Introduction
Esophageal carcinoma is one of the most common
malignant tumors in the world. It has a high incidence rate in
China, especially in Henan Province, with a poor prognosis
(1–3). In spite of significant advancement in
early diagnosis, surgical intervention as well as local and
systemic adjuvant therapies, the majority of cancer deaths are
attributable to tumor invasion and metastasis that are resistant to
available therapies (4,5). Notably, it was found that malignant
tumor cells increased their cell invasion and motility in a manner
similar to the epithelial-to-mesenchymal transition (EMT) (6).
EMT is a highly conserved process required for
embryonic development, tissue remodeling and wound repair, and is
also implicated in the progression of primary tumors through
invasion and metastasis (7–10). During the process of EMT, the
epithelial cells lose their epithelial characteristics, including
their polarity and specialized cell-cell contacts, are converted to
a mesenchymal phenotype and further acquire a migratory behavior,
allowing them to move away from their epithelial cell community and
to integrate into surrounding tissue, even at remote locations
(11). Therefore, EMT is thought to
play a fundamental role in the invasion and metastasis of cancer
cells (12). A hallmark of EMT is
the downregulation of epithelial markers including E-cadherin and
β-catenin and the upregulation of markers related to the
mesenchymal phenotype or fibrosis including vimentin, collagens and
fibronectin (11,13).
Numerous signaling pathways are involved in the
process of EMT in human cancer (14). Transforming growth factor (TGF)-β,
as a common and potent EMT-inducing signal (11,15),
can initiate cancer cells to undergo the EMT process and thus
develop an aggressive and invasive phenotype. It has been shown,
for example, that TGF-β can induce EMT in various types of human
cancer (16–18). However, the complex molecular
mechanisms underlying this TGF-β-induced EMT process are not yet
fully understood. The tumor suppressor gene PTEN, a negative
regulator of oncogenic PTEN/PI3K signaling pathway, can directly
antagonize PI3K signal, thereby negatively regulating aggressive
tumor behavior (19,20). Recent reports demonstrated that the
PTEN/PI3K signaling pathway contributes to EMT in cancer (21,22).
Since TGF-β and the PTEN/PI3K signaling pathway fulfill overlapping
roles in tumor progression, the study of the possible role of
PTEN/PI3K in TGF-β-induced EMT is warranted to shed light for
future research. To date, no data have been reported concerning the
influence of TGF-β on the PTEN/PI3K signaling pathway in the ESCC
cells and the role of this interaction on crucial characteristics
of tumor cells, such as invasion and migration.
In the present study, we provided insight into how
TGF-β1 and PTEN/PI3K act through multiple interconnected signaling
pathways to trigger events associated with EMT in esophageal
squamous cell carcinoma (ESCC). We examined the expression profile
of PTEN/PI3K signal in patients with ESCC and compared PTEN/PI3K
expression with E-cadherin/vimentin expression and
clinicopathological parameters (such as invasion depth and lymph
node metastasis), which are all associated with EMT and tumor
progression. Furthermore, we investigated the effect of TGF-β1 and
PTEN/PI3K on phenotypic and functional characteristics consistent
with EMT in ESCC cells and whether a crosstalk between TGF-β1 and
PTEN/PI3K signaling pathway is required to mediate this effect.
Materials and methods
Tissue samples
The present study included formalin-fixed and
paraffin-embedded (FFPE) tissue samples of 95 patients with
histopathologically confirmed ESCC from The First Affiliated
Hospital of Zhengzhou University, Henan, China. None of the
patients had received neoadjuvant radio-/chemotherapy. Clinical
samples used in this study were approved by the Committee for
Ethical Review of Research in our hospital. Clinical information on
the samples is summarized in Table
I.
 | Table IExpression of E-cadherin, vimentin,
PTEN and PI3K proteins in ESCC tissues and their correlation with
clinicopathological parameters. |
Table I
Expression of E-cadherin, vimentin,
PTEN and PI3K proteins in ESCC tissues and their correlation with
clinicopathological parameters.
| | E-cadherin | Vimentin | PTEN | PI3K |
|---|
| |
|
|
|
|
|---|
| n | − | + | P-value | − | + | P-value | − | + | P-value | − | + | P-value |
|---|
| Normal mucosa | 95 | 22 | 73 | 0.000 | 67 | 28 | 0.000 | 10 | 85 | 0.000 | 50 | 45 | 0.001 |
| ESCC | 95 | 57 | 38 | | 39 | 56 | | 48 | 47 | | 28 | 67 | |
| Gender |
| Male | 55 | 31 | 24 | 0.396 | 23 | 32 | 0.859 | 30 | 25 | 0.358 | 16 | 39 | 0.924 |
| Female | 40 | 26 | 14 | | 16 | 24 | | 18 | 22 | | 12 | 28 | |
| Age (years) |
| <60 | 42 | 28 | 14 | 0.238 | 20 | 22 | 0.247 | 25 | 17 | 0.118 | 13 | 29 | 0.778 |
| ≥60 | 53 | 29 | 24 | | 19 | 34 | | 23 | 30 | | 15 | 38 | |
| Histological
grade |
| I | 21 | 7 | 14 | 0.017 | 8 | 13 | 0.717 | 4 | 17 | 0.000 | 9 | 12 | 0.117 |
| II | 44 | 29 | 15 | | 20 | 24 | | 19 | 25 | | 14 | 30 | |
| III | 30 | 21 | 9 | | 11 | 19 | | 25 | 5 | | 5 | 25 | |
| Invasion depth |
| Superficial
muscularis | 24 | 6 | 18 | 0.000 | 18 | 6 | 0.000 | 6 | 18 | 0.004 | 15 | 9 | 0.000 |
| Deep
muscularis | 71 | 51 | 20 | | 21 | 50 | | 42 | 29 | | 13 | 58 | |
| Lymph node
metastasis |
| Yes | 30 | 23 | 7 | 0.024 | 10 | 20 | 0.299 | 27 | 3 | 0.000 | 4 | 26 | 0.019 |
| No | 65 | 34 | 31 | | 29 | 36 | | 21 | 44 | | 24 | 41 | |
Immunohistochemistry (IHC)
Sections of the 95 FFPE tissue specimens were
processed for PTEN (Zhongshan, Beijing, China; 1:100), PI3K
(Zhongshan; 1:100), E-cadherin (Zhongshan; 1:150) and vimentin
(Zhongshan; 1:100) staining according to previously established
protocols (23). Briefly, 5 μm
serial sections were deparaffinized, subsequently rehydrated in
gradients of ethanol, subjected to antigen retrieval and incubated
with primary antibodies overnight. Signal visualization was
developed by the avidin-biotin peroxidase method using DAB,
following immunohistochemical staining analysis performed by two
independent pathologists.
Evaluation of IHC staining
We assessed immunohistochemical staining only in
normal epithelial and cancer cells. PTEN, PI3K and vimentin
positive signals all showed brown-yellow granules in the cytoplasm,
whereas E-cadherin showed it in the membrane or cytoplasm. Under a
microscope at ×400 magnification, five fields of vision (FOVs) were
randomly selected (for each FOV, there were no fewer than 200
cells) and the results were interpreted in accordance with the
percentage of cells and depth of stain. (i) Scored in accordance
with the percentage of positive cells in like-kind cells: 1,
<30%; 2, 30–70%; 3, >70%. (ii) Scored in accordance with
depth of stain: 0, no cell coloration; 1, light yellow; 2, brown;
3, tan. The product of (i) and (ii) was used as the total
multiplied score, where 0–1 indicates a negative score (−) and ≥2 a
positive score (+) (23).
