Introduction
Estrogen receptors play a key role in breast cancer
progression (1). Seventy-five
percent of women diagnosed with breast cancer have estrogen and
progesterone receptor (ER, PR)-positive breast tumors (2,3).
Tamoxifen, a selective ER modulator, is the main five-year adjuvant
treatment for these patients (4).
However, one third of patients treated with tamoxifen will suffer
from a recurrence within 15 years (5). Thus, understanding the mechanisms
involved in the development of endocrine resistance is of great
clinical relevance.
ERs are steroid receptors that are located not only
in the nucleus, but also in the cytoplasm, the mitochondria, the
endoplasmic reticulum and the plasma membrane (6). Plasma membrane ERs were first
identified in endometrial cells (7). In breast cancer cells, the main plasma
membrane ER which constitute ~5–10% of the total ERs, is ER-α;
little ER-β is found at this location (8). Apart from the ERs, several other
membrane receptors have been proposed as mediators of the
non-genomic actions of estrogens (6,9,10).
Nonetheless, ER-α is believed to be the main receptor responsible
for rapid signaling in response to estrogens in established
experimental conditions in mammary cells (6). ER-α forms a ternary complex together
with the IGF-R1 and the adaptor protein SHC in the presence of
estradiol (10). Binding of
estradiol to ER-α leads to activation of SHC and the formation of
the complex followed by the phosphorylation of ER and the IGF-R1 by
the c-Src kinase (10). Other
authors have proposed a linear pathway implicating activation of
the IGF-R1 followed by activation of matrix metalloproteinases
(MMPs) which leads to the release of HB-EGF. EGF binds to its
receptor leading to activation of the MAPK/ERK 1/2 and PI3K/AKT
pathways downstream (11). Notably,
few publications to the best of our knowledge, have investigated
the effect of tamoxifen on plasma membrane ER and its subsequent
non-genomic actions (12).
We developed a mouse model of estrogen-dependent
breast cancer from a spontaneous tumor that arose in a female
BALB/c mouse in our animal facility (13). This tumor, called M05, is positive
for ER and PR and responds to tamoxifen within the first 9 passages
(13). From this tumor we
established the epithelial cell line LM05-E that is ER- and
PR-positive and is responsive to estradiol and tamoxifen as
determined by proliferation and apoptosis assays (14). In the present study we investigated
the non-genomic effects of estradiol and tamoxifen on the LM05-E
cell line. We additionally used MCF-7 cells as a control. We
established that, similar to estradiol, tamoxifen activates the
MAPK/ERK 1/2 pathway in breast cancer cells; however, we did not
find any effect of either estradiol or tamoxifen on the activation
status of PI3K/AKT. Short-term treatments with estradiol
stimulated, whereas tamoxifen inhibited, cell proliferation. Using
pharmacological inhibitors we showed that, as previously described,
the effect of estradiol is mediated by the MAPK/ERK 1/2 pathway.
Inhibition of this pathway did not affect the effect of tamoxifen.
Notably, however, blocking of PI3K/AKT signaling interfered with
the inhibitory effect of tamoxifen by reversing it. Analysis of the
involvement of the EGFR support previous findings that designate
this receptor as a mediator of the non-genomic effects of
estradiol. Unexpectedly, blocking EGFR also reversed the inhibitory
effect of tamoxifen. Finally, MMPs were confirmed as being involved
in the proliferative effect of estradiol but not on the inhibitory
effect of tamoxifen. These results demonstrated the novel
non-genomic effects of tamoxifen and revealed that pathways such as
EGFR upstream of PI3K/AKT are involved in inhibition of cell
proliferation. Consideration of these mechanisms should be taken
into account when analyzing strategies that aim at combining
endocrine therapy with specific signaling inhibitors.
Materials and methods
Cell culture
The MCF-7 and LM05-E cell lines were routinely
maintained in growth medium, consisting of DMEM/F12 medium
(Sigma-Aldrich), supplemented with 10% fetal calf serum (FCS;
GenSA, Buenos Aires, Argentina) and gentamicin, in a humidified 5%
CO2/air atmosphere. Serial passages were carried out by
treatment of 80% confluent monolayers with 0.25% trypsin
(Invitrogen) and 0.02% EDTA in Ca2+-free and
Mg2+-free PBS.
Reagents
Estradiol and 4-OH-tamoxifen (both from
Sigma-Aldrich) were prepared 1000X in absolute ethanol and used at
a final concentration of 10 nM and 1 μM, respectively. Controls
were subjected to the same dilution of absolute ethanol. The
MAPK/ERK 1/2 inhibitor PD98059 (10 μM), PI3K/AKT inhibitor LY294002
(10 μM), the matrix metalloproteinase inhibitor GM6001 (10 μM) and
the EGFR inhibitor AG1478 (6.4 μM) were all purchased from
Calbiochem, and Wortmannin (100 nM) was purchased from
Sigma-Aldrich. They were prepared in DMSO, in at least 1000X stock
solutions, and the equivalent dilutions of DMSO were used as
controls. We previously confirmed the effectiveness of these
inhibitors on LM05-E and MCF-7 cells (15).
Cell proliferation studies
Cells (3.0×104/well) were plated in
12-well plates in growth medium. The next day, cells were washed
twice with PBS and the medium was replaced with phenol red-free
DMEM/F12. After 48 h, cells were treated with either estradiol (10
nM) or 4-OH-tamoxifen (1 μM) (or vehicle) for up to 60 min and were
then washed 5 times with PBS as previously published elsewhere
(16). Subsequently, cells were
cultured for an additional 48 h in phenol red-free DMEM/F12
supplemented with 1% charcoal stripped serum. Cells were then
counted using a Neubauer chamber. To test the effects of the
pharmacological inhibitors on the proliferative response to both
estradiol and tamoxifen, starved cells were pretreated with the
corresponding inhibitor (or vehicle) for 1 h, and were then
subjected to either estradiol or 4-OH-tamoxifen exposure. All
experiments were carried out in triplicates and repeated at least
three times.
Western immunoblot assay
To evaluate the activation of the different
signaling pathways by estradiol and 4-OH-tamoxifen, LM05-E cells
(6.0×105) were plated on 60-mm plates in growth medium.
The next day, cells were washed twice and the medium was replaced
by phenol red-free medium. Cells were starved for 48 h and were
then subjected to treatment with estradiol (10 nM) or
4-OH-tamoxifen (1 μM) for the indicated periods of time. To examine
the effect of the specific inhibitors on the activation of the
signaling pathways, starved cells were pretreated for 1 h with the
inhibitors (or the corresponding dilution of DMSO) and were
subsequently treated with either estradiol or 4-OH-tamoxifen.
Protein extracts were prepared by homogenizing cells on ice in RIPA
buffer (50 mM Tris, pH 8.0 containing 150 mM NaCl, 0.1% SDS, 0.5%
deoxycholate and 1% NP-40) containing protease inhibitors (40 μm
phenylmethylsulfonyl fluoride, 5 μg/ml leupeptin, 50 μg/ml
aprotinin and 200 μM orthovanadate). Protein concentrations were
measured using the Bradford method. Samples were mixed with 4X
sample buffer containing β-mercaptoethanol and boiled for 2 min.
