Introduction
Poly(ADP-ribose) polymerase (PARP) enzyme inhibitors
are emerging as a valuable new drug class in the treatment of
cancer. PARP-1 is the founding member of a family of 18 PARP
members that have been identified thus far. PARP-1 functions as a
key molecule in the repair of DNA single-strand breaks (SSBs) via
the base excision DNA repair (BER) pathway (1). Inhibitors of PARP-1 have been shown to
enhance the cytotoxic effects of ionizing radiation and
DNA-damaging chemotherapeutic agents in vitro and in
vivo (2,3). Early preclinical and clinical trial
data suggest that PARP inhibitors may be used as chemo/radiotherapy
sensitizers and as single agents to selectively kill cancers
defective in DNA repair, specifically cancers with germ-line
mutations in breast cancer-associated BRCA1 and BRCA2
genes, a strategy known as ‘synthetic lethality’ (4–7). There
are currently at least eight PARP inhibitors being developed
(8), however, the development of
new drugs is extremely costly and there is a gap between the
resources invested in drug development and their translatability
into longer survival for cancer patients.
Repurposing existing drugs is another strategy for
drug development in which safety pharmacology studies have been
already done, which reduces the time and cost of approving the
compounds for clinical use (9,10).
Nicotinamide (NAM; pyridine-3-carboxylic acid) is a water-soluble
amide active form of vitamin B3 or niacin. NAM and niacin are
precursors for the synthesis of nicotinamide adenine dinucleotide
NAD+ and the phosphorylated derivative NADH+
(11). This drug has been used for
the treatment of pellagra, diabetes mellitus, acne and
schizophrenia and has been shown to have low toxicity even at high
doses (12). NAM was identified as
the first inhibitor of PARP, while the majority of PARP inhibitors
contain the NAM pharmacophore (13). Preclinical studies have shown that
NAM may exert antitumor effects and reverse chemotherapy resistance
in some models (14). In addition,
this drug is currently used as a component of accelerated
radiotherapy with carbogen and nicotinamide (ARCON), a therapeutic
strategy used in non-small cell lung cancer, head and neck and
bladder cancer (15–18). In the present study, the results
showed that NAM increased the cytotoxic effects of cisplatin and
radiation on breast cancer cells.
Materials and methods
Cell lines
The MDA-MB-436, MDA-MB-231 and MCF7 human breast
cancer cell lines were obtained from the American Type Culture
Collection (ATCC, Rockville, MD, USA). MDA-MB-436 cells harbor
5396+1G > A (spliced donor site of exon 20), a BRCA1 mutation.
MCF-7 and MDA-MB-231 cells express wild-type BRCA1 while MDA-MB-231
cells are hemizygous for BRCA1, with loss of one allele and
the remaining non-mutated allele containing two non-pathogenic
single-nucleotide polymorphisms. The cell lines were cultured in
DMEM/F-12 medium, 10% fetal bovine serum, penicillin (100 U/ml),
and streptomycin (1.0 mg/ml) at 37°C in a humidified atmosphere of
5% CO2 and 95% air.
PARP activity assay
PARP activity in vitro was assayed using the
Trevigen Universal Chemiluminescent PARP assay kit (Trevigen Inc.,
Gaithersburg, MD, USA) according to the manufacturer’s
instructions. Recombinant PARP was incubated for 1 h with different
NAM doses (0.05, 0.1, 0.2, 0.5, 0.75 and 1 mM) in the presence of
activated DNA. Chemiluminescent detection was performed as per the
manufacturer’s instructions. To assay endogenous PARP activity,
cells were grown in a 25-cm2 cell culture flask in
medium alone or with NAM for 72 h. The cells were washed once with
ice-cold PBS, lysed in 50 μl of PARP buffer containing 0.5 mol/l
NaCl, 1% (v/v) NP-40, and protease inhibitors (Sigma-Aldrich, St.
