Introduction
Cigarette smoking, which causes lung and other types
of cancer, is also a risk factor of both established tumor
metastasis and increases overall mortality in cancer patients
(1,2). In addition, smokers have increased
vulnerability to atherosclerosis and are predisposed to allergic
airway diseases (3,4). These studies suggest that by altering
cell viability, smoking enables tumor cells and vascular
endothelial cells to evade appropriate immune responses. Nicotine,
a major component of cigarette smoke, is widely accepted as a risk
factor for carcinogenesis and atherosclerosis (1). However, to date, little is known about
how and to what extent nicotine contributes to the adverse effects
of chronic tobacco use, apart from its psychoactive actions and
addictive properties. Although nicotine could promote lung cancer
development, reduce the efficacy of chemotherapeutic agents
(5) and activate hypoxia-inducible
factor-1α expression (6), the fast
synaptic transmission in key regions controlling behavior mediated
by nicotine via nicotine acetylcholine receptors (nAChRs) was also
reported (3), indicating that
nicotine might be a survival agonist against various stresses
inducing apoptosis (4). We
previously studied the biological roles of nicotine and found that
nicotine activates bone marrow-derived dendritic cells and
nicotine-treated dendritic cells have potential antitumor effects
(7–9). Hence, the exact effect of nicotine on
cell survival has not been fully characterized and remains
controversial.
nAChRs, which belong to a family of ionotropic
receptors consisting of α-subunits or a combination of α and
β-subunits, are widely distributed throughout the central and
peripheral nervous system (10). In
addition, non-neuronal cells such as monocytes, endothelial cells
and epithelial cells were also found to express nAChRs (11). Despite documented data showing that
α1, α3, α5 nAChRs play important roles in regulating cell
proliferation, apoptosis and facilitating tumor formation (12–14),
α7 nAChR, which is the main receptor of nACh in non-neuronal cells,
has acquired more attention in recent years. For example, α7 nAChR
was found to be involved in gastric and colon cancer migration
(15,16) and to also upregulate PPARβ/δ
expression in human lung carcinoma (17). The fact that inhibition of
α7-nicotinic receptor reduces tumorigenicity in A549 NSCLC
xenografts (18) and facilitates
lung cancer treatment (19)
indicated that α7 nAChR plays important roles in tumor
proliferation, angiogenesis and apoptosis (20–22).
To date, however, the exact role of α7 nAChR in nicotine-mediated
cell proliferation and anti-apoptotic effects has not been fully
elucidated, and is important for potential tumor therapeutic target
molecule discovery.
In the present study, to investigate the roles of α7
nAChR in nicotine-mediated cell proliferation and anti-apoptotic
effects, Raw264.7 and El4 cells were treated with nicotine and cell
proliferation was firstly determined by CCK-8 assay and western
blot analysis, respectively. Then, the roles of α7 nAChR in
nicotine-augmented cell proliferation and anti-apoptotic effects
were further explored by pre-incubation of cells with α7 nAChR
specific antagonist α-bungarotoxin and broad spectrum nicotinic
antagonist tubocurarine chloride. The effects of nicotine on
cisplatin-induced mitochondria translocation of Bax and the release
of cytochrome c from mitochondria were investigated by
mitochondria isolation and western blot analysis. Using kinase
inhibitors, the roles of Erk-JNK-p38 and PI3K-Akt phosphorylation
in nicotine-mediated cell proliferation and anti-apoptosis effects
were further investigated. The results showed that, firstly,
nicotine treatment clearly augmented cell viability, upregulated
Mcl-1 and Bcl-2 expression and decreased caspase-3 activation in
Raw264.7 and El4 cells. Secondly, the pretreatment of
α-bungarotoxin and tubocurarine chloride efficiently attenuated
nicotine-augmented cell proliferation, α7 nAChR upregulation and
abolished the inhibitory effects of nicotine on cisplatin-induced
caspase-3 activation. Further exploration demonstrated that
nicotine efficiently abolished cisplatin-promoted mitochondria
translocation of Bax and the release of cytochrome c.
Notably, both Erk-JNK-p38 and PI3K-Akt signaling pathways could be
activated by nicotine treatment in Raw264.7 and El4 cells. When
Erk-JNK and PI3K-Akt activities were inhibited, nicotine-augmented
cell proliferation ability and anti-apoptotic effects were reversed
accordingly. The results presented here indicate that nicotine
could achieve α7 nAChR-mediated proliferation and anti-apoptotic
effects by activating the Erk-JNK and PI3K-Akt pathways
respectively, providing potential molecules to deal with
tobacco-associated human diseases.
Materials and methods
Reagents
Reagents were purchased from the following
companies: nicotine, α-bungarotoxin and tubocurarine chloride were
obtained from Sigma-Aldrich (St. Louis, MO, USA). Cisplatin (DDP)
was purchased from Calbiochem (San Diego, CA, USA). Annexin V/PI
Apoptosis Detection kit was obtained from Promega (Madison, WI,
USA). Cell Counting Kit-8 (CCK-8) was from Dojindo Laboratories
(Kumamoto, Japan). p38 MAPK inhibitor SB203580, JNK MAPK inhibitor
SP600125, Erk1/2 inhibitor U0126, PI3K inhibitor LY294002 and Akt
inhibitor Wortmanin were from Cayman Chemical (Ann Arbor, MI, USA).
Antibodies to β-actin, Cox IV, PCNA, α7 nAChR, Bcl-2, Mcl-1, Bax,
cleaved caspase-3, cytochrome c, phospho-p38,
phospho-Erk1/2, phospho-Mek1/2, phospho-c-Raf, phospho-Msk,
phospho-p90Rsk, phospho-JNK were from Cell Signaling Technology
(Beverly, MA, USA). Mitochondria isolation kit for cultured cells
was from Thermo Fisher Scientific (Rockford, IL, USA). RPMI-1640
medium, Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine
serum (FBS) were purchased from HyClone (Logan, UT, USA).
Cell line
The Raw264.7 and El4, macrophage and T lymphoma cell
lines respectively, were obtained from Shanghai Cell Bank
(Shanghai, China). Cells were cultured in DMEM with 10% FBS at 37°C
in 5% CO2 and passed every 1–2 days to maintain
logarithmic growth. Cells were synchronized by serum starvation for
at least 12 h before the treatment of nicotine for indicated
periods or concentrations. To investigate the role of α7 nAChR in
nicotine-mediated proliferation and anti-apoptotic effect, the
cells were pretreated with 1 μg/ml α7 nAChR specific antagonist
α-bungarotoxin or broad spectrum nicotinic antagonist tubocurarine
chloride prior to the indicated nicotine treatment.
Cell apoptosis assay
Cell apoptosis assay was determined by flow
cytometry according to the method previously described (23). Briefly, 5×104 Raw264.7 or
El4 cells seeded in 24-well plates were pretreated with 10 μM
U0126, SB203580 or SP6001250 1 h prior to 24-h 10−7 M
nicotine stimulation and were further treated with 2 μg/ml
cisplatin for 17 h. Then, the cells were removed by trypsinization,
rinsed with PBS and re-suspended in binding buffer containing
Annexin V-FITC and propidium iodide (PI) for 20 min at room
temperature. The samples were analyzed on FACSCalibur and data were
analyzed with CellQuest software.
Cell proliferation assay
Cell proliferation assay was performed as previously
described (24). Briefly,
5×104 El4 or Raw264.7 cells were inoculated in 96-well
plate in 100 μl/well medium in a humidified incubator (37°C, 5%
CO2). Then, the cells were treated with 10 μM
LY294002/Wortmanin or 1 μg/ml tubocurarine chloride/α-bungarotoxin
60 min prior to 10−7 mol/l nicotine stimulation for
indicated periods. CCK-8 solutions (10 μl) were added to each well
of the plate and OD450 value was detected with the wavelength of
450 nm.