Fifty micrograms of each sample was then separated on SDS-PAGE Mini
gels (Bio-Rad) and transferred to PVDF membranes (Amersham
Biosciences, Uppsala, Sweden). The membranes were blocked overnight
in 5% fat-free milk, 0.1% Tween-20 in PBS at 4°C. Primary
antibodies were used at a 1/500–1/2,000 dilution in PBS containing
0.1% Tween-20 (PBST) and 2.5% fat-free milk, and were incubated at
4°C overnight. After washing with PBST, the membranes were
incubated with secondary antibodies at a 1/1,000 dilution for 1 h
at room temperature. Signals were detected with an enhanced
chemiluminescence kit (ECL; Amersham Biosciences). The following
primary antibodies were used: mouse anti-phospho-ERK 1/2, mouse
anti-ERK 1/2, rabbit anti-phospho-AKT and rabbit anti-AKT (all from
Santa Cruz Biotechnology). The following secondary antibodies were
used: donkey-anti rabbit HRP and goat anti-mouse HRP (Santa Cruz
Biotechnology). To evaluate the effects of the inhibitors on the
activation of the signaling pathways, the inhibitor and
vehicle-treated samples were run on the same gel using 15 lane
combs.
Statistical analysis
The statistical significance of differences between
the groups was calculated by applying one-way ANOVA, followed by
Tukey’s multiple comparisons test. A value of P<0.05 was
considered to indicate a statistically significant result.
Results
Activation of signaling pathways by
estradiol and 4-OH-tamoxifen in LM05-E cells
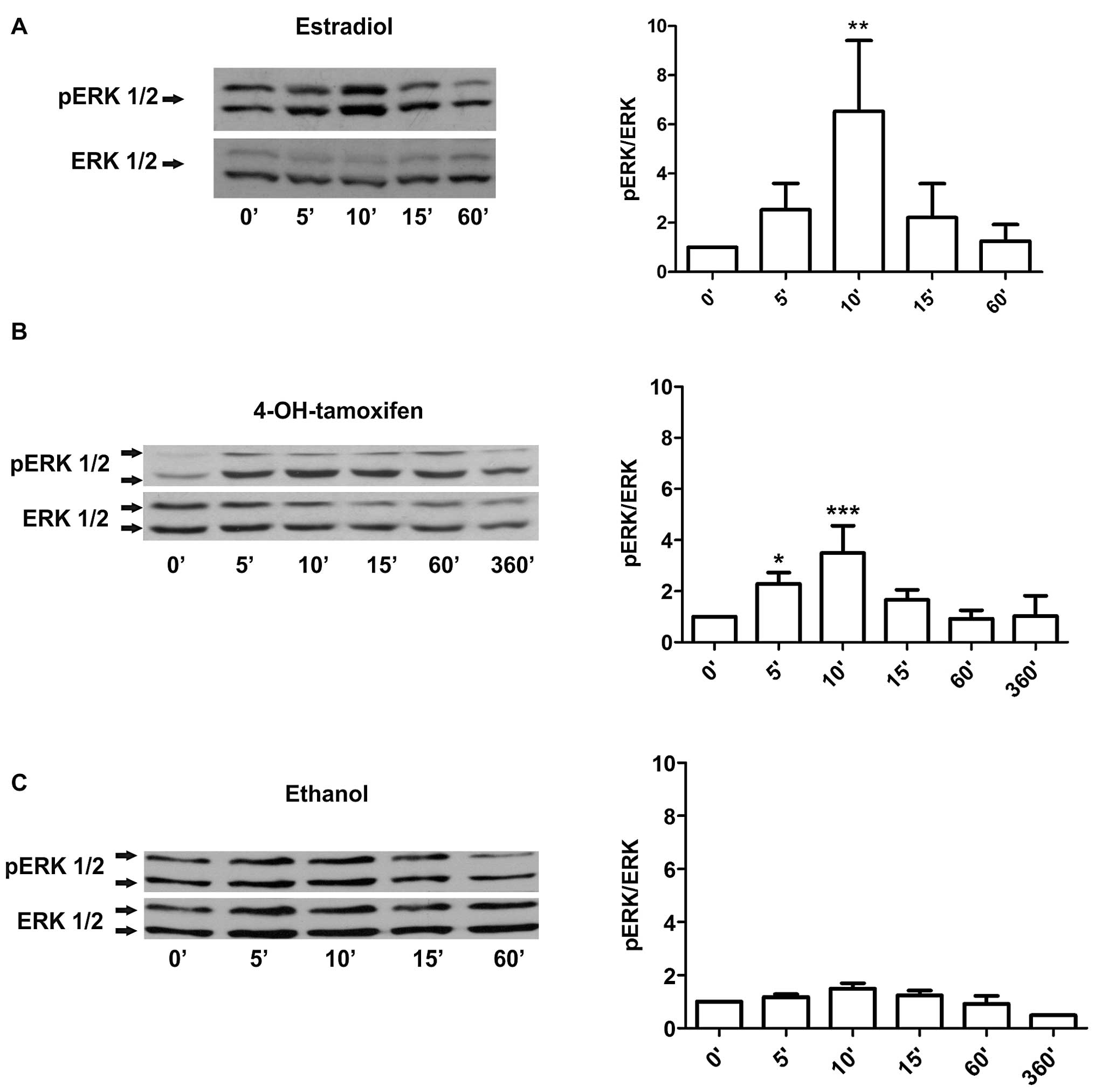
To determine the non-genomic responses of LM05-E
cells to 17-β-estradiol and 4-OH-tamoxifen, we first examined by
western blot analysis the activation of the MAPK/ERK signaling