Louis, MO, USA) on ice for 30 min with occasional vortexing. The
lysates were clarified by centrifugation at 14,000 rpm, at 4°C for
10 min. The protein concentration of the extracts was determined
using the Bradford protein dye reagent (Bio-Rad, Hercules, CA,
USA), and PARP activity was assayed using the Trevigen Universal
Chemiluminescent PARP assay kit according to the manufacturer’s
instructions, with modifications. The lysate (30 μg/well) was added
in duplicate to wells containing PARP buffer and PARP cocktail,
followed by incubation at room temperature for 1 h with NAM at
concentrations of 0.5, 0.75 and 1 mM. Activated DNA was added to
the standards but was omitted from the extracts. The plate was
washed three times with PBS and three times with PBS and 0.1%
Triton X-100, followed by incubation with streptavidin horseradish
peroxidase (1:1,000 dilution) in diluent buffer for 1 h. The plate
was washed again three times with PBS alone and three times with
PBS and 0.1% Triton X-100. Chemiluminescent detection was performed
as per the manufacturer’s instructions. The background reading was
subtracted from the readings of the samples, and PARP activity was
calculated using the standard curve obtained from readings of the
standards.
Immunofluorescence
The MCF-7, MDA-MB-436 and MDA-MB-231 cells were
grown in chamber slides (Sigma-Aldrich) at a 70% of confluence.
Immunostaining for poly (ADP-ribose) (PAR) was performed on cells
fixed in ice-cold 4% paraformaldehyde in PBS for 10 min. The cells
were fixed 1 h after treatment with NAM at concentrations of 0.5,
1, 5 and 10 mM and exposed to H2O2 (1 mmol/l,
10 min, 37°C). Controls were treated with medium alone. The primary
antibody used was anti-pADPR (H10, Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA). Poly ADP-ribosylation immunostaining was
developed following NAM treatment. The secondary antibody used was
the Alexa Fluor 488-conjugated goat anti-mouse IgG antibody
(Molecular Probes, Life Technologies, Carlsbad, CA, USA). Nuclear
counterstaining with DAPI was performed after removal of excess
secondary antibody. Immunostaining was visualized with a Carl Zeiss
confocal multiphotonic laser microscope 780 NLO (Hamburg,
Germany).
Cell viability assay
Cell viability was assessed by crystal violet
staining. Semi-confluent culture flasks were trypsinized and
2.5×104 cells were seeded in 12-well plates. After 24 h,
the cells were exposed to NAM (5, 10, 20, 40, 60, 80 and 100 mM)
and cisplatin (20, 40, 60 and 80 μM) at the indicated
concentrations. After 72 h, the cells were rinsed with PBS, fixed
in 2% formaldehyde for 5 min and stained with 1% crystal violet.
Relative cell viability was obtained by scanning with an ELISA
plate reader at 540 nm.
Data analysis of drug combination
Synergism or additivity was determined by
calculating the combination index (CI) using the equation: CIx =
(D1/Dx1) + (D2/Dx2) + a(D1)(D2)/(Dx1)(Dx2), where CIx is the CI
value for x% effect, Dx1 and Dx2 are the doses of agents 1 and 2
required to exert x% effect alone, and D1 and D2 are the doses of
agents 1 and 2 that elicit the same x% effect in combination with
the other agent, respectively. The factor indicates the type of
interaction: a =0 for mutually exclusive drugs (similar mechanisms
of action), and a =1 for mutually non-exclusive drugs (independent
modes of action), with the equation being resolved for a =1. A CI
of 1 indicates additivity, a CI of <1 synergism and a CI of
>1 antagonism.
siRNA transfection assay
Cells were seeded in 12-well plates Nunc (Thermo
Scientific™ Nunclon, Waltham, MA, USA) at 15×103
cells/well into 0.5 ml of Optimem (Applied Biosystems Life
Technologies, Carlsbad, CA, USA). After 24 h, the cells were
transfected with lipofectamine and PARP1 siRNA (cat no. 4390824;
Ambrion, Minneapolis, MN, USA) or RNAiMAX containing siRNA Scramble
(cat no. 4390844; Ambrion). Cisplatin was added for 24 h and the
cells were cultured at 37°C in humidified atmosphere containing 5%
CO2. After 72 h, the medium was aspirated and the cells
were processed for western blotting and cell viability.
Radiation and drug treatment
Ionizing radiation (IR) of cells was performed at
room temperature in culture medium, using a Theratron Phoenix
(60Co) irradiator (Best Theratronics Ltd., Ottawa, Ontario, Canada)
with an average energy of 1.25 MeV in a field of 20×20
cm2, to a distance isocenter of 80 cm. Cell lines were
irradiated at a range of 0.5–6 Gy in the presence of different
concentrations of NAM (0.5–20 mM). Control cells were treated with
medium only.