Mitochondria isolation
To explore the effect of nicotine on
cisplatin-induced mitochondrial translocation of Bax and the
release of cytochrome c, 2×107 Raw264.7 cells
were treated with 10−7 mol/l nicotine for 8 h prior to
cisplatin (1 μg/ml) treatment. The mitochondria isolation was
performed according to the standard procedure. Briefly, cell pellet
was harvested by 850 × g centrifuging and vortexed at medium speed
in 800 μl Mitochondria Isolation Reagent A. Then, 10 μl
Mitochondria Isolation Reagent B was added and incubated on ice for
5 min with further lysis by 800 μl Mitochondria Isolation Reagent
C. After 15 min 12,000 × g centrifugation, the pellet contained
isolated mitochondria and the supernatant contained cytosol
fraction. The Bax translocation and cytochrome c release
were determined by western blot analysis, respectively. β-actin and
Cox IV were used as internal control.
Western blot analysis
Proteins were obtained in lysis buffer as previously
described (7). For analysis of
PI3K-Akt, MAPK kinases phosphorylation and upregulation of Bcl-2,
Mcl-1, PCNA, α7 nAChR induced by nicotine stimulation,
2×107 Raw264.7 or El4 cells were treated with
10−7 mol/l nicotine stimulation for indicated periods.
To explore the role of α7 nAChR in nicotine-augmented cell
proliferation and anti-apoptotic effect, cells were treated with 1
μg/ml tubocurarine chloride or α-bungarotoxin 60 min prior to 24 h
10−7 mol/l nicotine stimulation. Proteins were loaded
onto SDS-PAGE gels for electrophoresis and then transferred onto
PVDF membranes. After blocking in 5% fat-free milk in TBST for 1.5
h, the membranes were incubated with primary antibodies at 4°C
overnight. Subsequently, the membranes were incubated with
corresponding HRP-conjugated secondary antibodies at room
temperature for 1.5 h. After washing six times with TBST (for 10
min each), bound antibodies were visualized using enhanced
chemiluminescence ECL. β-actin was used as loading control.
Statistical analysis
Each experiment was repeated at least 3 times and
confirmed that similar data were obtained. All data are expressed
as mean and standard errors. Statistical significance was tested
using one-way ANOVA with post Newman-Keuls test or two-way ANOVA
with post Bonferroni test. Statistical differences were considered
to be significant if P<0.05.
Results
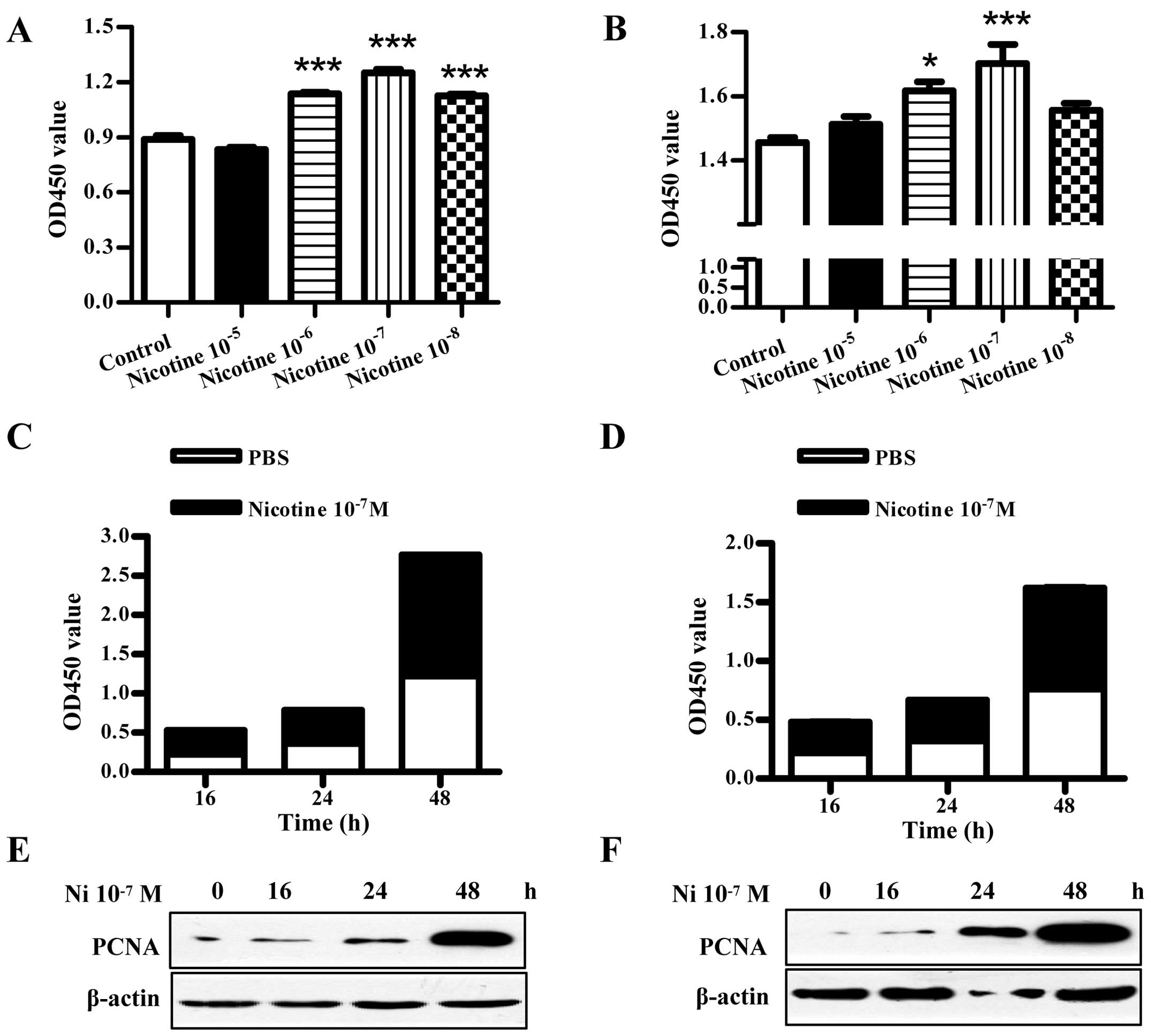
Nicotine treatment augments cell
proliferation abilities in Raw264.7 and El4 cells
Our previous studies showed that nicotine could
activate bone marrow-derived dendritic cells and nicotine-treated
dendritic cells have potential antitumor effects (7–9). To
explore the effect of nicotine on cell proliferation, both Raw264.7
and El4 cells were treated with 10−5–10−8
mol/l nicotine for 12 h or 10−7 mol/l nicotine for
indicated periods and cell viabilities, PCNA expression were
determined by CCK-8 assay and western blot analysis, respectively.
The results showed that compared to PBS-treated control,
10−6–10−8 mol/l nicotine treatment clearly
augmented Raw264.7 cell viability (***P<0.001,
one-way ANOVA with post Newman-Keuls test) (Fig. 1A). Meanwhile, 16, 24 and 48 h
10−7 mol/l nicotine treatment augmented Raw264.7 cell
proliferation to 137.5, 118.7 and 126.7% respectively (Fig. 1C). PCNA protein determination also
showed that nicotine treatment upregulated PCNA expression in
Raw264.7 cells (Fig. 1E). Cell
viability and PCNA expression investigations in El4 cells presented
similar results (Fig. 1B, D and F).
These results illustrate that nicotine augments cell proliferation
in both a concentration and period manner.