pathway. As previously shown (14),
short-term treatments with 10 nM estradiol led to a strong
phosphorylation of ERK 1/2 by 10 min that declined to basal levels
by 60 min (Fig. 1A). The same
analysis on cells treated with 1 μM 4-OH-tamoxifen showed a weaker
phosphorylation of ERK at 5–10 min with the signal declining by 60
min (Fig. 1B). Ethanol-treated
cells, in contrast, did not show a significant activation of ERK
1/2 under these experimental conditions (Fig. 1C). To further establish whether the
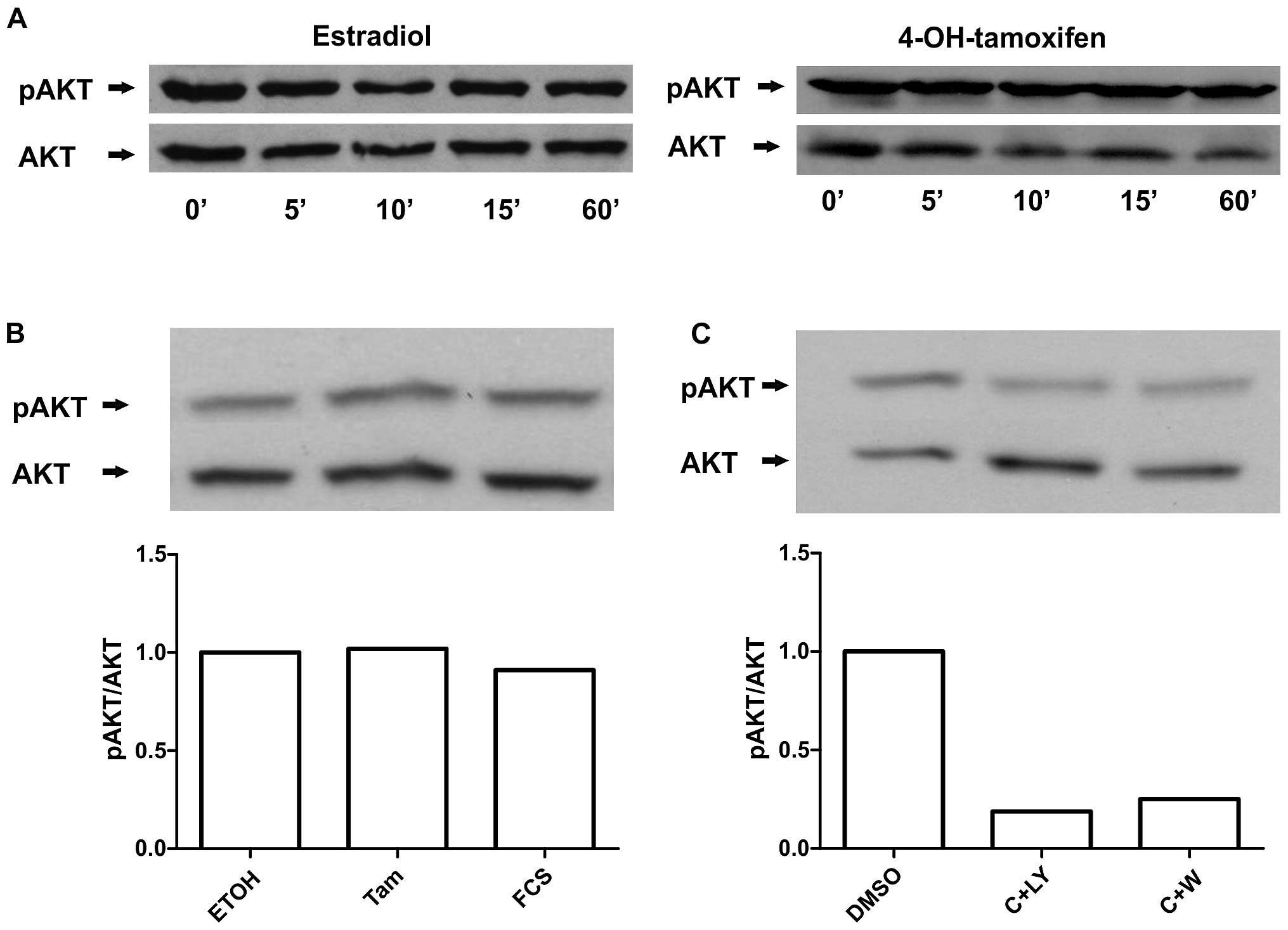
PI3K/AKT pathway is involved, western blots against phosphorylated
AKT were carried out on cells receiving the identical treatments.
Significant activation of AKT by estradiol or 4-OH-tamoxifen was
not detected under our the experimental conditions (Fig. 2A). Moreover, we tested 10% FCS as a
positive control and found that under these conditions no
significant AKT activation was observed (Fig. 2B). However, we observed that PI3K
inhibitors were able to reduce AKT phosphorylation (Fig. 2C). Thus, our results suggest that
estradiol and 4-OH-tamoxifen activate the MAPK/ERK 1/2 pathway in
LM05-E cells as previously reported by others in MCF-7 cells
(12) and that there was a high
basal activation of AKT in our model.
Proliferative response of LM05-E cells to
short-term treatments of estradiol and 4-OH-tamoxifen
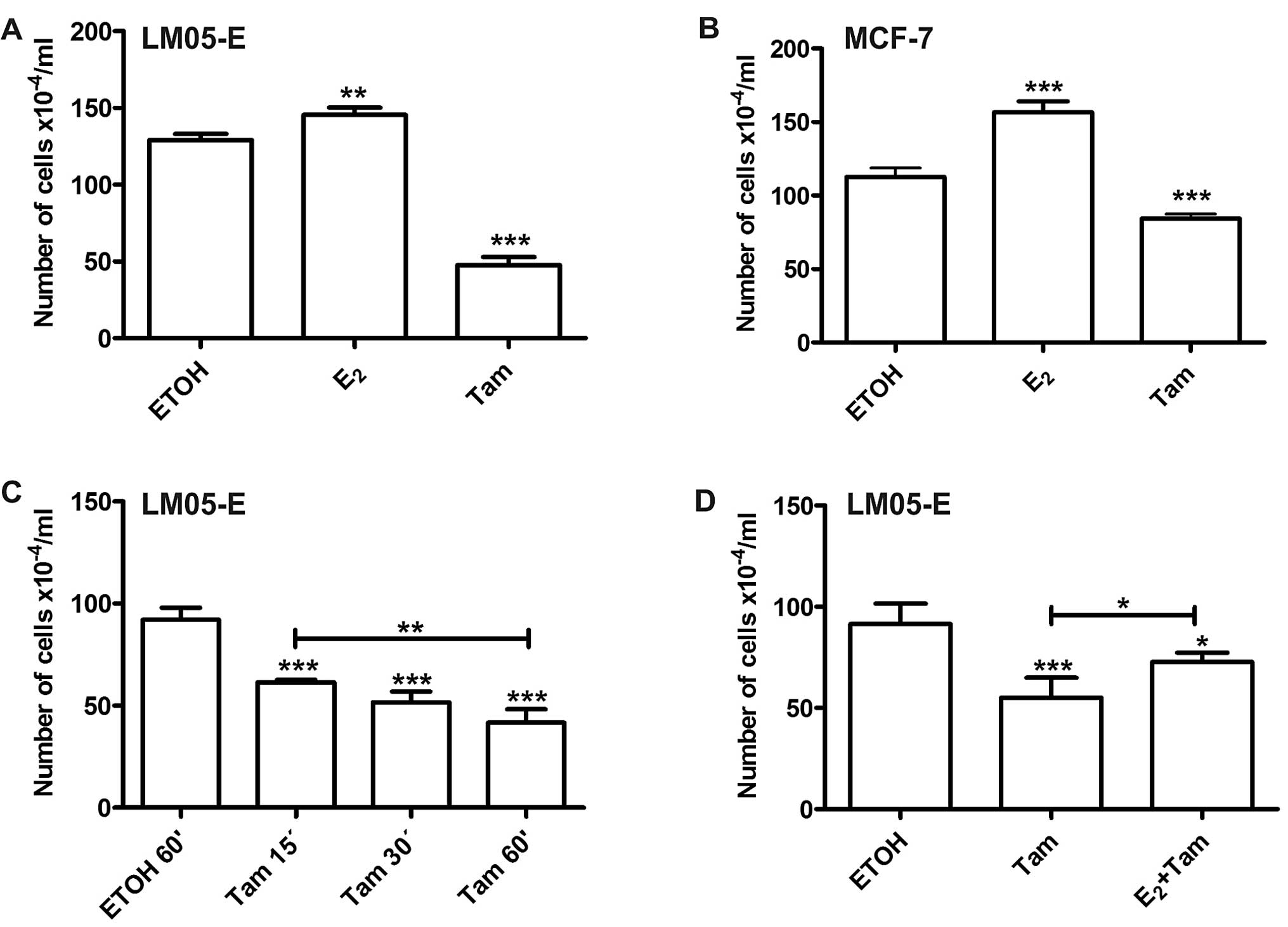
To investigate the proliferative response of LM05-E
cells to short-term treatments of 17-β-estradiol and
4-OH-tamoxifen, we treated cells that had been starved in