Clonogenic assay
Exponentially proliferating cells were plated into a
25-cm2 cell culture flask and incubated for 48 h to
allow cells to reach their optimum proliferation rate. NAM (0.5, 1,
5, 10 and 20 mM) was added to the dishes and incubated for 72 h and
the cells were irradiated. Control cells were not treated. Cells
were collected and cultured in drug-free medium in 60 mm Petri
dishes for up to 21 days, depending on the proliferation rate of
the individual cell line. Colonies were fixed in methanol and
acetic acid (3:1 v/v), stained with crystal violet and counted with
a stereoscopic microscopy [Leica Microsystems, (Schweiz) AG,
Heerbrugg, Switzerland]. Data are expressed as the percentage of
colonies in NAM-treated cultures compared with control cultures.
Lethal concentration 50 (LC50) was calculated for each
cell line in each independent experiment. Each assay was performed
in triplicate for each concentration. Plating efficiencies and the
surviving fractions were calculated.
Statistical analysis
Data are expressed as the mean ± SD values. For
statistical analysis the ANOVA and Bonferroni post-tests were used
to mediate GraphPad Prism 5. P<0.05 was considered to indicate a
statistically significant difference.
Results
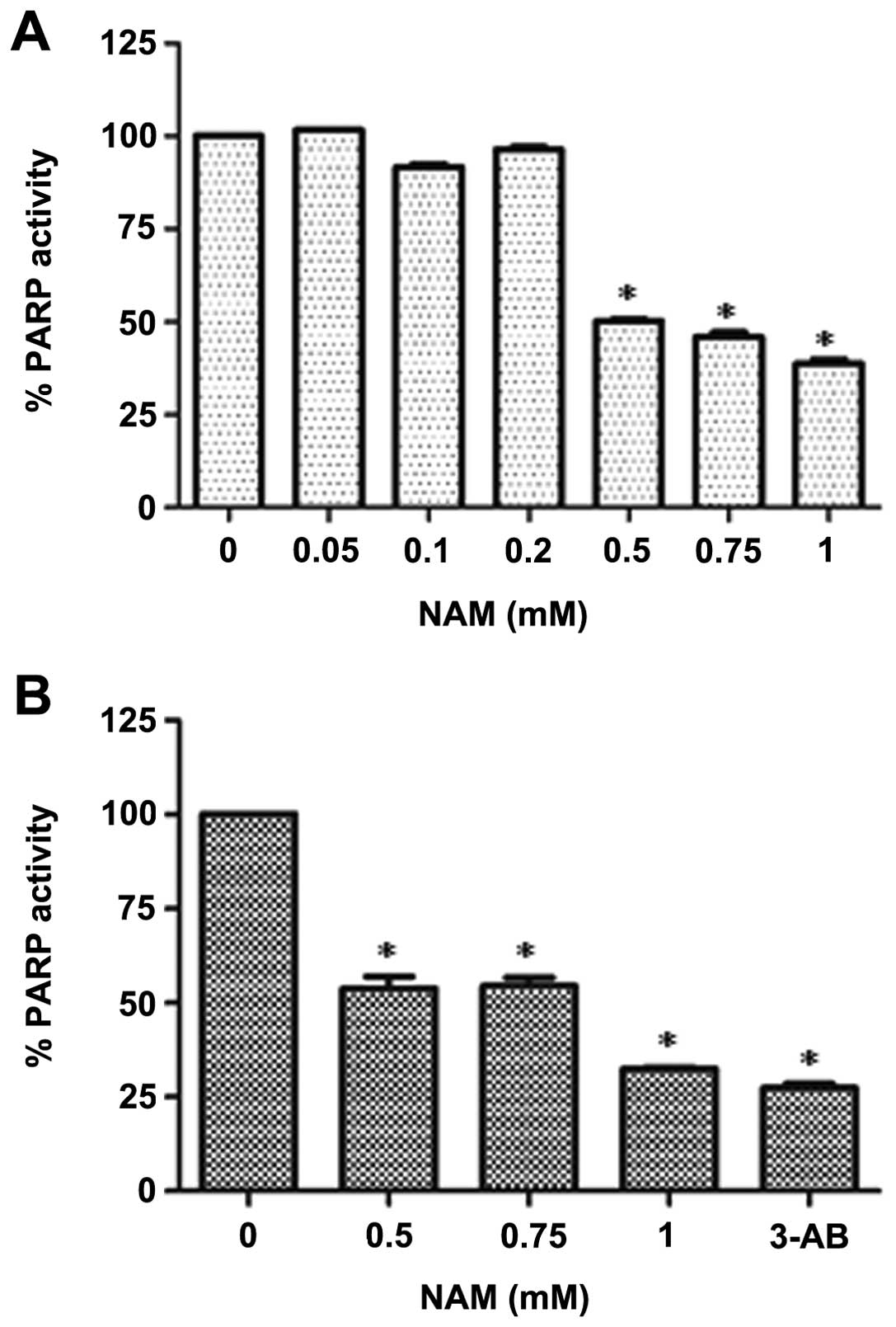
Nicotinamide decreases PARP activity
To examine whether NAM can inhibit PARP activity
in vitro, a cell-free PARP activity assay was performed.