Cell line
The human ESCC cell line EC-1, a gift from Professor
Shihua Cao (the University of Hong Kong), was cultured in RPMI-1640
medium (Gibco, USA) supplemented with 10% fetal bovine serum (FBS),
100 μg/ml streptomycin and 100 U/ml penicillin at 37°C in a
humidified incubator containing 5% CO2. The experiment
cells were in logarithmic phase.
pcDNA3.1-PTEN plasmid construction and
stable transfection
Human PTEN cDNA was subcloned into pcDNA3.1
(Invitrogen, USA) vector with primers as follows: forward,
5′-AAGCTTATGACAGCCATCATCAAAGAGAT-3′
(underlined, HindIII) and reverse, 5′-GGATCCGGAATAAAACGG GAAAGTGCC-3′
(underlined, BamHI). pcDNA3.1-PTEN and control empty
pcDNA3.1 vector were transfected into EC-1 cells using
Lipofectamine™ 2000 (Invitrogen) according to the manufacturer’s
protocol, respectively. G418 was used to select stable expression
clones.
Cell treatment
To evaluate the effect of TGF-β1 on EMT, EC-1 cells
were treated with 5 ng/ml of TGF-β1 (PeproTech, Rocky Hill, NJ,
USA) for different time intervals (0, 24 and 48 h). To analyze the
crosstalk of TGF-β1 and PTEN/PI3K pathway during the process of
EMT, EC-1 cells were stably transfected with pcDNA3.1-PTEN and
subsequently treated with 5 ng/ml of TGF-β1 for 48 h. After
different treatment, cells were observed and photographed for the
morphological alterations under a reversed light microscope and
analyzed for mRNA and protein levels of E-cadherin, vimentin, PTEN
and PI3K, cell motility and invasiveness as described below.
RNA preparation and quantitative
real-time PCR
Total cellular RNA was extracted from EC-1 cells
with different treatment by the TRIzol reagent (Invitrogen)
according to the manufacturer’s protocols. Total RNA (2 μg) was
used to generate the first strand of DNA using the TIANScript First
Strand cDNA Synthesis kit (Tiangen, China). Quantitative real-time
PCR (qPCR) was carried out with primers specific for PTEN (forward,
5′-ATACCAGGACCAGAGGAAACC-3′ and reverse,
5′-TTGTCATTATCCGCACGCTC-3′); PI3K (forward,
5′-TGTAGTGGTGGACGGCGAAGTA-3′ and reverse,
5′-GGGAGGTGTGTTGGTAATGTAGCA-3′); E-cadherin (forward,
5′-TGATTCTGCTGCTCTTGCTG and reverse, 5′-CAAAGTCCTGGTCCTCTTCTCC-3′);
vimentin (forward, 5′-AATGACCGCTTCGCCAACTA-3′ and reverse,
5′-GCTCCTGGATTTCCTCTTCG-3′) and β-actin (forward,
5′-CGGGAAATCGTGCGTGAC-3′ and reverse 5′-TGGAA GGTGGACAGCGAGG-3′).
All reactions were performed in triplicate on a 7300 real-time PCR
detection system (Applied Biosystems, Foster City, CA, USA), using
SYBR Green (Applied Biosystems) fluorescent dye and normalized to
β-actin mRNA levels.
Western blot analysis
EC-1 cells with different treatment were harvested
and lysed using the entire protein extraction reagent (KeyGen,
Nanjing, China) according to the manufacturer’s protocols. Thirty
micrograms of protein for each sample were electrophoresed through
a 10% SDS-PAGE gel and then electro-transferred to nitrocellulose
membranes (Yili, China) by a semi-dry transfer apparatus. The
membranes were incubated at 4°C overnight with the following
primary antibodies: PTEN (Zhongshan; 1:500), PI3K (Zhongshan;
1:500), E-cadherin (Zhongshan; 1:800), vimentin (Zhongshan; 1:1000)
and β-actin (Zhongshan; 1: 500). β-actin was measured to control
for equal loading. The experiments were performed in
triplicate.
Cell invasion assay
Tumor cell invasive ability was analyzed using an
invasion chamber (Oilin, China) with Matrigel-coated polycarbonate
membrane (8 μm pore size) in 24-well plates. EC-1 cells with
different treatment were seeded into the upper compartment of the
invasion chamber containing serum-free medium and RPMI-1640 with
10% FBS was added in the lower chamber as a chemoattractant. After
incubation at 37°C in 5% CO2 for 24 h, cells that
invaded through the Matrigel and adhered to the lower surface were
fixed in 95% alcohol, stained with HE and counted under a reversed
light microscope. Data represent the mean (±SD) of the number of
cells counted in five random FOVs at ×400 magnification in each of
3 independent experiments.
Scratch assay
Tumor cell migratory ability was measured by the
scratch assay. EC-1 cells transfected with or without pcDNA3.1-PTEN
were planted in 6-well plates and grown to 100% confluency,
respectively. A wound was then introduced to each well by
scratching the monolayer cells with a sterile 200 μl pipette tip.
The cultures were washed with PBS to remove detached cells.
Subsequently, cells were allowed to grow with or without treatment
of 5 ng/ml of TGF-β1 for an additional 48 h. At different time
intervals (0, 24 and 48 h), cell migration was photographed at a
×200 magnification under a reversed light microscope, quantitated
by measuring the width of the wounds at least five representative
fields and expressed as 1 minus the average percent of wound
closure by comparing with that at the zero time. Experiments were
performed with at least 6 replicates for each condition.
Statistical analysis
Data analyses were carried out with the Chi-square
test or t-test using SPSS version 13.0. In all statistical
analyses, P-value <0.05 was considered to indicate a
statistically significant difference and all P-values were
two-sided.
