and G, WTAPE1 potentiated the wtp53
DNA binding to both linear and nonlinear DNA structures, and
decreased APE1 attenuated the wtp53 DNA binding activities. We next
examined the effects of APE1 on reduced mutp53 DNA binding and
found that the reduced mutp53 also did not bind to linear DNA
(Fig. 3F), and the binding of
reduced mutp53 to nonlinear DNA was enhanced by WTAPE1 and
inhibited by Ad5/F35-siAPE1 infection (Fig. 3H).
Mutant p53 binds to DNA substrate in
vivo
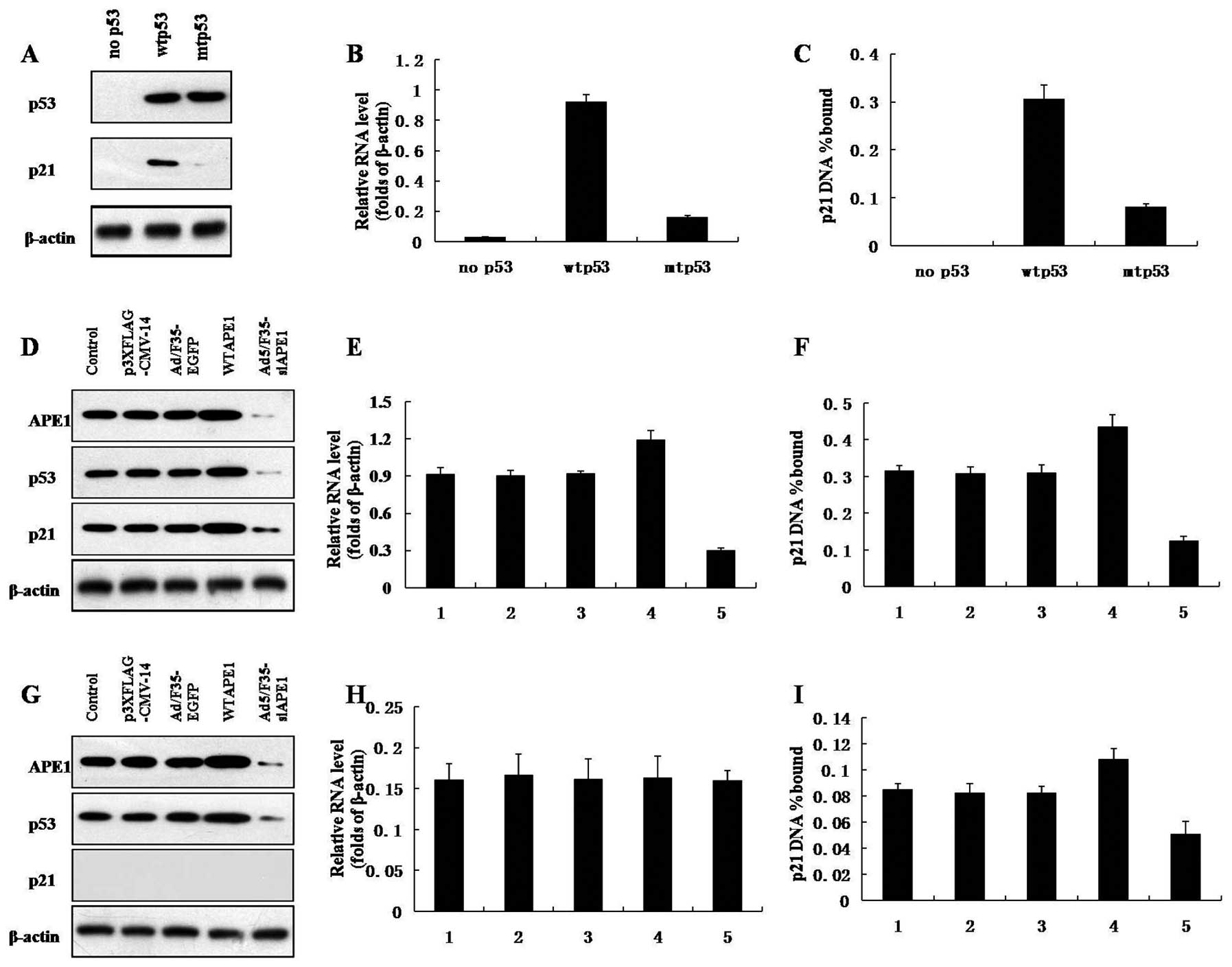
Western blotting showed that the cells expressed the
same amount of the different p53 alleles and confirmed that the p53
mutants are defective in induction of p21 (Fig. 4A). To demonstrate the DNA binding
activity of p53 in vivo, ChIP assays were performed on
Hep3B, HepG2 and MHCC97L cells. The amount of target DNA
precipitated is expressed as a percentage of the input target DNA.