serum-free medium for 48 h with 10 nM estradiol, 1 μM
4-OH-tamoxifen or vehicle for up to 1 h. The wells were
subsequently washed 5 times with PBS, and the medium was replaced
by phenol red-free DMEM/F12 supplemented with 1% charcoal stripped
serum as described in Materials and methods and published by others
(16). Forty-eight hours later,
cells were counted to establish the effect of the treatments on
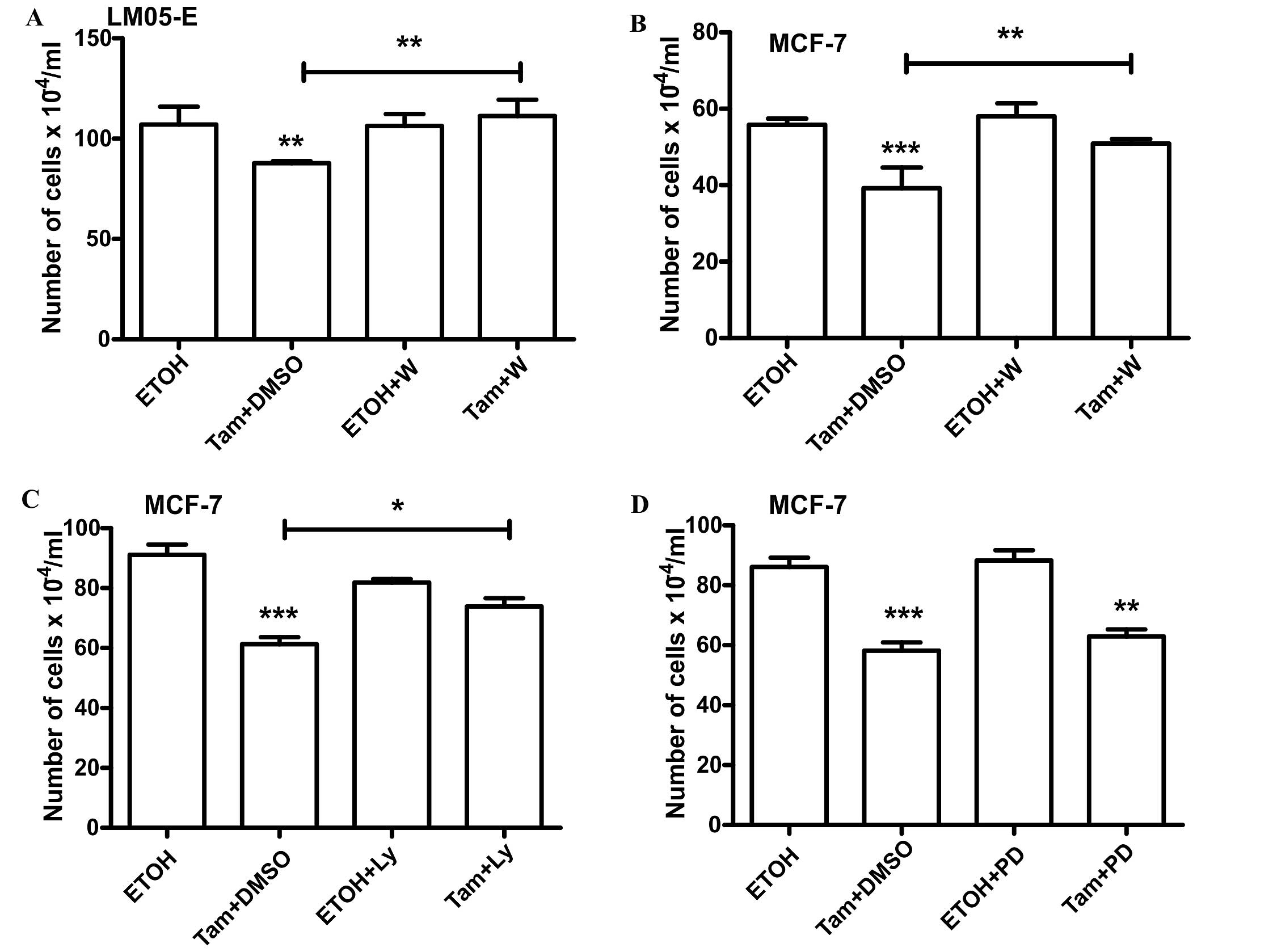
cell proliferation. Treatment with estradiol induced an increase in
the total number of cells, and 4-OH-tamoxifen, in contrast,
inhibited cell proliferation when compared to the vehicle-treated
control (Fig. 3A). Using the same
experimental setting, similar results were obtained with MCF-7
cells (Fig. 3B). Given that
treatment with 4-OH-tamoxifen reduced cell proliferation between 30
and 50%, we investigated whether significant results could be
obtained with shorter periods of treatment. The effect of
4-OH-tamoxifen continued to be significant with treatments as brief
as 15 min (Fig. 3C). To establish
whether the effects of the short-term treatments with
4-OH-tamoxifen were being mediated through the same receptor as
estradiol, LM05-E cells were pre-treated for 15 min with 10 nM
estradiol and subsequently 1 μM 4-OH-tamoxifen was added for 1 h.
Forty-eight hours later, the effect on cell proliferation showed
that estradiol only partially counteracted the effects of tamoxifen
(Fig. 3D), suggesting that the ER
is involved in this mechanism. However, it has also been proposed
by other authors, that tamoxifen may exert its effects through
ER-dependent and -independent mechanisms (17).