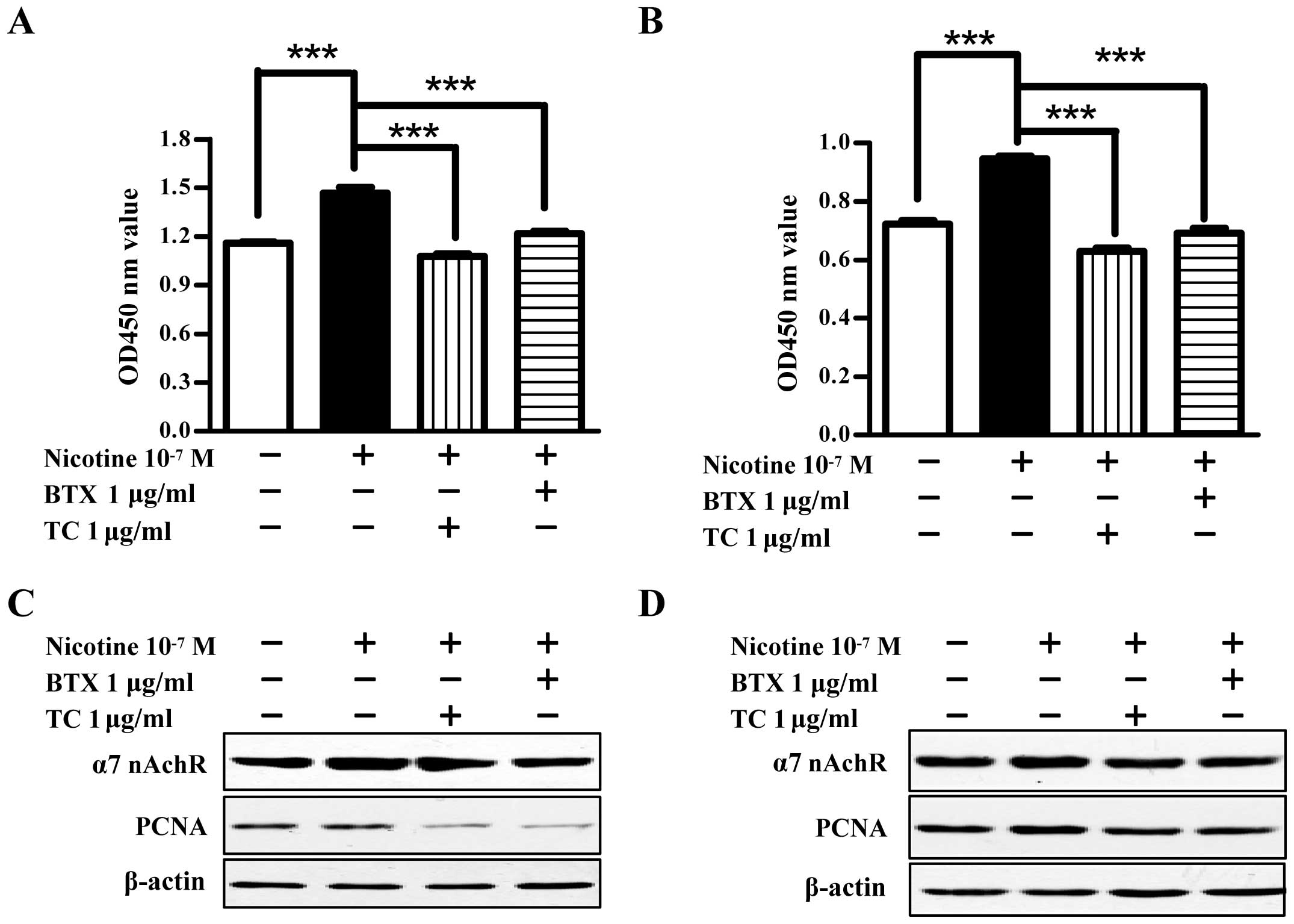
α7 nAChR is involved in
nicotine-augmented cell proliferation in both Raw264.7 and El4
cells
α7 nAChR, expressed in non-neuronal cells such as
monocytes and endothelial cells, is one of the most abundant
nicotinic acetylcholine receptors (11). To investigate the role of α7 nAChR
in nicotine-increased cell proliferation, Raw264.7 and El4 cells
were pretreated with α7 nAChR specific antagonist α-bungarotoxin or
non-specific antagonist tubocurarine chloride prior to nicotine
treatment. Then, cell viability, α7 nAChR and PCNA expression were
determined by CCK-8 assay and western blot analysis, respectively.
The results showed that compared with vehicle-treated cells,
nicotine treatment significantly enhanced cell viability in both
Raw264.7 and El4 cells (Fig. 2A and
B). The pretreatment of α-bungarotoxin or tubocurarine chloride
clearly abolished the effect of nicotine on cell viability. For
example, the pretreatment of α-bungarotoxin or tubocurarine
chloride achieved ~83 and 73% inhibitory rate, respectively, in
Raw264.7 cells (***P<0.001, one-way ANOVA with post
Newman-Keuls test). As nicotine significantly upregulated α7 nAChR
and PCNA expression in both Raw264.7 and El4 cells, the
preincubation of α-bungarotoxin or tubocurarine chloride abolished
the effect of nicotine on α7 nAChR and PCNA expression (Fig. 2C and D). These data indicate that
the upregulation of α7 nAChR induced by nicotine treatment
contributes to nicotine-enhanced cell proliferation abilities.
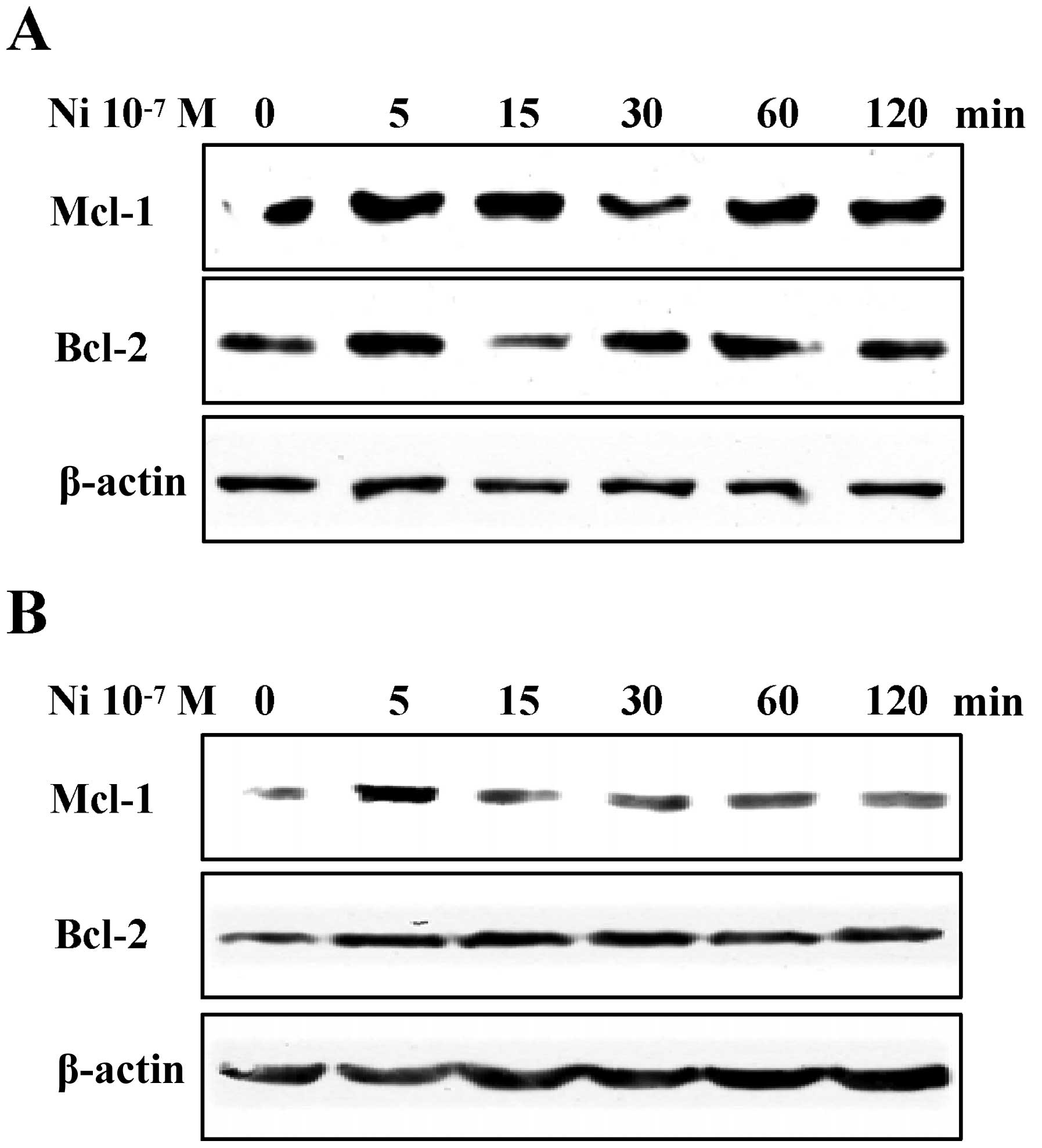
Nicotine treatment upregulates
anti-apoptotic protein Mcl-1 and Bcl-2 expression in Raw264.7 and
El4 cells
To explore the effect of nicotine on anti-apoptotic
protein Mcl-1 and Bcl-2 expression, Raw264.7 and El4 cells were
treated with 10−7M nicotine for indicated periods, the
expression of Mcl-1 and Bcl-2 was determined by western blot
analysis. The results showed that the expression of Mcl-1 and Bcl-2
was continuously augmented by nicotine treatment in both Raw264.7
(Fig. 3A) and El4 (Fig. 3B) cells, indicating that nicotine
treatment might achieve anti-apoptotic effects on these cells.