As shown in Fig. 4C, the p21
promoter precipitation and p21 mRNA level increased in wtp53 and
mutp53 cells, compared with that in p53-null cells (Fig. 4B and C). In addition, the p21
promoter precipitation and p21 mRNA expression in mutp53 cells was
much lower than that in wtp53 cells (Fig. 4B and C).
APE1 stimulates the mutp53 DNA binding to
p21 promoter in vivo
As shown in Fig. 4D,
a decrease in APE1, p53 and p21 protein expression was observed
after infection with Ad5/F35-siAPE1, and APE1, p53 and p21 protein
levels increased after the WTAPE1 infection in wtp53 cells.
Ad5/F35-siAPE1 also inhibited the protein expression of APE1 and
p53 in mutp53 cells, while WTAPE1 increased the APE1 and p53
protein levels (Fig. 4G). The p53
mutants were defective in induction of p21 protein in mutp53 cells
(Fig. 4G).
To test whether the DNA binding activity of mutp53
is regulated by APE1 in vivo, ChIP assays were performed. In
wtp53 cells, the WTAPE1 group showed significant increase in p21
mRNA level and p21 promoter precipitation compared with the
p3XFLAG-CMV-14 empty vector, while the p21 mRNA level and p21
promoter precipitation clearly decreased in the Ad5/F35-siAPE1
treatment group compared with the Ad5/F35-EGFP group (Fig. 4E and F). p21 promoter precipitation
and p21 mRNA expression were lower in mutp53 cells than in wtp53
cells. As shown in Fig. 4H, the p21
mRNA expression in mutp53 cells remained at lower levels and was
not affected by APE1. Of note, WTAPE1 increased the DNA binding of
mutp53 to p21 promoter, whereas silencing of APE1 inhibited the DNA
binding activity of mutp53 (Fig.
4I).
Discussion
In the present study, we first examined the
expression of APE1 and mutp53 in HCC tissues. Our data indicated
that the increased APE1 level, cytoplasmic localization of APE1 and
mutp53 expression were relevant to neoplastic alteration and poor
differentiation of HCC. Notably, we observed that reduced mutp53
bound efficiently to nonlinear DNA but not to linear DNA, and APE1
could enhance DNA binding activity of mutp53 to nonlinear DNA.
Recent studies demonstrated that elevated APE1
expression was observed in several human tumors, such as ovarian
cancer, cervical cancer, non-small cell lung cancer, osteosarcoma
and other tumors (6,21–24).
The increased APE1 expression was associated with chemo- and
radioresistance, whereas downregulation of APE1 can enhance tumor
sensitivity to chemo- and radiotherapy (5,6,25,26).
Moreover, the tumor cells showed cytoplasmic reactivity of APE1,
whereas the normal cells showed APE1 staining in the nucleus,
indicating that APE1 subcellular localization has a prognostic
value and correlates with aggressiveness (21,27–29).
In this study, the shifts of APE1 from nucleus to cytoplasm and
increased APE1 expression were correlated with neoplastic
alteration and a lower degree of differentiation, which is in line
with a previous study (8).
p53 is one of the most important tumor suppressor
genes in the genome and encodes a transcription regulatory protein
that helps preserve genomic integrity by its participation in
stress-response pathways and DNA repair pathways (30,31).