Results
A hallmark of EMT in ESCC tissues:
downregulation of epithelial marker E-cadherin and upregulation of
mesenchymal marker vimentin
To confirm the existence of EMT in ESCC, IHC was
used to detect protein expression levels of epithelial marker
E-cadherin and mesenchymal marker vimentin in ESCC tissues and
corresponding normal mucosa. Cytoplasmic or membranal E-cadherin
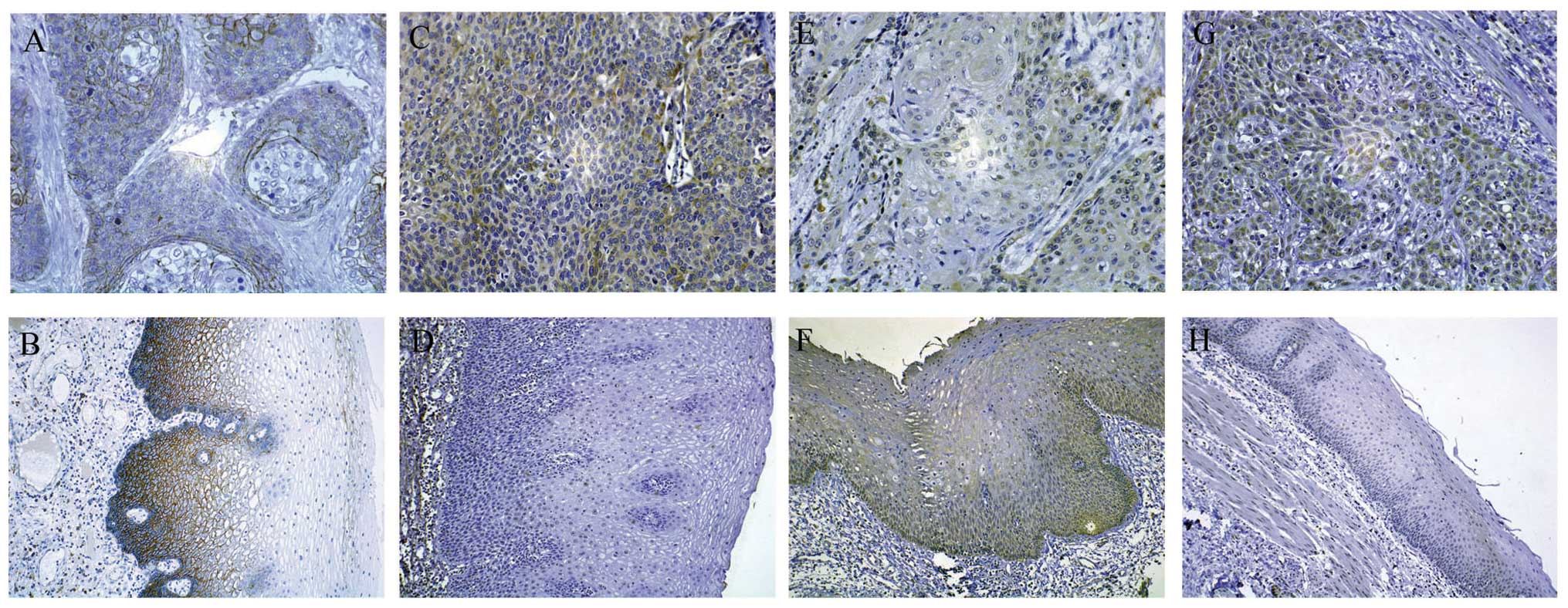
immunoreactivity (Fig. 1A and B)
was examined in 40.00% of ESCC tissues, significantly lower than
that in corresponding normal mucosa tissues (76.84%; P<0.01)
(Table I). However, cytoplasmic
vimentin immunoreactivity (Fig. 1C and
D) was examined in 58.95% of ESCC tissues, significantly higher
than that in corresponding normal mucosa tissues (29.47%;
P<0.01) (Table I). When we
assessed the correlation of E-cadherin with vimentin protein in
ESCC, a significantly negative correlation was found (P<0.01)
(Table II). In addition, in ESCC
tissues, E-cadherin expression was negatively correlated with
histological grade (P<0.05), invasion depth (P<0.01) and
lymph node metastasis (P<0.05) (Table I), whereas vimentin protein
expression was positively correlated with invasion depth
(P<0.01) (Table I). These data
demonstrated most ESCC tissues undergo EMT and acquire enhanced
ability of invasiveness and metastasis.
| Table IICorrelation of E-cadherin with
vimentin protein expression in ESCC tissues. |
Table II
Correlation of E-cadherin with
vimentin protein expression in ESCC tissues.
| Vimentin |
|---|
|
|
|---|
| E-cadherin | + | − | r | P-value |
|---|
| + | 10 | 28 | −0.542 | 0.000 |
| − | 46 | 11 | | |
The PTEN/PI3K signaling pathway is active
and correlates with EMT and tumor progression in ESCC tissues
To investigate the role of the PTEN/PI3K pathway in
ESCC, we examined the protein levels of PTEN and PI3K in ESCC
tissues in comparison with the corresponding normal esophageal
mucosa tissues by IHC. Cytoplasmic PTEN immunoreactivity (Fig. 1E and F) was examined in 49.47% of
ESCC tissues, significantly lower than that in corresponding normal
mucosa tissues (89.47%; P<0.01) (Table I). Moreover, cytoplasmic PI3K
immunoreactivity (Fig. 1G and H)
was examined in 70.53% of ESCC tissues, significantly higher than
that in corresponding normal mucosa tissues (47.37%; P<0.01)
(Table I). Furthermore, a
significantly negative correlation was found between the expression
of PTEN and PI3K protein in ESCC (P<0.01) (Table III). These data showed the
PTEN/PI3K pathway is active in most ESCC tissues.
| Table IIICorrelation of PTEN with PI3K protein
expression in ESCC tissues. |
Table III
Correlation of PTEN with PI3K protein
expression in ESCC tissues.
| PI3K |
|---|
|
|
|---|
| PTEN | + | - | r | P-value |
|---|
| + | 20 | 27 | −0.607 | 0.000 |
| − | 47 | 1 | | |
We also examined the correlation of PTEN and PI3K
protein expression with E-cadherin protein, vimentin protein and
clinicopathological features in ESCC. As would be expected from the
role of PTEN/PI3K signaling pathway in regulating EMT in ESCC, PTEN
expression in ESCC had a positive association with E-cadherin and a
negative association with vimentin (P<0.01) (Table IV), whereas PI3K expression
presented contrary results (P<0.05) (Table V). Furthermore, as shown in Table I, a negative correlation was found
between PTEN protein expression and histological grade (P<0.01),
invasion depth (P<0.01) and lymph node metastasis (P<0.01).
On the contrary, a positive correlation was found between PI3K
protein expression and invasion depth (P<0.01) and lymph node
metastasis (P<0.05). These data demonstrated that the PTEN/PI3K
pathway plays a pivotal role in the process of EMT and tumor
progression in ESCC. Additionally, our previous study also showed
that TGF-β1 correlates with EMT and tumor progression in ESCC
tissues, suggesting TGF-β1 and PTEN/PI3K signaling pathway fulfill
overlapping roles and there may be a crosstalk between them.
| Table IVCorrelation of PTEN with E-cadherin
and vimentin protein expression in ESCC tissues. |
Table IV
Correlation of PTEN with E-cadherin
and vimentin protein expression in ESCC tissues.
| E-cadherin | Vimentin |
|---|
|
|
|
|---|
| PTEN | + | − | r | P-value | + | − | r | P-value |
|---|
| + | 30 | 17 | 0.481 | 0.000 | 14 | 33 | −0.587 | 0.000 |
| − | 8 | 40 | | | 42 | 6 | | |
| Table VCorrelation of PI3K with E-cadherin
and vimentin protein expression in ESCC tissues. |
Table V
Correlation of PI3K with E-cadherin
and vimentin protein expression in ESCC tissues.