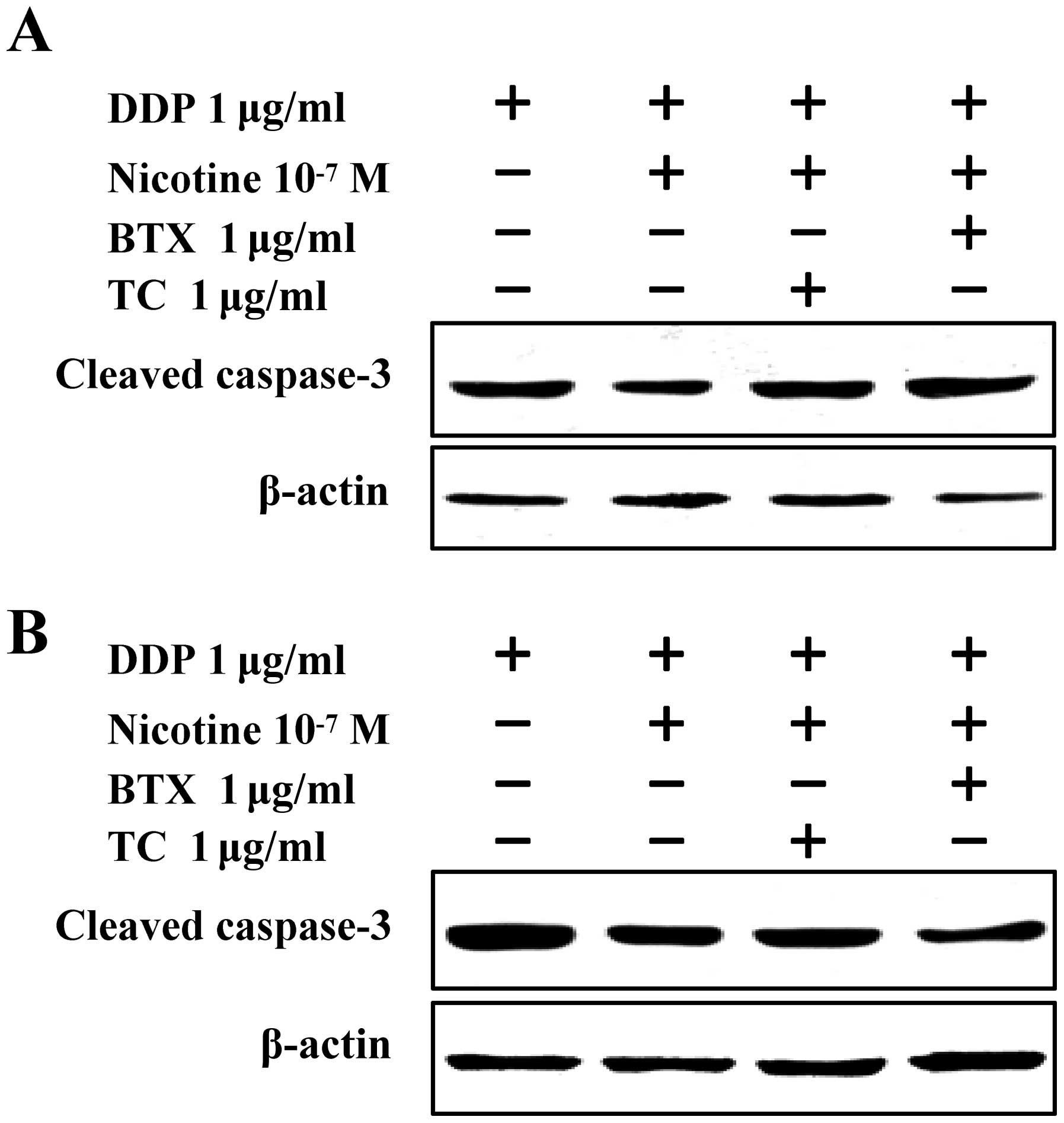
α7 nAChR is involved in
nicotine-inhibited cisplatin-induced caspase-3 activation in both
Raw264.7 and El4 cells
To investigate the role of α7 nAChR in
nicotine-mediated anti-apoptotic effects, Raw264.7 and El4 cells
were treated with α-bungarotoxin or tubocurarine chloride prior to
nicotine stimulation, the cleaved caspase-3 was determined by
western blot analysis. The results showed that compared with
cisplatin-treated cells, nicotine treatment significantly inhibited
cisplatin-induced cleaved caspase-3 in both Raw264.7 (Fig. 4A) and El4 (Fig. 4B) cells. Notably, the effect of
nicotine on cisplatin-induced caspase-3 activation was efficiently
abolished by the pretreatment of α-bungarotoxin or tubocurarine
chloride (Fig. 4), indicating that
the upregulation of α7 nAChR induced by nicotine contributed to
nicotine-mediated anti-apoptotic effects.