APE1 has been found to be a potent activator of p53 DNA binding,
which can enhance p53 DNA binding by redox-dependent and
-independent mechanisms (15,16,32,33).
Overexpression of APE1 in tumors is associated with increased
levels of p53, and they are independent predictors of prognosis and
poor response to chemotherapy (34). p53 is mutated in ~50% of human
cancer types including HCC (35),
and its mutations not only lose their tumor suppressive activities
but often gain additional oncogenic functions (13,14,36).
Our data showed that increased APE1 expression was associated with
mutp53 proteins, and the combination of higher APE1 expression and
mutp53 expression was correlated with a lower degree of tumor
differentiation, which might be a risk factor for HCC.
In addition to the DNA repair functions (37), APE1, as a redox factor, maintains
transcription factors in an active reduced state (38). In this role, APE1 stimulates the DNA
binding activity of numerous transcription factors, including AP-1,
NF-κB, HIF-1α, p53 and others (3,39–41).
Wtp53-SSDB can occur in different modes depending on the
conformation of p53-binding sites, either sequence-specific to
linear DNA, or sequence- and structure-specific to nonlinear DNA.
In contrast to SSDB to linear DNA, which is most probably mediated
solely by the p53 core DBD (42),
sequence-specific and DNA structure-dependent SSDB to nonlinear DNA
and non-SSDB modes of DNA interaction involve both the DBD and the
CRD (18,43–45).
The complex interactions of mutp53 with DNA were shown to require
both the mutp53 DBD and the intact p53 CRD (46). Although mutp53 has lost the
wtp53-SSDB and could not elicit the same transcriptional response
as wtp53, it requires the p53 DBD and the CRD for high-affinity
binding (47,48). Gohler et al revealed that the
specific and selective binding of mutp53 to nonlinear DNA provide
mutp53 proteins as DNA structure-specific DNA-binding (DSSB)
proteins (17). The CRD is
important for mediating stable complex formation of p53 with
nonlinear DNA in mutp53-DSSB (17)
and in wtp53-SSDB (18,44,45).
Similar to previous studies, we found that mutp53 bound efficiently
to nonlinear DNA, but not to linear DNA (17). Markedly, addition of DTT enhanced
the mutp53 DNA binding, requiring a reduced p53 status as well as
in wtp53 DNA binding (16).
APE1 is one of the cofactors positively influencing
DNA binding of p53 in vitro (15) and transcription in vivo
(33,49). As a redox factor, APE1 activates
wtp53 for SSDB by reducing disulfide bonds in the p53 DBD (15,49).
In addition, APE1 is also able to enhance DNA binding of reduced
wtp53 by a redox-independent manner, which facilitates the
formation of p53 tetramers from higher oligomeric forms as well as
from dimers. Previous studies revealed that APE1 enhanced wtp53 DNA
binding to both p53-RElin and p53-REstruct
(15,16). The redox-independent activation of
wtp53 is due to a regulatory interaction of APE1 with the p53 CRD,
as truncated p53 lacking the CRD could no longer be activated by
APE1 (15). Although a stable
interaction between p53 and APE1 could not be shown (15), a small fraction of the p53 and APE1
proteins (~5%) interacted in Far-Western and IP-Western assays
in vitro (33). Furthermore,
Tan et al showed that APE1 and p53 colocalize in vivo
(50), supporting the theory of a
specific, albeit physically weak, interaction of these proteins.
The 249th codon of p53 is mutated from AGG to AGT and the amino
acid from Arg to Ser (R249S) in >50% HCC patients in China,
which is intact as wtp53 proteins. As CRD is essential in DNA
structure-dependent binding of mutp53, which is intact in most of
the frequently encountered mutp53 proteins, we investigated whether
APE1 could regulate the DNA binding activity of mutp53 proteins.
Our on-array binding analyses showed that APE1 enhanced mutp53 DNA
binding to nonlinear DNA, but not to linear DNA. Notably, we
demonstrated that APE1 also enhanced the DNA binding activity of
mutp53 to p21 promoter in vivo. In addition, the p53 mutant
is defective in induction of p21, which is in accordance with
previous studies (33,49).