| E-cadherin | Vimentin |
|---|
|
|
|
|---|
| PI3K | + | − | r | P-value | + | − | r | P-value |
|---|
| + | 19 | 48 | −0.368 | 0.000 | 45 | 22 | 0.258 | 0.012 |
| − | 19 | 9 | | | 11 | 17 | | |
Treatment of EC-1 cells with TGF-β1
induces morphological and biochemical alterations consistent with
EMT and enhanced aberrant cell invasiveness and migration
TGF-β1 is a known inducer of EMT for a variety of
cancer cells (16–18). In the present study, EC-1 cells
belonging to human ESCC cell line were tested for their response to
TGF-β1. After treatment with 5 ng/ml of TGF-β1, cell morphology was
observed under a reversed light microscope at different time
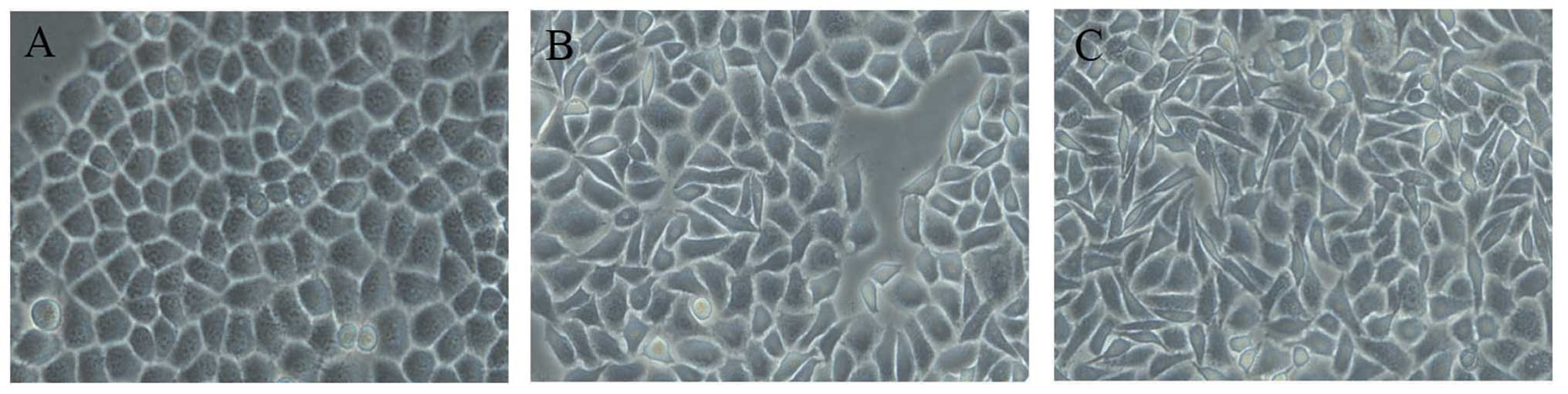
intervals (0, 24 and 48 h). Compared to the untreated EC-1 cells
(Fig. 2A), cells treated with
TGF-β1 were scattered, elongated and spindle-shaped, i.e.,
characteristic of fibroblasts (Fig. 2B
and C). Notably, this morphological alteration was
time-dependent and occurred at 24 h (Fig. 2B), but became apparent at 48 h
(Fig. 2C).
In line with the morphological alteration, EMT was
characterized by suppression of epithelial marker E-cadherin and
elevation of mesenchymal marker vimentin. Quantitative real-time
PCR and western blot analysis showed that TGF-β1 treatment induced
a significant decrease in E-cadherin, but a significant increase in
vimentin at both mRNA and protein levels in EC-1 cells as compared
with those in untreated cells (P<0.01) (Fig. 3). Moreover, as shown in Fig. 3, treatment with TGF-β1 displayed a
significantly time-dependent change of E-cadherin and vimentin at
mRNA and protein levels (P<0.05).
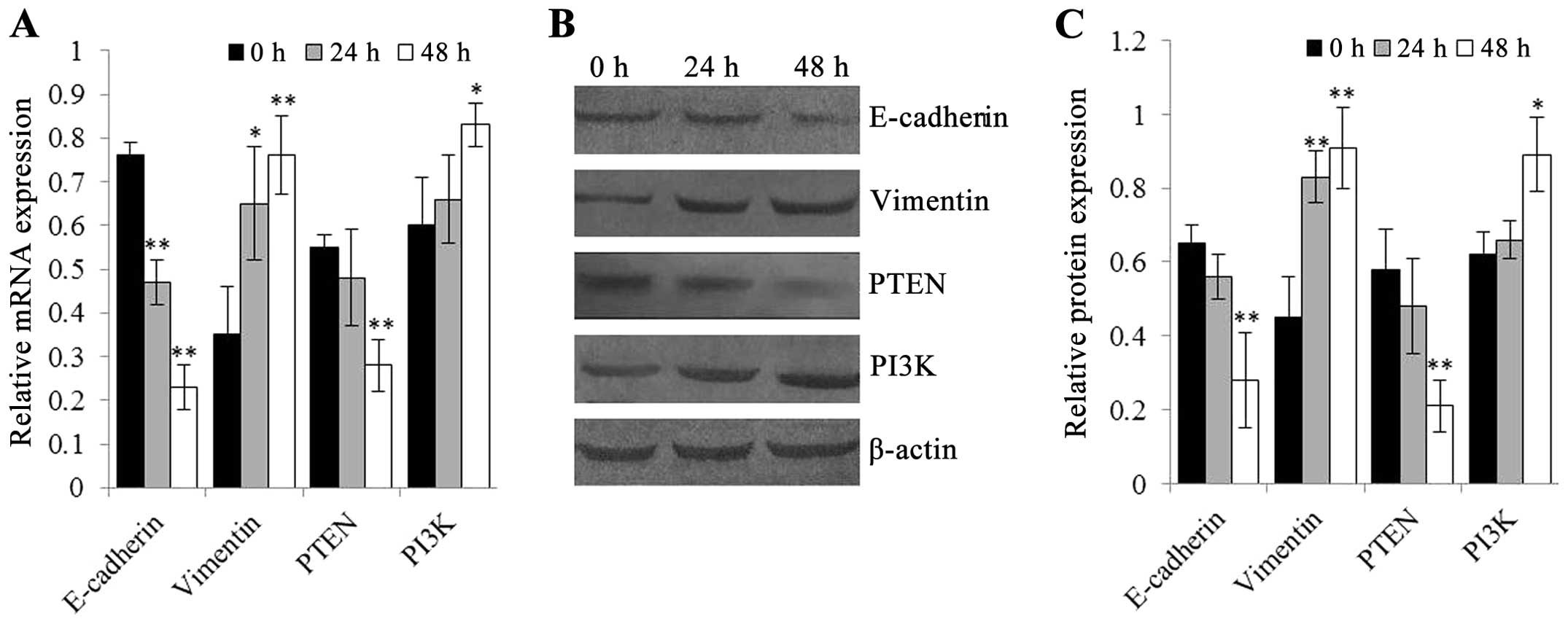 | Figure 3Quantitative real-time PCR and
western blot analysis were used to detect mRNA and protein levels
of E-cadherin, vimentin, PTEN and PI3K in EC-1 cells after
treatment with 5 ng/ml of TGF-β1 for different time intervals (0,
24 and 48 h), respectively. In EC-1 cells, treatment with 5 ng/ml
of TGF-β1 reduced the epithelial marker E-cadherin but increased
the mesenchymal marker vimentin mRNA and protein expression, which
are the hallmark of EMT. Furthermore, this TGF-β1-induced
alteration of E-cadherin and vimentin occurred in a time-dependent
manner. In addition, TGF-β1 treatment decreased PTEN but increased
PI3K mRNA and protein expression, leading to PTEN/PI3K signal
activation. (A) mRNA levels of E-cadherin, vimentin, PTEN and PI3K
in EC-1 cells treated with 5 ng/ml of TGF-β1 for different time
intervals (0, 24 and 48 h). (B and C) Protein levels of E-cadherin,
vimentin, PTEN and PI3K in EC-1 cells treated with 5 ng/ml of
TGF-β1 for different time intervals (0, 24 and 48 h). β-actin was
used as internal control. *P<0.05 and
**P<0.01, compared to the untreated EC-1 cells
(treatment with 5 ng/ml of TGF-β1 for 0 h). |
We next examined the effect of TGF-β1 on cell
invasiveness and migration by using invasion and scratch assay,
respectively. The invasion assay demonstrated that EC-1 cells
treated with 5 ng/ml of TGF-β1 exhibited a significant increase in
the number of cells that traversed the membrane toward a
chemoattractant in a time-dependent manner as compared with
untreated cells (P<0.01) (Fig.