The MAPK/ERK pathway is involved in the
proliferative response to estradiol whereas the PI3K/AKT pathway
mediates the inhibitory effect of 4-OH-tamoxifen
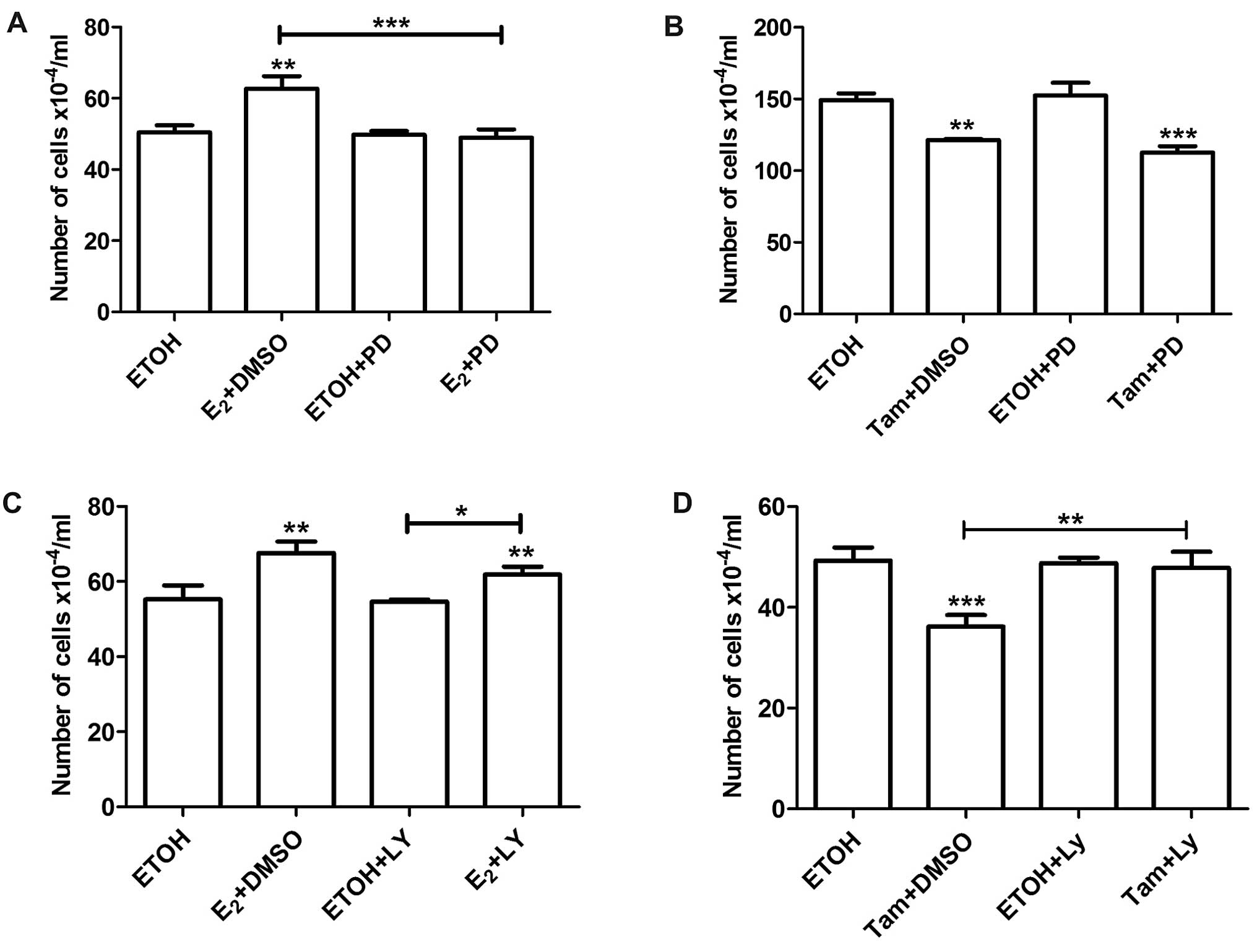
We established above that short-term treatments with
estradiol stimulated while 4-OH-tamoxifen inhibited the
proliferation of LM05-E cells; however, both effects were
accompanied by an increase in ERK phosphorylation and no changes in
AKT phosphorylation. To further establish the involvement of the
downstream signaling pathways, we pre-treated LM05-E cells with the
MEK inhibitor PD98059 (10 μM) or the PI3K inhibitors LY294002 (10
μM) and Wortmannin (100 nM) and then subjected them to short pulses
of estradiol and 4-OH-tamoxifen as explained above. Pre-treatment
with PD98059 abrogated the stimulatory effects of estradiol on cell
proliferation (Fig. 4A), but did
not affect the inhibitory effect of 4-OH-tamoxifen (Fig. 4B). On the other hand, LY294002 did
not affect the stimulatory effect of estradiol (Fig. 4C). However, inhibition of the
PI3K/AKT pathway with either LY294002 or Wortmannin reversed the
inhibitory effect of 4-OH-tamoxifen (Figs. 4D and 5A). Pretreatment with Wortmaninn or
LY294002 also reversed the inhibitory effect of 4-OH-tamoxifen on
MCF-7 cells (Fig. 5B and C);
however, the MEK inhibitor, as in the case of LM05-E cells, did not
have an impact on the effect of the anti-estrogen (Fig. 5D). Our results thus suggest that the
MAPK/ERK pathway plays a key role in mediating the stimulatory
effect of estradiol whereas the PI3K/AKT pathway is involved in the
inhibitory effect of 4-OH-tamoxifen.