In conclusion, APE1 was able to stimulate DNA
binding activity of mutp53, and the expression of APE1 and mutp53
was correlated with carcinogenesis and progression of HCC, which
indicates that APE1 and p53 may be potential molecular therapeutic
targets of HCC. The present study may contribute to a better
understanding of the transcriptional regulation of p53 by APE1 and
may, therefore, be the basis for the design of new clinical
trials.
Acknowledgements
Grant support was provided by the National Natural
Science Foundation of China (no. 30872975).
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Fleck O and Nielsen O: DNA repair. J Cell
Sci. 117:515–517. 2004. View Article : Google Scholar
|
|
3
|
Evans AR, Limp-Foster M and Kelley MR:
Going APE over ref-1. Mutat Res. 461:83–108. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang D, Xiang DB, Yang XQ, et al: APE1
overexpression is associated with cisplatin resistance in non-small
cell lung cancer and targeted inhibition of APE1 enhances the
activity of cisplatin in A549 cells. Lung Cancer. 66:298–304. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koukourakis MI, Giatromanolaki A,
Kakolyris S, et al: Nuclear expression of human
apurinic/apyrimidinic endonuclease (HAP1/Ref-1) in head-and-neck
cancer is associated with resistance to chemoradiotherapy and poor
outcome. Int J Radiat Oncol Biol Phys. 50:27–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang D, Luo M and Kelley MR: Human
apurinic endonuclease 1 (APE1) expression and prognostic
significance in osteosarcoma: enhanced sensitivity of osteosarcoma
to DNA damaging agents using silencing RNA APE1 expression
inhibition. Mol Cancer Ther. 3:679–686. 2004.
|
|
7
|
Xiang DB, Chen ZT, Wang D, et al: Chimeric
adenoviral vector Ad5/F35-mediated APE1 siRNA enhances sensitivity
of human colorectal cancer cells to radiotherapy in vitro and in
vivo. Cancer Gene Ther. 15:625–635. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Di Maso V, Avellini C, Crocè LS, et al:
Subcellular localization of APE1/Ref-1 in human hepatocellular
carcinoma: possible prognostic significance. Mol Med. 13:89–96.
2007.PubMed/NCBI
|
|
9
|
Puglisi F, Barbone F, Tell G, et al:
Prognostic role of Ape/Ref-1 subcellular expression in stage I–III
breast carcinomas. Oncol Rep. 9:11–17. 2002.PubMed/NCBI
|
|
10
|
Helton ES and Chen X: p53 modulation of
the DNA damage response. J Cell Biochem. 100:883–896. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zurer I, Hofseth LJ, Cohen Y, et al: The
role of p53 in base excision repair following genotoxic stress.
Carcinogenesis. 25:11–19. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Strano S, Dell’Orso S, Di Agostino S,
Fontemaggi G, Sacchi A and Blandino G: Mutant p53: an oncogenic
transcription factor. Oncogene. 26:2212–2219. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McClendon AK, Dean JL, Ertel A, et al: RB
and p53 cooperate to prevent liver tumorigenesis in response to
tissue damage. Gastroenterology. 141:1439–1450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Itoh T, Shiro T, Seki T, et al:
Relationship between p53 overexpression and the proliferative
activity in hepatocellular carcinoma. Int J Mol Med. 6:137–142.
2000.PubMed/NCBI
|
|
15
|
Jayaraman L, Murthy KG, Zhu C, Curran T,
Xanthoudakis S and Prives C: Identification of redox/repair protein
Ref-1 as a potent activator of p53. Genes Dev. 11:558–570. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hanson S, Kim E and Deppert W: Redox
factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting
p53 tetramerization. Oncogene. 24:1641–1647. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gohler T, Jager S, Warnecke G, Yasuda H,
Kim E and Deppert W: Mutant p53 proteins bind DNA in a DNA
structure-selective mode. Nucleic Acids Res. 33:1087–1100. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gohler T, Reimann M, Cherny D, et al:
Specific interaction of p53 with target binding sites is determined
by DNA conformation and is regulated by the C-terminal domain. J
Biol Chem. 277:41192–41203. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hainaut P and Milner J: Redox modulation
of p53 conformation and sequence-specific DNA binding in vitro.