4A). Additionally, the scratch assay showed that EC-1 cells
treated with 5 ng/ml of TGF-β1 resulted in a significant increase
in the tumor cell migration distance as compared with untreated
cells (P<0.01) (Fig. 4B and
C).
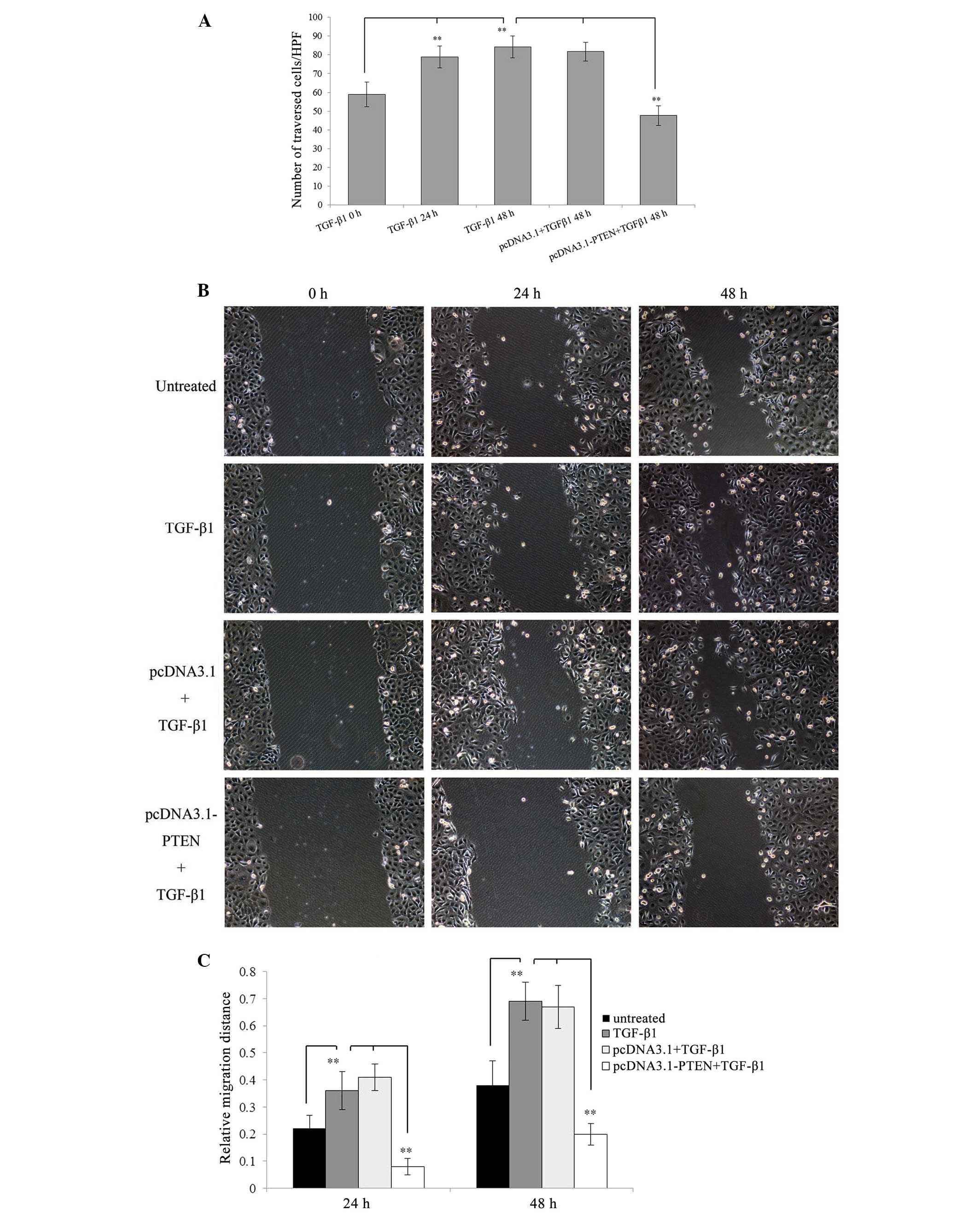 | Figure 4Invasiveness and migration of EC-1
cells are enhanced by TGF-β1 stimulation, but are partly inhibited
by pcDNA3.1-PTEN transfection. For the invasion assay, EC-1 cells
untransfected and stably transfected with pcDNA3.1 or pcDNA3.1-PTEN
were treated with 5 ng/ml of TGF-β1 for different time intervals
(0, 24 and 48 h), then seeded in the upper invasion chamber and
incubated for an additional 24 h, respectively. After incubation,
cells that invaded through the Matrigel were fixed, stained and
counted under a reversed light microscope at a ×200 magnification.
For the scratch assay, EC-1 cells untransfected and stably
transfected with pcDNA3.1 or pcDNA3.1-PTEN were planted in 6-well
plates, grown to 100% confluency and then scratched with a sterile
200 μl pipette tip, respectively. After making the scratch, cells
were treated with 5 ng/ml of TGF-β1 for different time intervals
(0, 24 and 48 h) and cell migration distances were quantitated by
measuring the width of the wounds at a ×200 magnification under a
reversed light microscope. (A) The number of traversed cells is
presented as mean ± SD. (B) Images of EC-1 cell migration. (C) The
relative migration distance is presented as 1 minus the average
percent of wound closure by comparing with that at the zero time.
**P<0.01. |
Taken together, these data suggest that TGF-β1
stimulation promotes EC-1 cells to undergo EMT and thus develop an
aggressive and invasive phenotype.
TGF-β1 stimulation activates PTEN/PI3K
signaling and TGF-β1-induced EMT is partly prevented by inhibition
of the PTEN/PI3K signaling pathway in EC-1 cells
To analyze the crosstalk between TGF-β1 and
PTEN/PI3K signaling pathway during the process of EMT in ESCC
cells, we first tested whether TGF-β1 treatment activates the
PTEN/PI3K pathway. As shown in Fig.