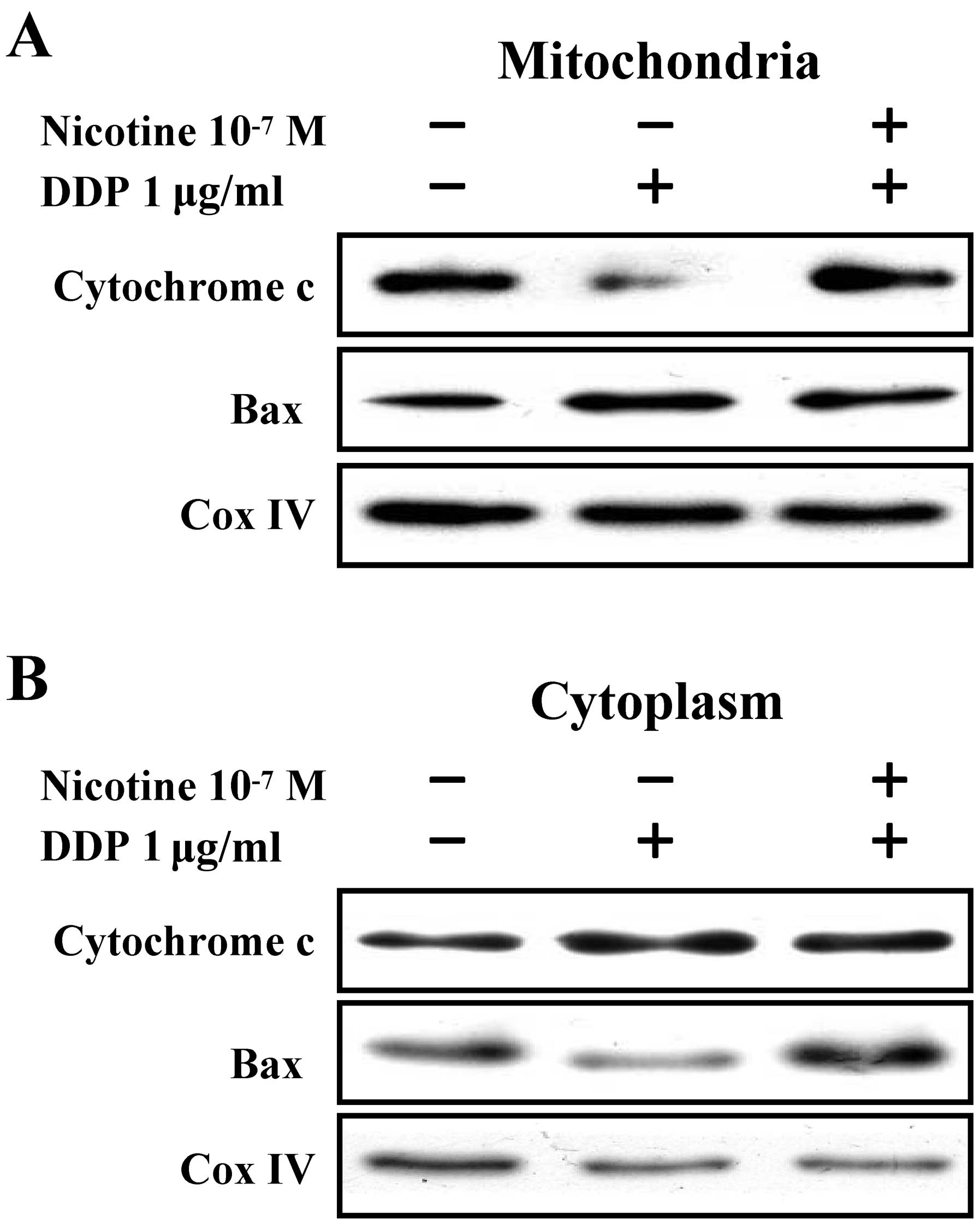
The mitochondrial translocation of Bax is
involved in nicotine-inhibited cisplatin-induced cytochrome c
release in Raw264.7 cells
To investigate the role of Bax translocation in
nicotine-mediated anti-apoptotic effects, Raw264.7 cells were
stimulated with nicotine prior to cisplatin treatment and
mitochondria/cytosol fraction was extracted. The mitochondrial
translocation of Bax and the release of cytochrome c were
determined by western blot analysis. The results showed that the
decreased cytochrome c level and increased Bax existence in
mitochondria was derived from cisplatin treatment (Fig. 5A). On the other hand, cisplatin
stimulation was found to augment cytochrome c level and
decrease Bax expression in cytoplasm (Fig. 5B), indicating that mitochondrial
translocation of Bax was involved in cisplatin-induced cytochrome
c release in Raw264.7 cells. The pretreatment of nicotine
abolished cisplatin’s effects on Bax translocation and cytochrome
c release (Fig. 5).
Therefore, nicotine achieved anti-apoptotic effects by attenuating
Bax translocation from cytoplasm to mitochondria.
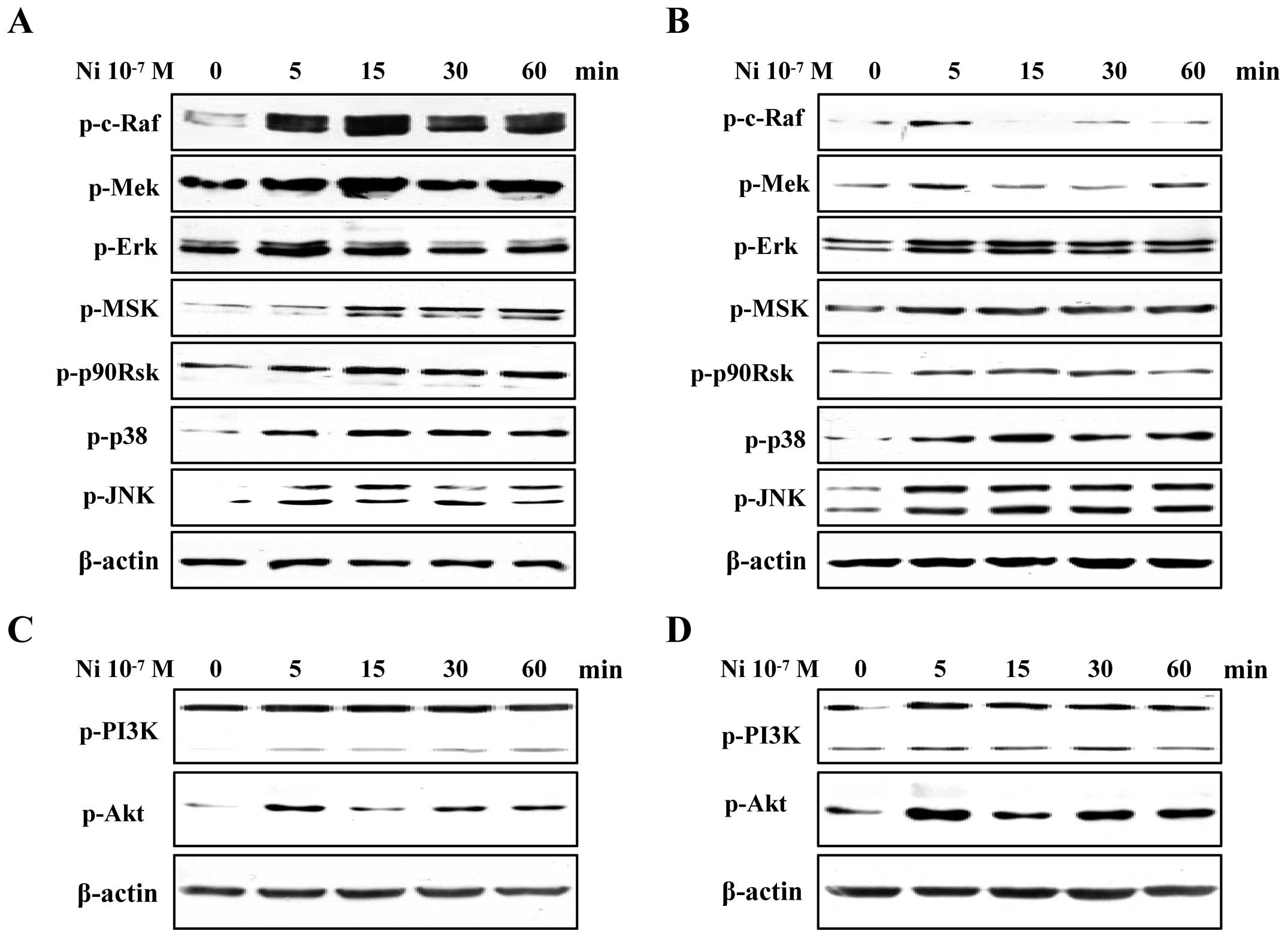
Nicotine activates MAPK and PI3K-Akt
pathways in both Raw264.7 and El4 cells
Erk1/2-p38-JNK and PI3K-Akt pathways are reported to
be involved in regulating anti-apoptosis protein expression
(25–27) and promoting lung and colon cancer
cell proliferation (27,28), respectively. To investigate the
roles of Erk1/2-p38-JNK and PI3K-Akt pathways in nicotine-mediated
anti-apoptotic effects and promoting cell proliferation, Raw264.7
and El4 cells were treated with nicotine and the effects of
nicotine on Erk1/2-p38-JNK and PI3K-Akt pathway activation was
determined by western blot analysis. The results showed that
Erk1/2-p38-JNK kinases and other components of these pathways, such
as c-Raf, Mek, MSK and p90Rsk, were clearly activated by nicotine
(10−7 M) treatment in both Raw264.7 (Fig. 6A) and El4 (Fig. 6B) cells. Meanwhile, the augmented
phosphorylation levels of PI3K p55 (Tyr199), PI3K p85 (Tyr458) and
Akt were visible within 5 min and continued to 60 min in both
Raw264.7 (Fig. 6C) and El4 cells
(Fig. 6D). These results indicate
that nicotine treatment activated the MAPK and PI3K-Akt
pathways.