Cancer Res. 53:4469–4473. 1993.PubMed/NCBI
|
|
20
|
Delphin C, Cahen P, Lawrence JJ and
Baudier J: Characterization of baculovirus recombinant wild-type
p53. Dimerization of p53 is required for high-affinity DNA binding
and cysteine oxidation inhibits p53 DNA binding. Eur J Biochem.
223:683–692. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang S, Irani K, Heffron SE, Jurnak F and
Meyskens FL Jr: Alterations in the expression of the
apurinic/apyrimidinic endonuclease-1/redox factor-1 (APE/Ref-1) in
human melanoma and identification of the therapeutic potential of
resveratrol as an APE/Ref-1 inhibitor. Mol Cancer Ther.
4:1923–1935. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Robertson KA, Bullock HA, Xu Y, et al:
Altered expression of Ape1/ref-1 in germ cell tumors and
overexpression in NT2 cells confers resistance to bleomycin and
radiation. Cancer Res. 61:2220–2225. 2001.PubMed/NCBI
|
|
23
|
Bobola MS, Blank A, Berger MS, Stevens BA
and Silber JR: Apurinic/apyrimidinic endonuclease activity is
elevated in human adult gliomas. Clin Cancer Res. 7:3510–3518.
2001.PubMed/NCBI
|
|
24
|
Moore DH, Michael H, Tritt R, Parsons SH
and Kelley MR: Alterations in the expression of the DNA
repair/redox enzyme APE/ref-1 in epithelial ovarian cancers. Clin
Cancer Res. 6:602–609. 2000.PubMed/NCBI
|
|
25
|
Wilson DM III, Bennett RA, Marquis JC,
Ansari P and Demple B: Trans-complementation by human apurinic
endonuclease (Ape) of hypersensitivity to DNA damage and
spontaneous mutator phenotype in apn1-yeast. Nucleic Acids Res.
23:5027–5033. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hansen WK, Deutsch WA, Yacoub A, Xu Y,
Williams DA and Kelley MR: Creation of a fully functional human
chimeric DNA repair protein. Combining O6-methylguanine DNA
methyltransferase (MGMT) and AP endonuclease (APE/redox effector
factor 1 (Ref 1)) DNA repair proteins. J Biol Chem. 273:756–762.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Minisini AM, Di Loreto C, Mansutti M, et
al: Topoisomerase IIalpha and APE/ref-1 are associated with
pathologic response to primary anthracycline-based chemotherapy for
breast cancer. Cancer Lett. 224:133–139. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Puglisi F, Aprile G, Minisini AM, et al:
Prognostic significance of Ape1/ref-1 subcellular localization in
non-small cell lung carcinomas. Anticancer Res. 21:4041–4049.
2001.PubMed/NCBI
|
|
29
|
Zhang Y, Wang J, Xiang D, Wang D and Xin
X: Alterations in the expression of the apurinic/apyrimidinic
endonuclease-1/redox factor-1 (APE1/Ref-1) in human ovarian cancer
and indentification of the therapeutic potential of APE1/Ref-1
inhibitor. Int J Oncol. 35:1069–1079. 2009.PubMed/NCBI
|
|
30
|
Muller PA, Vousden KH and Norman JC: p53
and its mutants in tumor cell migration and invasion. J Cell Biol.