3, EC-1 cells treated with 5 ng/ml of TGF-β1 for 48 h had lower
PTEN but higher PI3K expression at both mRNA and protein levels as
compared with the untreated cells (P<0.05), indicating TGF-β1
treatment could activate the PTEN/PI3K signaling pathway.
Furthermore, we inhibited the PTEN/PI3K signaling
pathway by pcDNA3.1-PTEN transfection for the purpose of
demonstrating that TGF-β1-induced EMT is driven by the PTEN/PI3K
signaling pathway. As shown in Fig.
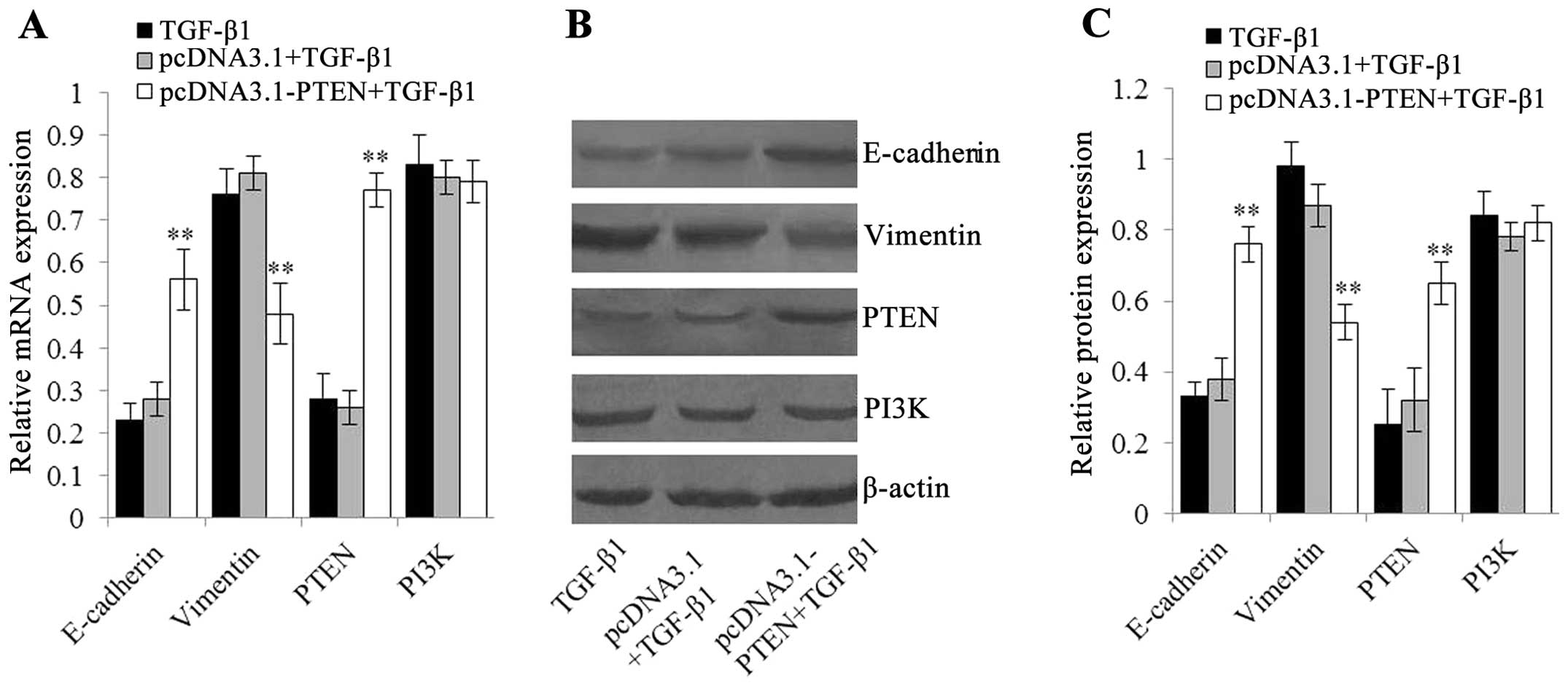
5, after treatment with 5 ng/ml of TGF-β1 for 48 h, EC-1 cells
transfected with pcDNA3.1-PTEN had higher PTEN mRNA and protein
expression as compared with cells untransfected or transfected with
pcDNA3.1 empty vector (P<0.01), demonstrating the PTEN/PI3K
pathway was inhibited by pcDNA3.1-PTEN directly. Moreover,
accompanied by PTEN/PI3K signal inhibition, mRNA and protein levels
of E-cadherin in EC-1 cells transfected with pcDNA3.1-PTEN were
clearly higher than those in cells untransfected or transfected
with pcDNA3.1 empty vector, whereas vimentin expression showed
contrary results (P<0.01) (Fig.
5). These data suggested inhibition of the PTEN/PI3K signaling
pathway by pcDNA3.1-PTEN prevents TGF-β1-induced EMT to some
extent.
The PTEN/PI3K pathway modulates
TGF-β1-induced tumor cell invasiveness and migration
To examine whether the PTEN/PI3K pathway also
modulated TGF-β1-induced tumor cell invasiveness and migration, we
measured these two parameters after inhibition of the PTEN/PI3K
signaling pathway by pcDNA3.1-PTEN in EC-1 cells. Invasion and
scratch assay showed that, after treatment with 5 ng/ml of TGF-β1
for 48 h, EC-1 cells transfected with pcDNA3.1-PTEN exhibited a
significant decrease in the number of traversed cells and the cell
migration distance as compared with cells untransfected or
transfected with pcDNA3.1 empty vector (P<0.01) (Fig. 4A–C). These data demonstrate that
inhibition of the PTEN/PI3K signaling pathway by pcDNA3.1-PTEN
counteracts TGF-β1-induced tumor cell invasiveness and
migration.
Discussion
Although numerous reports have implicated EMT in the
progression of primary tumors towards invasion and metastasis
(8–10), the detailed molecular mechanisms and
networks involved in TGF-β and the PTEN/PI3K signaling pathway in
EMT and progression of ESCC remain poorly understood.
EMT can be induced or regulated by various growth
and differentiation factors including TGF-β, growth factors that
act through receptor tyrosine kinases, such as FGF and HGF, and
Notch and Wnt proteins (14). Among
these, TGF-β has received much attention as a major inducer of EMT
during cancer progression (24). In
the literature TGF-β1 was shown to induce EMT in oral (16), liver (17) and pancreatic cancer (18). In the present study, we stimulated
EC-1 cells with 5 ng/ml of TGF-β1 for up to 48 h for the purpose of
confirming TGF-β1-induced EMT. The results showed that TGF-β1
stimulation induced EC-1 cells to undergo a transition from the
epithelial to the spindle-like mesenchymal morphology. This change
was accompanied by the loss of E-cadherin and the gain of vimentin.
In addition, the cells obtained increased ability of invasion and
migration. Similar results were obtained for another ESCC cell line
(EC9706) in a previous study (25).
Thus, TGF-β1 is also a key factor in EMT induction of ESCC cell
lines.