Recombinant PARP was incubated for 1 h at different concentrations
of NAM (50, 100, 200, 500, 750, and 1,000 μM). A significant
dose-dependent decrease of PARP activity, starting at 500 μM was
observed (Fig. 1A). We also
examined whether NAM similarly inhibits PARP cell activity in
MDA-MB-436 breast cancer cells deficient in BRCA1. Whole-cell
lysates were incubated in the presence of nicotinamide adenine
dinucleotide NAD+ and a conjugated histone acceptor
protein, and in the absence of activated DNA, allowing us to
determine already activated PARP. A significant decrease in PARP
activity was observed (Fig.
1B).
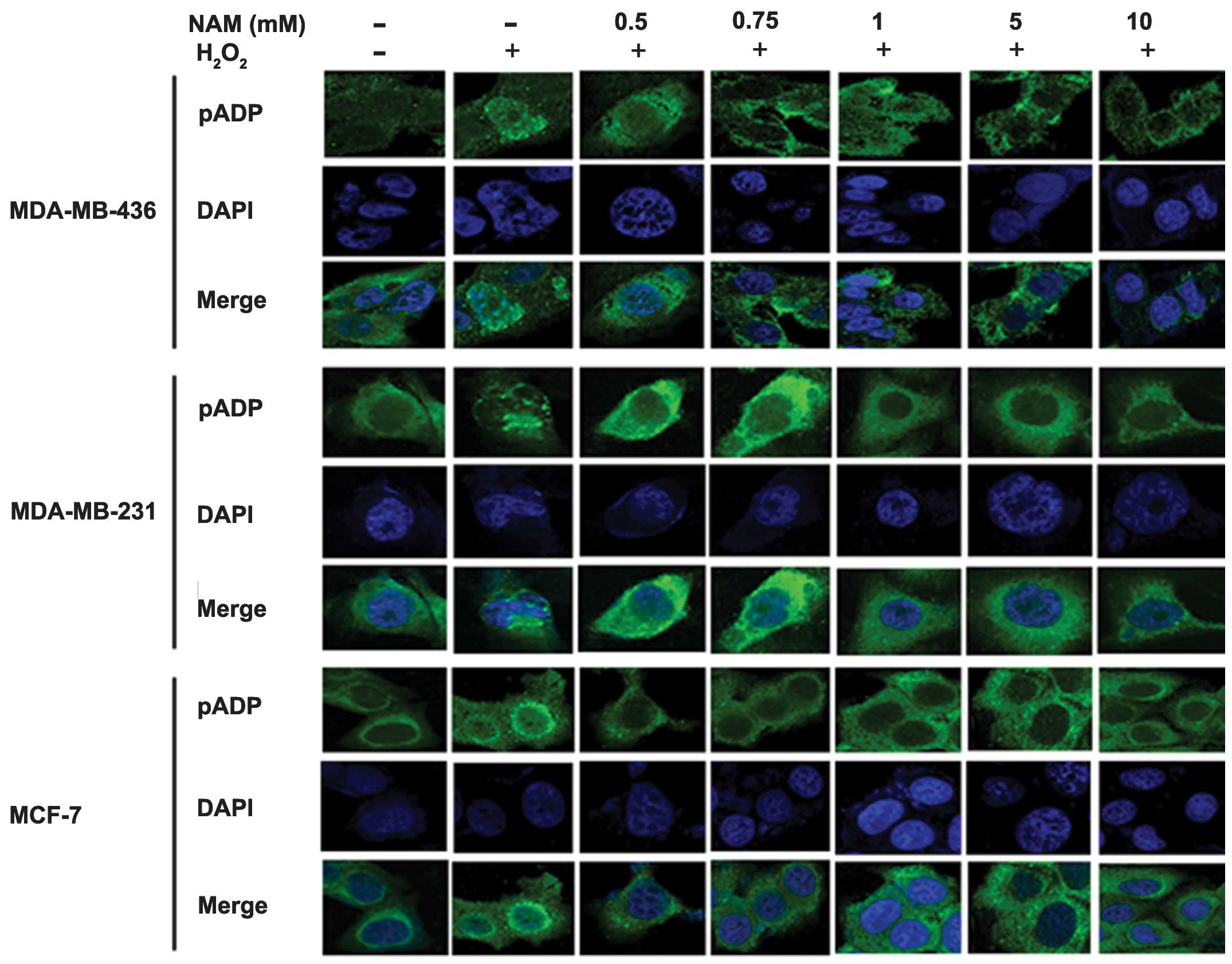
Nicotinamide reduces poly
ADP-ribosylation in the presence of DNA damage
To confirm that NAM inhibits poly-ADP-ribosylation,
we induced DNA damage with H2O2 (10 mmol/l
for 10 min) leading to poly-ADP ribosylation in the three cell
lines after treatment with 0.5, 0.75, 1, 5 and 10 mM of NAM. We
performed a series of immunofluorescent stainings using anti-pADPR.