EGFR mediates the downstream effects of
both estradiol and 4-OH-tamoxifen, while MMPs are only involved in
the effects of estradiol
It has been previously shown in MCF-7 cells that
binding of estradiol to membrane ER leads to the shedding of HB-EGF
by MMPs and the subsequent activation of the MAPK/ERK pathway
(11). To investigate whether EGFR
is involved in the proliferative response to short-term treatment
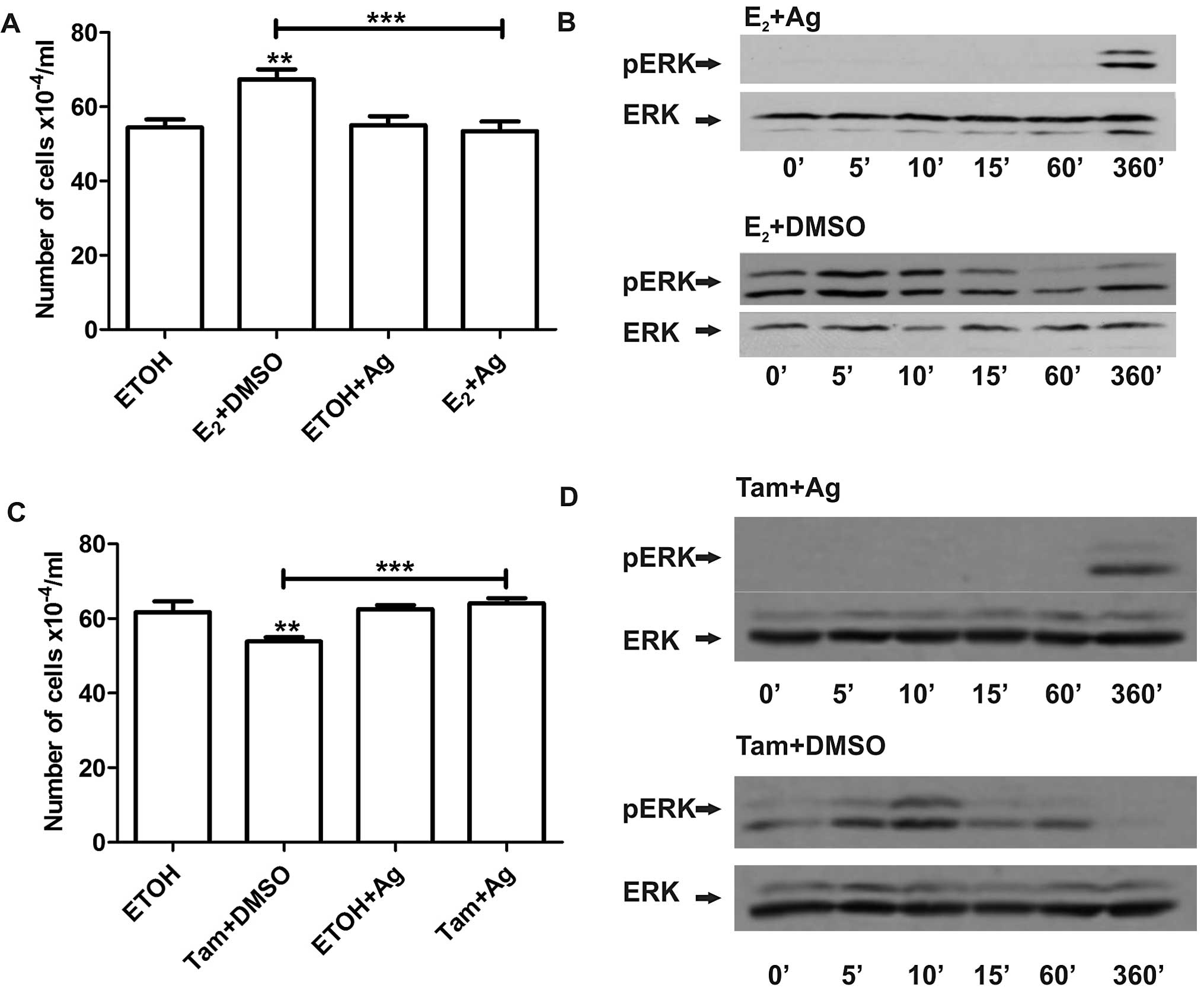
with estradiol or 4-OH-tamoxifen in LM05-E cells, we pre-incubated
the cells with the EGFR inhibitor AG1478 (6.4 μM) and then
incubated the cells with estradiol or 4-OH-tamoxifen for 1 h. The
EGFR inhibitor completely blocked the increase in cell number
induced by estradiol (Fig. 6A).
Western blot analysis showed that inhibition of EGFR led to reduced
activation of MAPK/ERK (Fig. 6B).
Moreover, it abrogated the inhibitory effect of 4-OH-tamoxifen in
both LM05-E (Fig. 6C) and MCF-7
cells (data not shown) as previously revealed (12). Western blot analysis showed that
4-OH-tamoxifen induced MAPK/ERK 1/2 activation was also inhibited
(Fig. 6D) suggesting that EGFR
provides a point of convergence for both the stimulatory and
inhibitory effects of these compounds, even though MAPK/ERK 1/2
appears to play a key role only in estradiol-induced cell
proliferation.
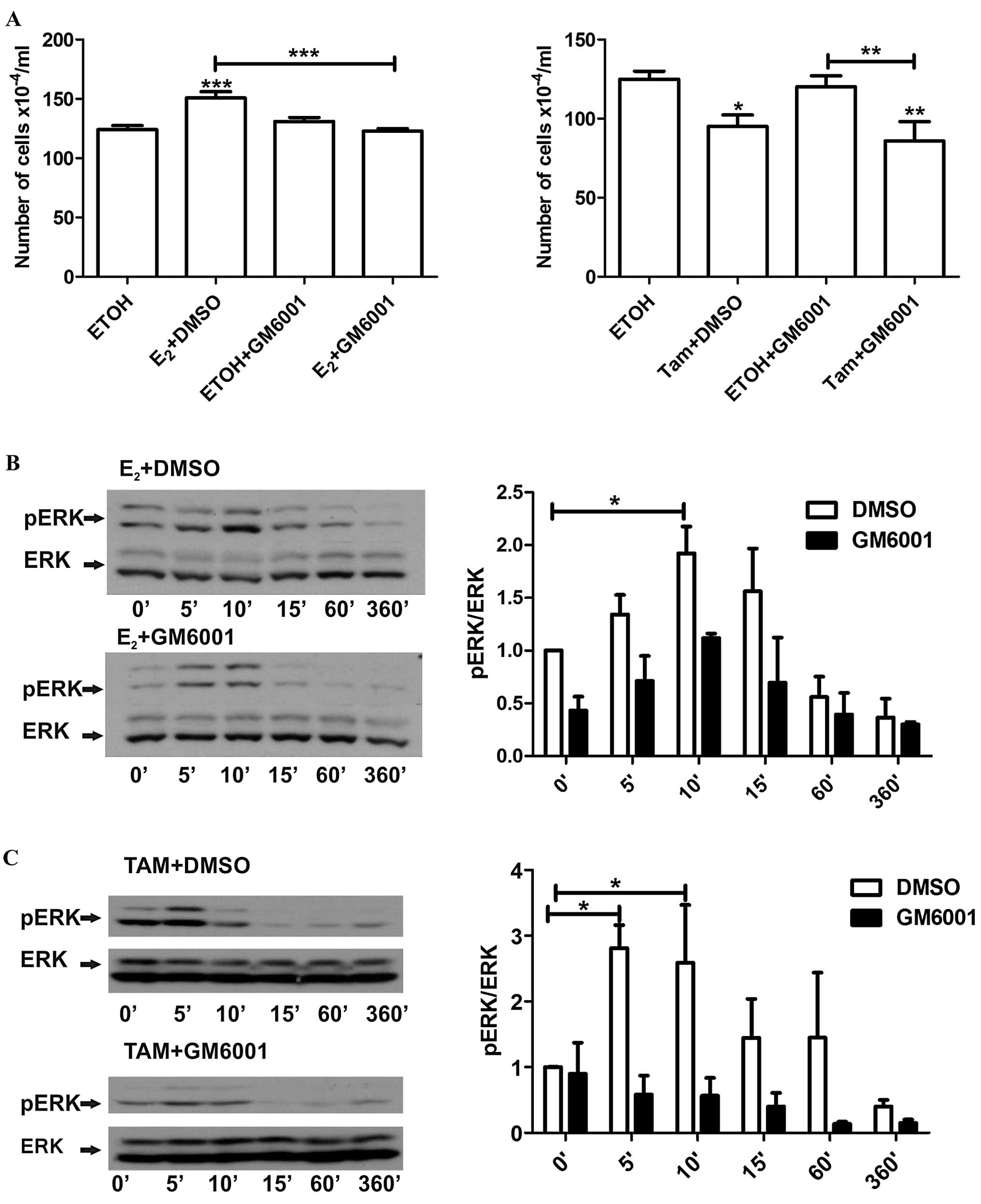
To determine whether shedding of HB-EGF is also a
common event leading to these distinct effects, LM05-E cells were
pre-incubated with the MMP inhibitor GM6001 (10 μM) and then
treated with either estradiol or 4-OH-tamoxifen. GM6001, as did
AG1478, led to a reversal of the stimulatory effect of estradiol,
but did not affect the inhibitory effect of 4-OH-tamoxifen
(Fig. 7A). Moreover, as noted in
the western blot analysis, the MMP inhibitor consistently delayed
the phosphorylation of ERK in the cells treated with estradiol
(Fig. 7B); notably, this was also
the case for the cells treated with 4-OH-tamoxifen (Fig. 7C). Thus, our results showed that
similar to MCF-7 cells, the non-genomic effects of estradiol
involve MMP activity and the participation of EGFR. Moreover, our
results suggest that 4-OH-tamoxifen also requires an active EGFR to
exert its biological effects even though MMP activity does not
appear to play a role.