Another notable factor involved in EMT is the
PTEN/PI3K signaling pathway. The tumor suppressor gene PTEN is a
dual protein/lipid phosphatase whose main substrate is PIP3, a
product of PI3K (26). PTEN
directly antagonizes PI3K function via abrogation of PIP3-mediated
activation of downstream signaling events and ultimately
participates in the regulation of the cell cycle, proliferation,
apoptosis, cell adhesion and cancer progression (6,19,27).
In human tumors, PTEN activity is lost by mutation, deletion or
promoter methylation silencing at high frequency (26,28)
and PI3K is constitutively activated in the loss of PTEN function
(29,30). Therefore, the PTEN/PI3K signaling
pathway is frequently activated in a variety of human malignant
tumors (31–33). Furthermore, activated PTEN/PI3K
signaling pathway has also been identified as a central feature of
EMT in tumor cells (21,34). In the present study, PTEN/PI3K
signal was activated in ESCC and the activation of the PTEN/PI3K
signaling pathway was negatively correlated with EMT marker
E-cadherin, but was positively correlated with EMT marker vimentin
and strongly contributed to tumor differentiation, invasion depth
and lymph node in ESCC, suggesting the PTEN/PI3K signaling pathway
plays a potential role in EMT and tumor progression. Indeed, in
ESCC, in addition to the absence of functional PTEN, PI3K would be
activated and would promote a series of EMT-inducing transcription
factors, such as Snail (35).
Upregulation of Snail, together with other transcription factors,
leads to downregulation of EMT hallmark E-cadherin by inhibiting
E-cadherin transcription (36,37).
Thus, ESCC cells undergo EMT and acquire enhanced invasive and
migratory ability.
Although TGF-β1 and PTEN/PI3K signaling pathway
fulfill overlapping roles in EMT, a little known coordination
mechanism has been proposed to explain their relationship in EMT to
date. It has been reported that TGF-β induces EMT by Smad-mediated
transcription regulation (11,14,15,38,39)
and, moreover, TGF-β/BMP-SMAD4 signaling pathway promotes prostate
cancer EMT and progression by PTEN deletion (40). Furthermore, in mammary epithelial
cells, TGF-β can activate PI3K during EMT (41). Therefore, it is reasonable to
postulate TGF-β1-induced EMT is driven through the PTEN/PI3K
signaling pathway. In the present study, we first hypothesized that
the PTEN/PI3K signaling pathway is a downstream target of TGF-β1
signal and examined PTEN and PI3K expression levels in EC-1 cells
after treatment with TGF-β1. Western blot analysis and qPCR showed
the protein and mRNA levels of PTEN were decreased, but PI3K was
increased, indicating that downregulation of PTEN and upregulation
of PI3K is influenced by TGF-β1 stimulation (42). Although TGF-β1 also leads to the
downregulation of E-cadherin and upregulation of vimentin in EC-1
cells according to our previous results, it is unclear if this
observed dysregulation of PTEN/PI3K and E-cadherin/vimentin through
TGF-β1 stimulation is caused by a parallel mechanism or by a serial
mechanism. Herein, we next inhibited PTEN/PI3K signaling pathway by
pc-DNA3.1-PTEN application to confirm TGF-β1-induced EMT via
PTEN/PI3K pathway. As expected, our data showed that the
enhancement of PTEN level by pc-DNA3.1-PTEN in TGF-β1-treated EC-1
cells upregulated TGF-β1-inhibited E-cadherin expression, but
downregulated TGF-β1-promoted vimentin expression, affirming the
hypothesis that TGF-β1-induced EMT is driven by the PTEN/PI3K
signaling pathway. Additional evidence for the modulation of
TGF-β1-induced tumor cell invasiveness and migration by PTEN/PI3K
signaling pathway comes from the invasion and scratch assay in
vitro, which showed the inhibition of the PTEN/PI3K signaling
pathway by pcDNA3.1-PTEN counteracts TGF-β1-induced tumor cell
invasiveness and migration. TGF-β1 signal can regulate the cellular
process by binding and phosphorylating TGF-β receptors type I (TβR
I) and type II (TβR II) (43), the
activated TβR I and TβR II then bind to PI3K regulatory subunit
p85, resulting in PI3K activation (44). In the presence of PTEN, activated
PI3K function was antagonized and therefore TGF-β1-induced EMT and
EMT-associated mobility and invasiveness would be inhibited.
In conclusion, the present study suggests that
TGF-β1 and PTEN/PI3K signaling pathway contribute to EMT, and the
PTEN/PI3K signaling pathway is a key regulator of TGF-β1-induced
EMT in ESCC. Disruption of the PTEN/PI3K pathway involved in
TGF-β1-induced EMT may provide possible routes for therapeutic
intervention of ESCC.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant no. 81071970) and the
Medical Science and Technology Program of Henan Province (grant no.
112102310195). We thank Professor Shihua Cao (the University of
Hong Kong) for providing the EC-1 cell line.
References
|
1
|
Pennathur A, Gibson MK, Jobe BA and
Luketich JD: Oesophageal carcinoma. Lancet. 381:400–412. 2013.
View Article : Google Scholar
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
3
|
Ribeiro U Jr, Posner MC, Safatle-Ribeiro
AV and Reynolds JC: Risk factors for squamous cell carcinoma of the
oesophagus. Br J Surg. 83:1174–1185. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lu JC, Tao H, Zhang YQ, Zha WW, Qian PD,
Li F and Xu KX: Extent of prophylactic postoperative radiotherapy
after radical surgery of thoracic esophageal squamous cell
carcinoma. Dis Esophagus. 21:502–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ismail NI, Kaur G, Hashim H and Hassan MS:
S100A4 overexpression proves to be independent marker for breast
cancer progression. Cancer Cell Int. 8:122008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang H, Quah SY, Dong JM, Manser E, Tang
JP and Zeng Q: PRL-3 down-regulates PTEN expression and signals
through PI3K to promote epithelial-mesenchymal transition. Cancer
Res. 67:2922–2926. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thiery JP, Acloque H, Huang RY and Nieto
MA: Epithelial-mesenchymal transitions in development and disease.
Cell. 139:871–890. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Birchmeier C, Birchmeier W and
Brand-Saberi B: Epithelial-mesenchymal transitions in cancer
progression. Acta Anat (Basel). 156:217–226. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hay ED: An overview of
epithelio-mesenchymal transformation. Acta Anat (Basel). 154:8–20.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu J, Lamouille S and Derynck R:
TGF-β-induced epithelial to mesenchymal transition. Cell Res.
19:156–172. 2009.
|
|
12
|
Boyer B, Valles AM and Edme N: Induction
and regulation of epithelial-mesenchymal transitions. Biochem
Pharmacol. 60:1091–1099. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee JM, Dedhar S, Kalluri R and Thompson
EW: The epithelial-mesenchymal transition: new insights in
signaling, development, and disease. J Cell Biol. 172:973–981.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar
|
|
15
|
Zavadil J and Bottinger EP: TGF-β and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005.
|
|
16
|
Quan J, Elhousiny M, Johnson NW and Gao J:
Transforming growth factor-β1 treatment of oral cancer induces
epithelial-mesenchymal transition and promotes bone invasion via
enhanced activity of osteoclasts. Clin Exp Metastasis. 30:659–670.