Fig. 2 shows that poly-ADP
ribosylation was detected in the nuclei of cells treated with
H2O2. Its effect was reduced as a consequence
of NAM treatment in a dose-dependent manner.
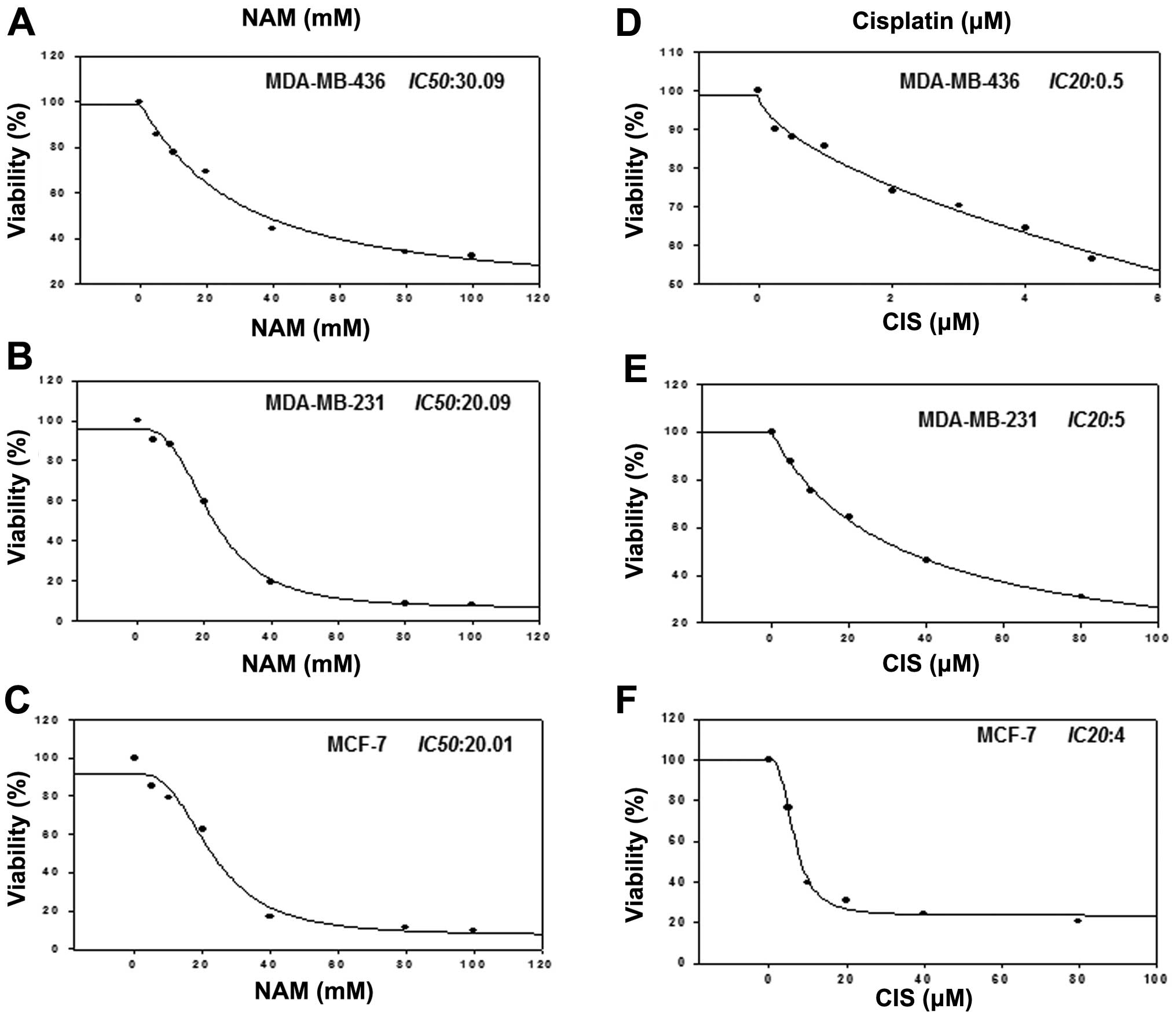
Growth inhibition by nicotinamide and
cisplatin
To determine whether PARP inhibition with NAM
sensitizes cell to cisplatin-induced death, we treated MDA-MB-436,
MDA-MB-231 and MCF-7 cell lines with cisplatin (0–100 μM) and NAM
(5, 10, 20, 40, 60, 80 and 100 mM) and determined the
IC50 value. Fig. 3A–C
shows the IC50 value for NAM in each cell line (20–30
mM). On the other hand, because it has been reported that deficient
BRCA1 cells are more sensitive to agents that induce DNA damage, we
analyzed the effect of cisplatin in three cell lines to obtain the
IC20 value through a dose response curve. The results
demonstrated that MDA-MB-436 cells (Fig. 3D–F), deficient of BRCA1 were more