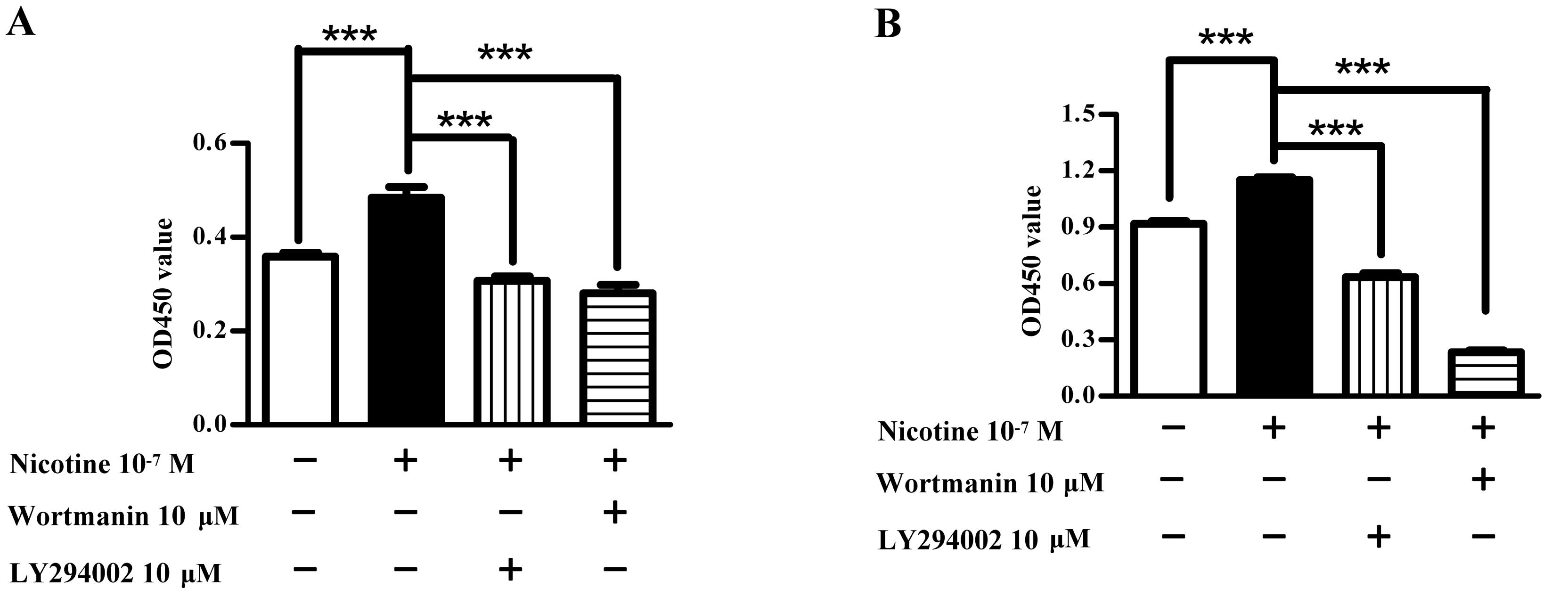
The PI3K-Akt pathway is involved in
nicotine-augmented cell proliferation
To investigate the role of the PI3K-Akt pathway in
nicotine-augmented cell proliferation, Raw264.7 and El4 cells were
pretreated with PI3K-Akt kinase inhibitor prior to nicotine
stimulation and cell viabilities were determined by CCK-8 assay.
The results showed that while nicotine obviously augmented both
Raw264.7 and El4 cell proliferation, the inhibition of PI3K kinase
activities which was derived from LY294002 pretreatment achieved
36.6 and 44.9% inhibitory rates in Raw264.7 (Fig. 7A) and El4 (Fig. 7B) cells, respectively. Meanwhile,
the usage of Wortmanin which is an inhibitor of Akt kinase also
abolished the effects of nicotine on cell proliferation, which
revealed 42.1 and 79.5% inhibitory rates in Raw264.7 (Fig. 7A) and El4 (Fig. 7B) cells, respectively
(***P<0.001, one-way ANOVA with post Newman-Keuls
test) (Fig. 7). Hence,
nicotine-induced PI3K-Akt activation is involved in the nicotine
effects on cell proliferation.
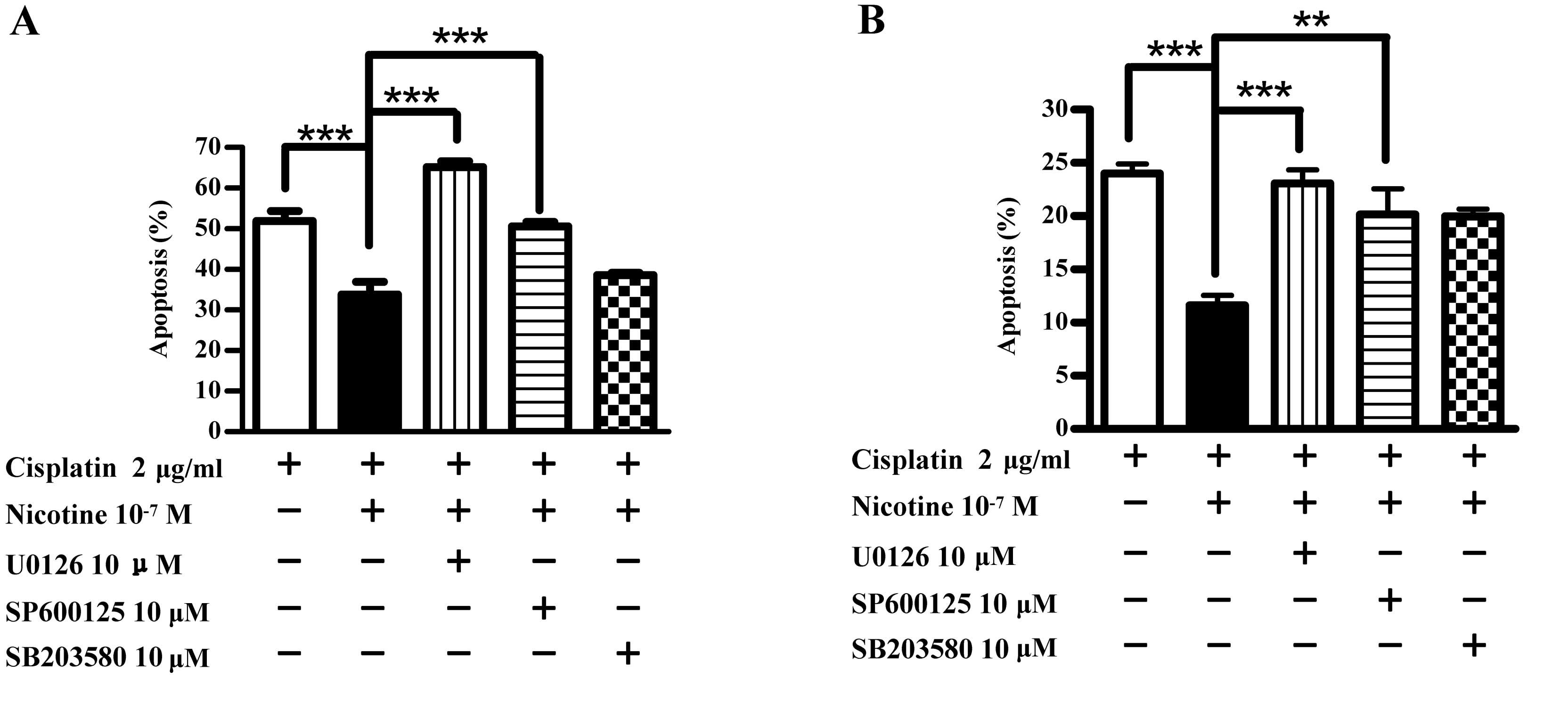
The Erk-JNK pathway is involved in
nicotine-mediated anti-apoptotic effects
To explore the role of Erk-p38-JNK MAPK pathway in
nicotine-augmented anti-apoptotic effects, Raw264.7 and EL4 cells
were pretreated with kinase inhibitors prior to nicotine
stimulation and cell apoptosis was determined by flow cytometry.