192:209–218. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Norbury CJ and Zhivotovsky B: DNA
damage-induced apoptosis. Oncogene. 23:2797–2808. 2004. View Article : Google Scholar
|
|
32
|
Yu Y, Li CY and Little JB: Abrogation of
p53 function by HPV16 E6 gene delays apoptosis and enhances
mutagenesis but does not alter radiosensitivity in TK6 human
lymphoblast cells. Oncogene. 14:1661–1667. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gaiddon C, Moorthy NC and Prives C: Ref-1
regulates the transactivation and pro-apoptotic functions of p53 in
vivo. EMBO J. 18:5609–5621. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Couture C, Raybaud-Diogene H, Tetu B, et
al: p53 and Ki-67 as markers of radioresistance in head and neck
carcinoma. Cancer. 94:713–722. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Puisieux A, Ponchel F and Ozturk M: p53 as
a growth suppressor gene in HBV-related hepatocellular carcinoma
cells. Oncogene. 8:487–490. 1993.PubMed/NCBI
|
|
36
|
Cadwell C and Zambetti GP: The effects of
wild-type p53 tumor suppressor activity and mutant p53
gain-of-function on cell growth. Gene. 277:15–30. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Demple B and Sung JS: Molecular and
biological roles of Ape1 protein in mammalian base excision repair.
DNA Repair. 4:1442–1449. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kelley MR, Georgiadis MM and Fishel ML:
APE1/Ref-1 role in redox signaling: translational applications of
targeting the redox function of the DNA repair/redox protein
APE1/Ref-1. Curr Mol Pharmacol. 5:36–53. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tell G, Damante G, Caldwell D and Kelley
MR: The intracellular localization of APE1/Ref-1: more than a
passive phenomenon? Antioxid Redox Signal. 7:367–384. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xanthoudakis S, Miao GG and Curran T: The
redox and DNA-repair activities of Ref-1 are encoded by
nonoverlapping domains. Proc Natl Acad Sci USA. 91:23–27. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Walker LJ, Robson CN, Black E, Gillespie D
and Hickson ID: Identification of residues in the human DNA repair
enzyme HAP1 (Ref-1) that are essential for redox regulation of Jun
DNA binding. Mol Cell Biol. 13:5370–5376. 1993.PubMed/NCBI
|
|
42
|
Anderson ME, Woelker B, Reed M, Wang P and
Tegtmeyer P: Reciprocal interference between the sequence-specific
core and nonspecific C-terminal DNA binding domains of p53:
implications for regulation. Mol Cell Biol. 17:6255–6264.
1997.PubMed/NCBI
|
|
43
|
Yakovleva T, Pramanik A, Terenius L,
Ekstrom TJ and Bakalkin G: p53 latency - out of the blind alley.
Trends Biochem Sci. 27:612–618. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Espinosa JM and Emerson BM:
Transcriptional regulation by p53 through intrinsic DNA/chromatin
binding and site-directed cofactor recruitment. Mol Cell. 8:57–69.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
McKinney K and Prives C: Efficient
specific DNA binding by p53 requires both its central and
C-terminal domains as revealed by studies with high-mobility group
1 protein. Mol Cell Biol. 22:6797–6808. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Muller BF, Paulsen D and Deppert W:
Specific binding of MAR/SAR DNA-elements by mutant p53. Oncogene.
12:1941–1952. 1996.PubMed/NCBI
|
|
47
|
Lanyi A, Deb D, Seymour RC, Ludes-Meyers
JH, Subler MA and Deb S: ‘Gain of function’ phenotype of
tumor-derived mutant p53 requires the
oligomerization/nonsequence-specific nucleic acid-binding domain.
Oncogene. 16:3169–3176. 1998.
|
|
48
|
Frazier MW, He X, Wang J, Gu Z, Cleveland
JL and Zambetti GP: Activation of c-myc gene expression by
tumor-derived p53 mutants requires a discrete C-terminal domain.
Mol Cell Biol. 18:3735–3743. 1998.
|
|
49
|
Ueno M, Masutani H, Arai RJ, et al:
Thioredoxin-dependent redox regulation of p53-mediated p21
activation. J Biol Chem. 274:35809–35815. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tan Z, Sankar R, Tu W, et al:
Immunohistochemical study of p53-associated proteins in rat brain
following lithium-pilocarpine status epilepticus. Brain Res.
929:129–138. 2002. View Article : Google Scholar : PubMed/NCBI
|