sensitive than MDA-MB-231 and MCF-7 cells.
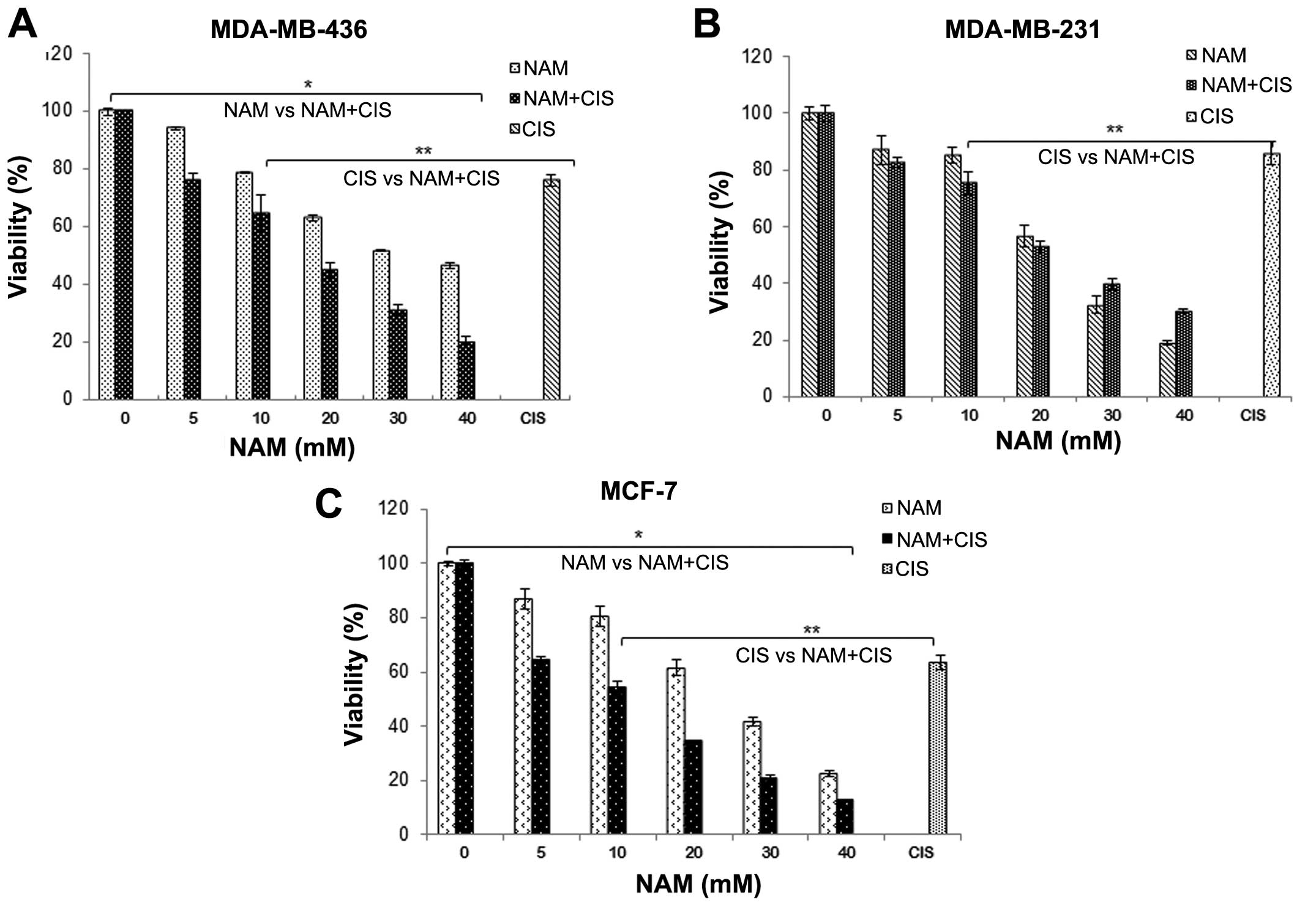
Nicotinamide sensitizes breast cancer
cells to cisplatin toxicity
We also analyzed the effect of co-treatment of NAM
and cisplatin using a lower dose of cisplatin (IC20) and
varying doses of NAM (5, 10, 20 and 40 mM). The results
demonstrated that a combination of NAM and cisplatin significantly
decreased the viability of MDA-MB-436 and MCF-7 cells as compared
to NAM alone (P<0.001, Fig. 4A and
C), while MDA-MB-231 cells showed no significant differences
(Fig. 4B). On the other hand, when
comparing the effect of NAM and cisplatin on the viability of
breast cancer cells, we observed a significantly higher decrease
compared to cisplatin alone (P<0.001). To verify these results
we determined the CI (combination index) using the median-effect
method which revealed a clear synergistic interaction between NAM