Discussion
In the present study, we investigated the
non-genomic responses of murine LM05-E breast cancer cells to
estradiol and 4-OH-tamoxifen. Through proliferation assays, based
on a previously published method established by Vallejo et
al (16) and western blot
analysis, we determined that, as previously shown for MCF-7 and
T47D cells, short-term exposure to estradiol and 4-OH-tamoxifen led
to the phosphorylation of MAPK/ERK 1/2. As expected, treatment with
estradiol had a positive impact on cell number at 48 h, whereas
4-OH-tamoxifen had an inhibitory effect. Estradiol partially
counteracted the inhibitory effect of 4-OH-tamoxifen suggesting
that the ER is at least partially involved in tamoxifen
membrane-initiated signaling. Analysis of the active involvement of
the signaling pathways on the non-genomic responses using specific
inhibitors revealed that MAPK/ERK 1/2 mediates the proliferative
response to estradiol, but not the inhibitory effect of
4-OH-tamoxifen. On the other hand, we did not find an involvement
of PI3K/AKT in the response to estradiol; surprisingly, however,
LY294002 reversed the inhibitory effect of 4-OH-tamoxifen,
suggesting that PI3K/AKT mediates the inhibitory effect of
tamoxifen. Moreover, the PI3K inhibitor Wortmannin also abolished
the inhibitory effect of tamoxifen, indicating that it is most
probably due to PI3K inhibition and not other off-targets of
LY294002, such as CK2 or GSK3β (18). Blocking EGFR strongly inhibited the
activation of MAPK/ERK 1/2 induced by both estradiol and
4-OH-tamoxifen. With regards to proliferation, EGFR mediated the
stimulatory effect of estradiol, as previously shown by others,
and, as in the case of the PI3K/AKT pathway, its constraint
neutralized the inhibitory effect of 4-OH-tamoxifen. Finally, we
found that the MMP inhibitor GM6001 delayed the activation of ERK
1/2 by estradiol and 4-OH-tamoxifen but had an impact only on the
proliferative effect of estradiol.
Previously, several studies demonstrated that
short-term treatment with estradiol led to the activation of the
MAPK/ERK 1/2 pathway in human breast cancer cells and other cell
types such as endothelial cells (11,19).
Few authors have investigated the non-genomic effects of
4-OH-tamoxifen and reported rapid activation of MAPK/ERK 1/2 by
this SERM (12). Using LM05-E
cells, we found that similar results were obtained regarding the
MAPK/ERK 1/2 pathway, with rapid MMP-dependent phosphorylation
following both estradiol and 4-OH-tamoxifen treatments. We did not
detect, however, any changes in AKT phosphorylation for either
treatment or FCS pulses. This may be due to a high basal activation
of AKT in our model. Nevertheless, activation of AKT by estradiol
has been described for MCF-7 cells by Razandi et al
(11). Together with the activation
of the signaling pathways, we investigated the impact of each of
these on cell proliferation as a measure of the long-term impact on
cell behavior. As expected, 48 h after exposure to estradiol, a
statistically significant increase in cell number was detected;
conversely a decrease was found in culture plates treated with
4-OH-tamoxifen. Blocking the MAPK/ERK 1/2 pathway had a direct
impact on the increase in cell number induced by estradiol,
however, it did not affect the inhibitory effect of 4-OH-tamoxifen.
This is contrary to the findings reported by Zheng et al
(12) who described a reduction in
cell death induced by 4-OH-tamoxifen in cells pre-treated with
PD98059. However, the concentrations of both 4-OH-tamoxifen and the
MAPK/ERK inhibitors used by the other authors were higher than
those used in the present study (5 and 20 μM, respectively), and
the number of dead cells were counted up to 60 min after the
initiation of the treatments as their end point. In contrast, we
evaluated the effect of the treatments on cell number 48 h later.
Most interesting, however, was the finding that blocking PI3K/AKT
counteracted the effect of 4-OH-tamoxifen. As mentioned above, we
did not find a statistically significant change in AKT
phosphorylation following treatment with either estradiol or
4-OH-tamoxifen on LM05-E cells, and LY294002 did not affect the
increase in cell number induced by estradiol. In addition, we found
that both LY294002 and Wortmannin reduced basal AKT activity. Other
studies found increased phosphorylation of AKT as a result of
non-genomic effects of estradiol (11,19).
Notably, PI3K/AKT inhibition has been shown to promote resistance
to chemotherapeutic drugs in MDA-MB-231 breast cancer cells as well
as in other cell types such as DU-145 prostate, HCT-116 colon and
A-549 lung carcinoma cells (20).
The authors found that this effect was only present when the cells
were pre-incubated with the PI3K/AKT inhibitor. On the contrary,
when the timing was changed, with cells being exposed to LY294002
after treatment with chemotherapeutic drugs, cell survival was
decreased. Based on the same line of evidence, we found that
inhibition of EGFR also reversed the inhibitory effect of
4-OH-tamoxifen but additionally had an impact on the proliferative
effect of estradiol. Western blot analysis showed that AG1478
completely blocked the MAPK/ERK 1/2 pathway and this could account
for the interference of the proliferative effect of estradiol.
However, we did not find any change in the status of the PI3K/AKT
pathway (data not shown) in the presence of AG1478 suggesting that
some other pathway downstream of EGFR was accountable for the
observed effect. Other authors have recently shown that inhibition
of EGFR by various inhibitors increases ErbB3 protein levels in
breast cancer cells and downregulates FAS which is involved in
apoptotic cell death (21). On the
other hand, HER2/HER3 heterodimer formation has been shown to be
involved in EGFR inhibitor resistance in lung cancer cells
(22).