The results showed that while nicotine pretreatment decreased
cisplatin-induced apoptosis from 51.2 to 33.8% and 24.5 to 12.6% in
Raw264.7 (Fig. 8A) and El4
(Fig. 8B) cells, respectively, the
usages of Erk kinase U0126 and JNK kinase SP600125 clearly
abolished the effects of nicotine on cell anti-apoptosis in both
Raw264.7 (Fig. 8A) and El4
(Fig. 8B) cells, respectively
(***P<0.001, one-way ANOVA with post Newman-Keuls
test) (Fig. 8). The inhibition of
p38 kinase activities by the usage of SB203580 did not reverse
nicotine’s effects on cell apoptosis, indicating that p38
phosphorylation is not associated with nicotine’s anti-apoptotic
function. These results indicate that the Erk-JNK MAPK pathway is
involved in nicotine-augmented anti-apoptotic effects.
Discussion
In recent years, our studies have focused on the
effect of neurotransmitter on immune cells and we have found that
nicotine, an acetylcholine agonist, could activate bone
marrow-derived dendritic cells and have potential antitumor effects
(7–9). Further exploration showed that the
biological effect of nicotine on lymphocyte is dependent on
nicotine dose, duration of exposure and lipopolysaccharide existing
in experiment system (23).
However, other studies indicated that inhibition of α7 nACh
mediated signaling could reduce tumorigenicity (18) and facilitate lung cancer treatment
(19). Hence, the exact roles of α7
nACh in acetylcholine-mediated biological characterizations remain
controversial and require further investigation. In the present
study, Erk-JNK and PI3K-Akt signaling pathways were found to be
respectively responsible for α7 nACh-mediated cell proliferation
and anti-apoptotic effects which were supported by the following
data; firstly, nicotine-augmented cell viabilities, upregulated α7
nACh and PCNA expression were obviously abrogated by the
pretreatment of α7 nAChR antagonist α-bungarotoxin and tubocurarine
chloride; secondly, nicotine attenuated cisplatin-induced caspase-3
activation and cytochrome c release from mitochondria were
reversed by the treatment of α-bungarotoxin or tubocurarine
chloride. Further studies showed that nicotine could efficiently
activate the Erk-JNK-p38 and PI3K-Akt signaling pathway. The
inhibition of Erk-JNK MAPK activities significantly decreased
nicotine-mediated anti-apoptotic effects. Of note, the
downregulation of nicotine-augmented proliferation abilities was
achieved by the usage of PI3K-Akt kinase inhibitor.
AChRs, which is an integral membrane protein that
responds to the binding of acetylcholine, can be classified as
nAChR and mAChR according to their relative affinities and
sensitivities to nicotine or muscarine. As nAChRs are widely
expressed by nervous system (10)
and non-neuronal cells (11), α1,
α3, α5 nAChRs has been found to regulate cell proliferation and
apoptosis (12–14). In the present study, the
pretreatment of α7 nAChR specific antagonist α-bungarotoxin or
non-specific antagonist tubocurarine chloride not only abrogated
nicotine-augmented α7 nAChR upregulation, cell proliferation
(Fig. 2) but also reversed
nicotine-mediated anti-apoptotic effects (Fig. 4), indicating that α7 nAChR might be
mainly acetylcholine receptor in Raw264.7 and El4 cells. However,
it should be noted that α7 nAChR, which was found to be involved in
tumor migration (15,16) and PPARβ/δ expression (17), has also been documented to
facilitate dendritic cell-mediated antitumor immune response
(7–9). Hence, the exact roles of α7 nAChR and
other AChRs in nicotine-mediated proliferation and anti-apoptotic
effects require further investigation.
Bcl-2 and Mcl-1, which belong to the Bcl-2 family,
are located in the outer mitochondrial membrane and protect cells
against a variety of apoptotic stimuli (29). In the present study, both Bcl-2 and
Mcl-1 expression was upregulated by nicotine treatment in both
Raw264.7 and El4 cells (Fig. 3).
Hence, nicotine clearly inhibited cisplatin-induced mitochondrial
dependent caspase-3 activation (Fig.
4) and cytochrome c release (Fig. 5). The decreased Bax level in
mitochondria and increased Bax level in cytoplasm induced by
nicotine stimulation (Fig. 5)
indicated that Bax translocation from cytoplasm to mitochondria
might be a key step involved in nicotine-mediated anti-apoptotic
effects. Upon apoptotic signalling, Bak/Bax translocated to the
mitochondrial outer membrane, inserted its transmembrane domain,
oligomerized and released cytochrome c (30). Cytochrome c then binds to and
penetrates lipid structures which contain the inner mitochondrial
membrane lipid cardiolipin, leading to protein conformational
changes and increased peroxidase activity (31). Hence, the exact mechanisms of Bax in
nicotine decreased cytochrome c release remain to be further
examined.
Erk-JNK, one of the vital signal transduction
pathways transmitting and converting stress signaling into
apoptosis signaling (32), has been
reported to upregulate Fas ligand expression (33) and is necessary for mitochondrial
mediated apoptosis (34).
Meanwhile, JNK activation playing an anti-apoptotic function was
also documented (35). In the
present study, the inhibition of Erk-JNK kinase abrogated
nicotine-mediated anti-apoptotic effects in both Raw264.7 and El4
cells (Fig. 8), indicating that
Erk-JNK phosphorylation facilitated cell survival in Raw264.7 and
El4 cells. Although p38 could be efficiently activated (Fig. 6), the preincubation of SB203580
could not reverse nicotine-mediated anti-apoptotic effects,
indicating that p38 phosphorylation is not associated with the
anti-apoptotic function of nicotine.
Collectively, our study revealed that nicotine could
achieve α7 nAChR-mediated proliferation effects by activating the
PI3K-Akt pathway. Nicotine’s anti-apoptotic effect might result
from Bcl-2 family protein upregulation and Erk-JNK MAPK kinase
phosphorylation, which provides potential molecules to deal with
nicotine-associated human diseases.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (No. 81273203), the Natural
Science Foundation of Xiamen (No. 3502Z20104002) and a grant from
the National Laboratory for Oncogenes and Related Genes of China
(90-08-02).
References
|
1
|
Printz C: Gap narrows in African American
smoking-related cancers, increases in breast and colorectal
cancers. Cancer. 117:23572011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li S, Peng Q, Chen Y, You J, Chen Z, Deng
Y, Lao X, Wu H, Qin X and Zeng Z: DNA repair gene XRCC1
polymorphisms, smoking, and bladder cancer risk: a meta-analysis.
PLoS One. 8:e734482013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Albuquerque EX, Pereira EF, Alkondon M and
Rogers SW: Mammalian nicotinic acetylcholine receptors: from
structure to function. Physiol Rev. 89:73–120. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cardinale A, Nastrucci C, Cesario A and
Russo P: Nicotine: specific role in angiogenesis, proliferation and
apoptosis. Crit Rev Toxicol. 42:68–89. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tournier JM and Birembaut P: Nicotinic
acetylcholine receptors and predisposition to lung cancer. Curr
Opin Oncol. 23:83–87. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo L, Li L, Wang W, Pan Z, Zhou Q and Wu
Z: Mitochondrial reactive oxygen species mediates nicotine-induced
hypoxia-inducible factor-1α expression in human non-small cell lung
cancer cells. Biochim Biophys Acta. 1822:852–861. 2012.PubMed/NCBI
|
|
7
|
Jin HJ, Li HT, Sui HX, Xue MQ, Wang YN,
Wang JX and Gao FG: Nicotine stimulated bone marrow-derived
dendritic cells could augment HBV specific CTL priming by
activating PI3K-Akt pathway. Immunol Lett. 146:40–49. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin HJ, Sui HX, Wang YN and Gao FG:
Nicotine up-regulated 4-1BBL expression by activating Mek-PI3K
pathway augments the efficacy of bone marrow-derived dendritic cell
vaccination. J lin Immunol. 33:246–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao FG, Wan DF and Gu JR: Ex vivo
nicotine stimulation augments the efficacy of therapeutic bone
marrow-derived dendritic cell vaccination. Clin Cancer Res.