and cisplatin (CI <1) in MDA-MB-436 and MCF-7 cells (Table I), whereas MDA-MB-231 cells failed
to show a synergistic interaction.
 | Table ICombination index of nicotinamide and
cisplatin. |
Table I
Combination index of nicotinamide and
cisplatin.
| Cell line | Doses (mM) | | | | | | |
|---|
| Drugs in
combination | Drugs alone | Control growth | Combination index
(Cix) | Interaction |
|---|
| Nicotinamide
(D1) | Cisplatin (D2) | Nicotinamide
(Dx1) | Cisplatin
(Dx2) | (x%) | (ICx) | |
|---|
| MDA-MB-436 | 5 | 0.0005 | 11.1 | 0.0018 | 76 | 0.73 | Synergistic |
| 10 | 0.0005 | 18.65 | 0.0041 | 64 | 0.66 | Synergistic |
| 20 | 0.0005 | 39.14 | 0.11 | 45 | 0.52 | Synergistic |
| 30 | 0.0005 | 121 | 0.12 | 31 | 0.25 | Synergistic |
| 40 | 0.0005 | 122 | 0.13 | 19 | 0.33 | Synergistic |
| MDA-MB-231 | 5 | 0.005 | 7.33 | 0.0082 | 82 | 1.29 | |
| 10 | 0.005 | 10.03 | 0.0133 | 75 | 1.37 | |
| 20 | 0.005 | 20.27 | 0.0345 | 52 | 1.13 | |