2013.
|
|
17
|
Reichl P, Haider C, Grubinger M and
Mikulits W: TGF-β in epithelial to mesenchymal transition and
metastasis of liver carcinoma. Curr Pharm Des. 18:4135–4147.
2012.
|
|
18
|
Ellenrieder V, Hendler SF, Boeck W,
Seufferlein T, Menke A, Ruhland C, Adler G and Gress TM:
Transforming growth factor β1 treatment leads to an
epithelial-mesenchymal transdifferentiation of pancreatic cancer
cells requiring extracellular signal-regulated kinase 2 activation.
Cancer Res. 61:4222–4228. 2001.
|
|
19
|
Zhang S and Yu D: PI(3)king apart PTEN’s
role in cancer. Clin Cancer Res. 16:4325–4330. 2010.PubMed/NCBI
|
|
20
|
Cantley LC and Neel BG: New insights into
tumor suppression: PTEN suppresses tumor formation by restraining
the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA.
96:4240–4245. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wallin JJ, Guan J, Edgar KA, Zhou W,
Francis R, Torres AC, Haverty PM, Eastham-Anderson J, Arena S,
Bardelli A, et al: Active PI3K pathway causes an invasive phenotype
which can be reversed or promoted by blocking the pathway at
divergent nodes. PLoS One. 7:e364022012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karamitopoulou E: Tumor budding cells,
cancer stem cells and epithelial-mesenchymal transition-type cells
in pancreatic cancer. Front Oncol. 2:2092013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang HY, Zheng XZ, Wang XH, Xuan XY, Wang
F and Li SS: S100A4 mediated cell invasion and metastasis of
esophageal squamous cell carcinoma via the regulation of MMP-2 and
E-cadherin activity. Mol Biol Rep. 39:199–208. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wendt MK, Allington TM and Schiemann WP:
Mechanisms of the epithelial-mesenchymal transition by TGF-β.
Future Oncol. 5:1145–1168. 2009.
|
|
25
|
Sun Y, Li SS, Wang XH, Wang XJ and Yan AH:
Transforming growth factor beta1 regulation of
epithelial-mesenchymal transition in esophagus squamous cell
carcinoma. Zhonghua Bing Li Xue Za Zhi. 37:542–548. 2008.(In
Chinese).
|
|
26
|
Carnero A, Blanco-Aparicio C, Renner O,
Link W and Leal JF: The PTEN/PI3K/AKT signalling pathway in cancer,
therapeutic implications. Curr Cancer Drug Targets. 8:187–198.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Leslie NR, Yang X, Downes CP and Weijer
CJ: PtdIns(3,4,5)P(3)-dependent and -independent roles for PTEN in
the control of cell migration. Curr Biol. 17:115–125. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Inoue K, Fry EA and Taneja P: Recent
progress in mouse models for tumor suppressor genes and its
implications in human cancer. Clin Med Insights Oncol. 7:103–122.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Simpson L and Parsons R: PTEN: life as a
tumor suppressor. Exp Cell Res. 264:29–41. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cully M, You H, Levine AJ and Mak TW:
Beyond PTEN mutations: the PI3K pathway as an integrator of
multiple inputs during tumorigenesis. Nat Rev Cancer. 6:184–192.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li Y, Gao L, Luo X, Wang L, Gao X, Wang W,
Sun J, Dou L, Li J, Xu C, et al: Epigenetic silencing of
microRNA-193a contributes to leukemogenesis in t(8;21) acute
myeloid leukemia by activating the PTEN/PI3K signal pathway. Blood.
121:499–509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mulholland DJ, Kobayashi N, Ruscetti M,
Zhi A, Tran LM, Huang J, Gleave M and Wu H: Pten loss and RAS/MAPK
activation cooperate to promote EMT and metastasis initiated from
prostate cancer stem/progenitor cells. Cancer Res. 72:1878–1889.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bian Y, Hall B, Sun ZJ, Molinolo A, Chen
W, Gutkind JS, Waes CV and Kulkarni AB: Loss of TGF-β signaling and
PTEN promotes head and neck squamous cell carcinoma through
cellular senescence evasion and cancer-related inflammation.
Oncogene. 31:3322–3332. 2012.
|
|
34
|
Larue L and Bellacosa A:
Epithelial-mesenchymal transition in development and cancer: role
of phosphatidylinositol 3′ kinase/AKT pathways. Oncogene.
24:7443–7454. 2005.
|
|
35
|
Chen KC, Chen CY, Lin CJ, Yang TY, Chen
TH, Wu LC and Wu CC: Luteolin attenuates TGF-β1-induced
epithelial-mesenchymal transition of lung cancer cells by
interfering in the PI3K/Akt-NF-κB-Snail pathway. Life Sci.
93:924–933. 2013.PubMed/NCBI
|
|
36
|
Barrallo-Gimeno A and Nieto MA: The Snail
genes as inducers of cell movement and survival: implications in
development and cancer. Development. 132:3151–3161. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cano A, Perez-Moreno MA, Rodrigo I,
Locascio A, Blanco MJ, del Barrio MG, Portillo F and Nieto MA: The
transcription factor snail controls epithelial-mesenchymal
transitions by repressing E-cadherin expression. Nat Cell Biol.
2:76–83. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moustakas A and Heldin CH: Induction of
epithelial-mesenchymal transition by transforming growth factor β.
Semin Cancer Biol. 22:446–454. 2012.
|
|
39
|
Lv ZD, Kong B, Li JG, Qu HL, Wang XG, Cao
WH, Liu XY, Wang Y, Yang ZC, Xu HM and Wang HB: Transforming growth
factor-β 1 enhances the invasiveness of breast cancer cells by
inducing a Smad2-dependent epithelial-to-mesenchymal transition.
Oncol Rep. 29:219–225. 2013.
|
|
40
|
Ju X, Casimiro MC, Gormley M, Meng H, Jiao
X, Katiyar S, Crosariol M, Chen K, Wang M, Quong AA, et al:
Identification of a cyclin D1 network in prostate cancer that
antagonizes epithelial-mesenchymal restraint. Cancer Res.
74:508–519. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bakin AV, Tomlinson AK, Bhowmick NA, Moses
HL and Arteaga CL: Phosphatidylinositol 3-kinase function is
required for transforming growth factor β-mediated epithelial to
mesenchymal transition and cell migration. J Biol Chem.
275:36803–36810. 2000.
|
|
42
|
Li DM and Sun H: TEP1, encoded by a
candidate tumor suppressor locus, is a novel protein tyrosine
phosphatase regulated by transforming growth factor β. Cancer Res.
57:2124–2129. 1997.PubMed/NCBI
|
|
43
|
Blobe GC, Schiemann WP and Lodish HF: Role
of transforming growth factor β in human disease. N Engl J Med.
342:1350–1358. 2000.
|
|
44
|
Krymskaya VP, Hoffman R, Eszterhas A,
Ciocca V and Panettieri RA Jr: TGF-β 1 modulates EGF-stimulated
phosphatidylinositol 3-kinase activity in human airway smooth
muscle cells. Am J Physiol. 273:L1220–L1227. 1997.
|