Analysis of the effect of the MMP inhibitor GM6001
on cells treated with estradiol and 4-OH-tamoxifen showed that only
in the case of estradiol did GM6001 have an effect. However,
western blot analysis revealed that the MMP inhibitor delayed the
phosphorylation of MAPK/ERK 1/2 induced by both estradiol and
4-OH-tamoxifen. The fact that at the proliferative level only cells
treated with estradiol were affected supports our previous
conclusion of the involvement of the MAPK/ERK 1/2 pathway as a
mediator of the effect of estradiol, but not of tamoxifen.
Our results demonstrated that the selective estrogen
receptor modulator 4-OH-tamoxifen stimulates non-genomic signaling
in murine breast cancer cells. Even though the MAPK/ERK 1/2 pathway
is activated as with estradiol, it does not appear to be a mediator
of the effect of tamoxifen. Moreover, the PI3K/AKT pathway does not
appear to be altered by tamoxifen treatment; however, its
inhibition reverses the inhibitory effect of tamoxifen as does the
blockage of the EGFR. These results suggest that caution should be
exercised when considering the possibility of combining
anti-estrogens with signaling pathway inhibitors as the latter may
interfere with the non-genomic effects of tamoxifen and counteract
part of its inhibitory effect.
Acknowledgements
The present study is supported by grants from the
Susan G. Komen Foundation (BCTR0600341), Préstamo BID PICT
2008-0325 and the Florencio Fiorini Foundation to M.S., and grants
UBACYT 2011–2014 (M0243) and Préstamo BID PICT 2010-01296 to
E.B.K.J.
References
|
1
|
Russo J and Russo IH: The role of estrogen
in the initiation of breast cancer. J Steroid Biochem Mol Biol.
102:89–96. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harvey JM, Clark GM, Osborne CK and Allred
DC: Estrogen receptor status by immunohistochemistry is superior to
the ligand-binding assay for predicting response to adjuvant
endocrine therapy in breast cancer. J Clin Oncol. 17:1474–1481.
1999.PubMed/NCBI
|
|
3
|
Graham JD, Yeates C, Balleine RL, et al:
Characterization of progesterone receptor A and B expression in
human breast cancer. Cancer Res. 55:5063–5068. 1995.PubMed/NCBI
|
|
4
|
Sengupta S and Jordan VC: Selective
estrogen modulators as an anticancer tool: mechanisms of efficiency
and resistance. Adv Exp Med Biol. 630:206–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Musgrove EA and Sutherland RL: Biological
determinants of endocrine resistance in breast cancer. Nat Rev
Cancer. 9:631–643. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Levin ER and Pietras RJ: Estrogen
receptors outside the nucleus in breast cancer. Breast Cancer Res
Treat. 108:351–361. 2008. View Article : Google Scholar
|
|
7
|
Pietras RJ and Szego CM: Specific binding
sites for oestrogen at the outer surfaces of isolated endometrial
cells. Nature. 265:69–72. 1977. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pedram A, Razandi M and Levin ER: Nature
of functional estrogen receptors at the plasma membrane. Mol
Endocrinol. 20:1996–2009. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Toran-Allerand CD, Guan X, MacLusky NJ, et
al: ER-X: a novel, plasma membrane-associated, putative estrogen
receptor that is regulated during development and after ischemic
brain injury. J Neurosci. 22:8391–8401. 2002.PubMed/NCBI
|
|
10
|
Song RX, Fan P, Yue W, Chen Y and Santen
RJ: Role of receptor complexes in the extranuclear actions of
estrogen receptor α in breast cancer. Endocr Relat Cancer. 13(Suppl
1): S3–S13. 2006. View Article : Google Scholar
|
|
11
|
Razandi M, Pedram A, Park ST and Levin ER:
Proximal events in signaling by plasma membrane estrogen receptors.
J Biol Chem. 278:2701–2712. 2003. View Article : Google Scholar
|
|
12
|
Zheng A, Kallio A and Harkonen P:
Tamoxifen-induced rapid death of MCF-7 breast cancer cells is
mediated via extracellularly signal-regulated kinase signaling and
can be abrogated by estrogen. Endocrinology. 148:2764–2777. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Simian M, Manzur T, Rodriguez V, de Kier
Joffe EB and Klein S: A spontaneous estrogen-dependent,
tamoxifen-sensitive mouse mammary tumor: a new model system to
study hormone-responsiveness in immune competent mice. Breast
Cancer Res Treat. 113:1–8. 2009. View Article : Google Scholar
|
|
14
|
Pontiggia O, Rodriguez V, Fabris V, et al:
Establishment of an in vitro estrogen-dependent mouse mammary tumor
model: a new tool to understand estrogen responsiveness and
development of tamoxifen resistance in the context of
stromal-epithelial interactions. Breast Cancer Res Treat.
116:247–255. 2009. View Article : Google Scholar
|
|
15
|
Pontiggia O, Sampayo R, Raffo D, et al:
The tumor microenvironment modulates tamoxifen resistance in breast
cancer: a role for soluble stromal factors and fibronectin through
β1 integrin. Breast Cancer Res Treat. 133:459–471. 2012. View Article : Google Scholar
|
|
16
|
Vallejo G, Ballare C, Baranao JL, Beato M
and Saragueta P: Progestin activation of nongenomic pathways via
cross talk of progesterone receptor with estrogen receptor β
induces proliferation of endometrial stromal cells. Mol Endocrinol.
19:3023–3037. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ferlini C, Scambia G, Marone M, et al:
Tamoxifen induces oxidative stress and apoptosis in oestrogen
receptor-negative human cancer cell lines. Br J Cancer. 79:257–263.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gharbi SI, Zvelebil MJ, Shuttleworth SJ,
et al: Exploring the specificity of the PI3K family inhibitor
LY294002. Biochem J. 404:15–21. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stice JP, Mbai FN, Chen L and Knowlton AA:
Rapid activation of nuclear factor κB by 17β-estradiol and
selective estrogen receptor modulators: pathways mediating cellular
protection. Shock. 38:128–136. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McDonald GT, Sullivan R, Pare GC and
Graham CH: Inhibition of phosphatidylinositol 3-kinase promotes
tumor cell resistance to chemotherapeutic agents via a mechanism
involving delay in cell cycle progression. Exp Cell Res.
316:3197–3206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Grovdal LM, Kim J, Holst MR, Knudsen SL,
Grandal MV and van Deurs B: EGF receptor inhibitors increase ErbB3
mRNA and protein levels in breast cancer cells. Cell Signal.
24:296–301. 2012. View Article : Google Scholar
|
|
22
|
Hirata A, Hosoi F, Miyagawa M, et al: HER2
overexpression increases sensitivity to gefitinib, an epidermal
growth factor receptor tyrosine kinase inhibitor, through
inhibition of HER2/HER3 heterodimer formation in lung cancer cells.
Cancer Res. 65:4253–4260. 2005. View Article : Google Scholar : PubMed/NCBI
|