13:3706–3712. 2007. View Article : Google Scholar
|
|
10
|
Changeux JP: The nicotinic acetylcholine
receptor: the founding father of the pentameric ligand-gated ion
channel superfamily. J Biol Chem. 287:40207–40215. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Matteoli G and Boeckxstaens GE: The vagal
innervation of the gut and immune homeostasis. Gut. 62:1214–1222.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Al-Wadei MH, Al-Wadei HA and Schuller HM:
Pancreatic cancer cells and normal pancreatic duct epithelial cells
express an autocrine catecholamine loop that is activated by
nicotinic acetylcholine receptors α3, α5, and α7. Mol Cancer Res.
10:239–249. 2012.PubMed/NCBI
|
|
13
|
Al-Wadei MH, Al-Wadei HA and Schuller HM:
Effects of chronic nicotine on the autocrine regulation of
pancreatic cancer cells and pancreatic duct epithelial cells by
stimulatory and inhibitory neurotransmitters. Carcinogenesis.
33:1745–1753. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang S, Takayama K, Tanaka K, Takeshita M,
Nakagaki N, Ijichi K, Li H and Nakanishi Y: Nicotine induces
resistance to epidermal growth factor receptor tyrosine kinase
inhibitor by α1 nicotinic acetylcholine receptor-mediated
activation in PC9 cells. J Thorac Oncol. 8:719–725. 2013.
|
|
15
|
Lien YC, Wang W, Kuo LJ, Liu JJ, Wei PL,
Ho YS, Ting WC, Wu CH and Chang YJ: Nicotine promotes cell
migration through alpha7 nicotinic acetylcholine receptor in
gastric cancer cells. Ann Surg Oncol. 18:2671–2679. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wei PL, Kuo LJ, Huang MT, Ting WC, Ho YS,
Wang W, An J and Chang YJ: Nicotine enhances colon cancer cell
migration by induction of fibronectin. Ann Surg Oncol.
18:1782–1790. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun X, Ritzenthaler JD, Zhong X, Zheng Y,
Roman J and Han S: Nicotine stimulates PPARβ/δ expression in human
lung carcinoma cells through activation of PI3K/mTOR and
suppression of AP-2α. Cancer Res. 69:6445–6453. 2009.
|
|
18
|
Paleari L, Sessa F, Catassi A, Servent D,
Mourier G, Doria-Miglietta G, Ognio E, Cilli M, Dominioni L,
Paolucci M, Calcaterra A, Cesario A, Margaritora S, Granone P and
Russo P: Inhibition of non-neuronal α7-nicotinic receptor reduces
tumorigenicity in A549 NSCLC xenografts. Int J Cancer. 125:199–211.
2009.
|
|
19
|
Paleari L, Negri E, Catassi A, Cilli M,
Servent D, D’Angelillo R, Cesario A, Russo P and Fini M: Inhibition
of nonneuronal α7-nicotinic receptor for lung cancer treatment. Am
J Respir Crit Care Med. 179:1141–1150. 2009.
|
|
20
|
Shi D, Guo W, Chen W, Fu L, Wang J, Tian
Y, Xiao X, Kang T, Huang W and Deng W: Nicotine promotes
proliferation of human nasopharyngeal carcinoma cells by regulating
α7AChR, ERK, HIF-1α and VEGF/PEDF signaling. PLoS One.
7:e438982012.PubMed/NCBI
|
|
21
|
Cesario A, Russo P, Nastrucci C and
Granone P: Is α7-nAChR a possible target for lung cancer and
malignant pleural mesothelioma treatment? Curr Drug Targets.
13:688–694. 2012.
|
|
22
|
Brown KC, Lau JK, Dom AM, Witte TR, Luo H,
Crabtree CM, Shah YH, Shiflett BS, Marcelo AJ, Proper NA, Hardman
WE, Egleton RD, Chen YC, Mangiarua EI and Dasgupta P: MG624, an
α7-nAChR antagonist, inhibits angiogenesis via the Egr-1/FGF2
pathway. Angiogenesis. 15:99–114. 2012.
|
|
23
|
Hu SX, Sui HX, Jin HJ, Ni XY, Liu XX, Xue
MQ, Zhang Y and Gao FG: Lipopolysaccharide and dose of nicotine
determine the effects of nicotine on murine bone marrow-derived
dendritic cells. Mol Med Rep. 5:1005–1010. 2012.PubMed/NCBI
|
|
24
|
Zhang SH and Huang Q: Etoposide induces
apoptosis via the mitochondrial- and caspase-dependent pathways and
in non-cancer stem cells in Panc-1 pancreatic cancer cells. Oncol
Rep. 30:2765–2770. 2013.PubMed/NCBI
|
|
25
|
Park HY, Kim GY, Kwon TK, Hwang HJ, Kim
ND, Yoo YH and Choi YH: Apoptosis induction of human leukemia U937
cells by 7,8-dihydroxyflavone hydrate through modulation of the
Bcl-2 family of proteins and the MAPKs signaling pathway. Mutat
Res. 751:101–108. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu X, Jiang Y, Shan PF, Shen J, Liang QH,
Cui RR, Liu Y, Liu GY, Wu SS, Lu Q, Xie H, Liu YS, Yuan LQ and Liao
EY: Vaspin attenuates the apoptosis of human osteoblasts through
ERK signaling pathway. Amino Acids. 44:961–968. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chi J, Zhu Y, Fu Y, Liu Y, Zhang X, Han L,
Yin X and Zhao D: Cyclosporin A induces apoptosis in H9c2
cardiomyoblast cells through calcium-sensing receptor-mediated
activation of the ERK MAPK and p38 MAPK pathways. Mol Cell Biochem.
367:227–236. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cucina A, Dinicola S, Coluccia P, Proietti
S, D’Anselmi F, Pasqualato A and Bizzarri M: Nicotine stimulates
proliferation and inhibits apoptosis in colon cancer cell lines
through activation of survival pathways. J Surg Res. 178:233–241.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kale J, Liu Q, Leber B and Andrews DW:
Shedding light on apoptosis at subcellular membranes. Cell.
151:1179–1184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferrer PE, Frederick P, Gulbis JM, Dewson
G and Kluck RM: Translocation of a Bak C-terminus mutant from
cytosol to mitochondria to mediate cytochrome C release:
implications for Bak and Bax apoptotic function. PLoS One.
7:e315102012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bergstrom CL, Beales PA, Lv Y, Vanderlick
TK and Groves JT: Cytochrome c causes pore formation in
cardiolipin-containing membranes. Proc Natl Acad Sci USA.
110:6269–6274. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Chen G, Zhao J, Nie X, Wan C, Liu J,
Duan Z and Xu G: 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) induces
microglial nitric oxide production and subsequent rat primary
cortical neuron apoptosis through p38/JNK MAPK pathway. Toxicology.
312:132–141. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faris M, Kokot N, Latinis K, Kasibhatla S,
Green DR, Koretzky GA and Nel A: The c-Jun N-terminal kinase
cascade plays a role in stress-induced apoptosis in Jurkat cells by
up-regulating Fas ligand expression. J Immunol. 160:134–144.
1998.PubMed/NCBI
|
|
35
|
Minamino T, Christou H, Hsieh CM, Liu Y,
Dhawan V, Abraham NG, Perrella MA, Mitsialis SA and Kourembanas S:
Targeted expression of heme oxygenase-1 prevents the pulmonary
inflammatory and vascular responses to hypoxia. Proc Natl Acad Sci
USA. 98:8798–8803. 2001. View Article : Google Scholar : PubMed/NCBI
|