| 30 | 0.005 | 28.35 | 0.0516 | 39 | 1.16 | |
| 40 | 0.005 | 35.9 | 30.03 | 30 | 1.28 | |
| MCF-7 | 5 | 0.004 | 14.34 | 0.0055 | 64 | 1.08 | |
| 10 | 0.004 | 19.73 | 0.0082 | 54 | 0.99 | |
| 20 | 0.004 | 34.28 | 0.0166 | 34 | 0.82 | Synergistic |
| 30 | 0.004 | 51 | 0.15 | 20 | 0.62 | Synergistic |
| 40 | 0.004 | 66 | 0.2 | 12 | 0.63 | Synergistic |
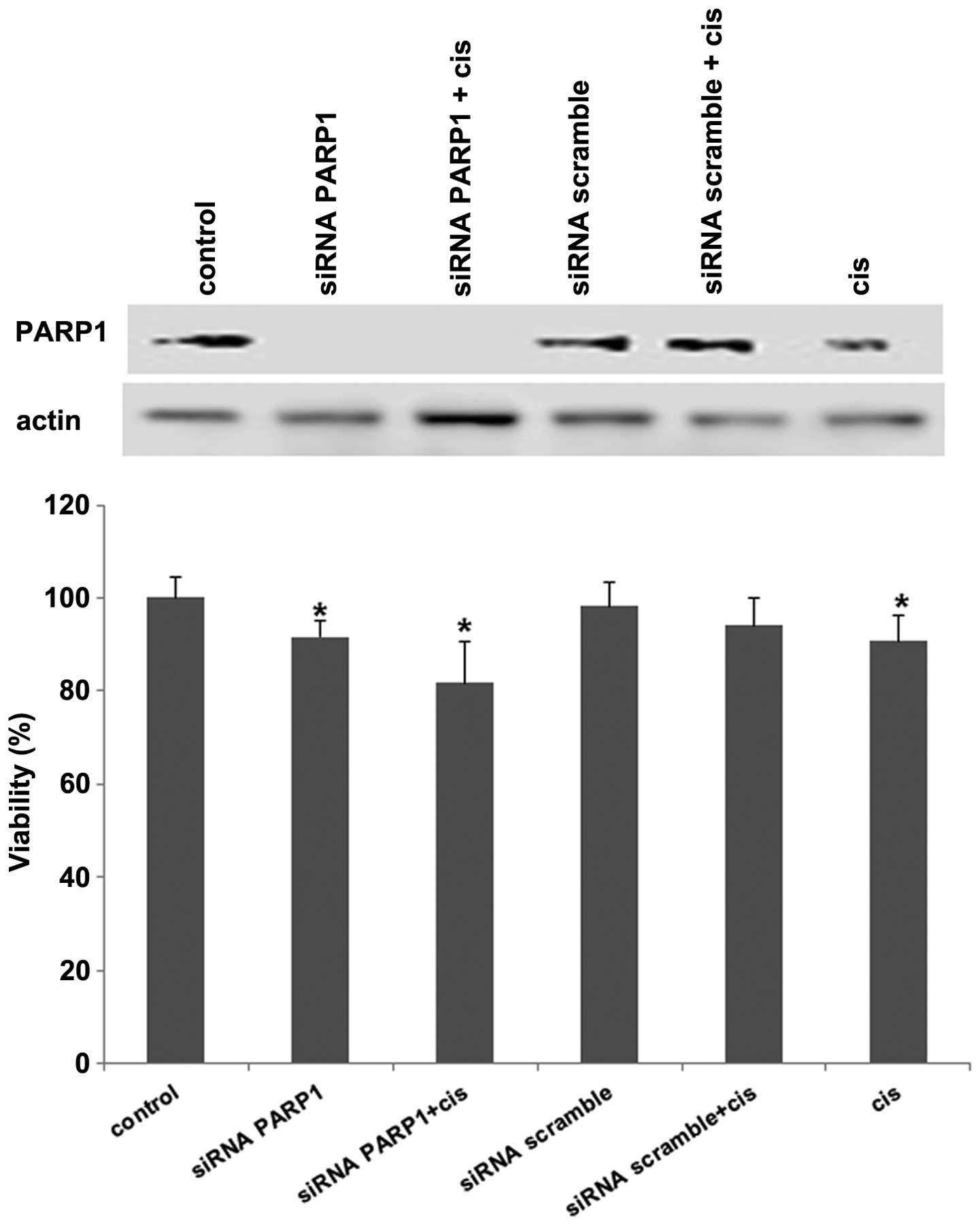
Growth inhibition by PARP1 knockdown is
increased by cisplatin
To determine to what extent the downregulation of
PARP1 inhibited cell growth, MDA-MB-436 were subjected to siRNA
against PARP1. The results showed that at 72 h there was complete
knockdown of mRNA of PARP1 and cell growth was inhibited by 10%.
When cisplatin was added this inhibition increased as compared to
cisplatin alone or PARP1 inhbition. These differences were
statistically significant. No growth effects were observed for
scramble siRNA with or without cisplatin (Fig. 5).
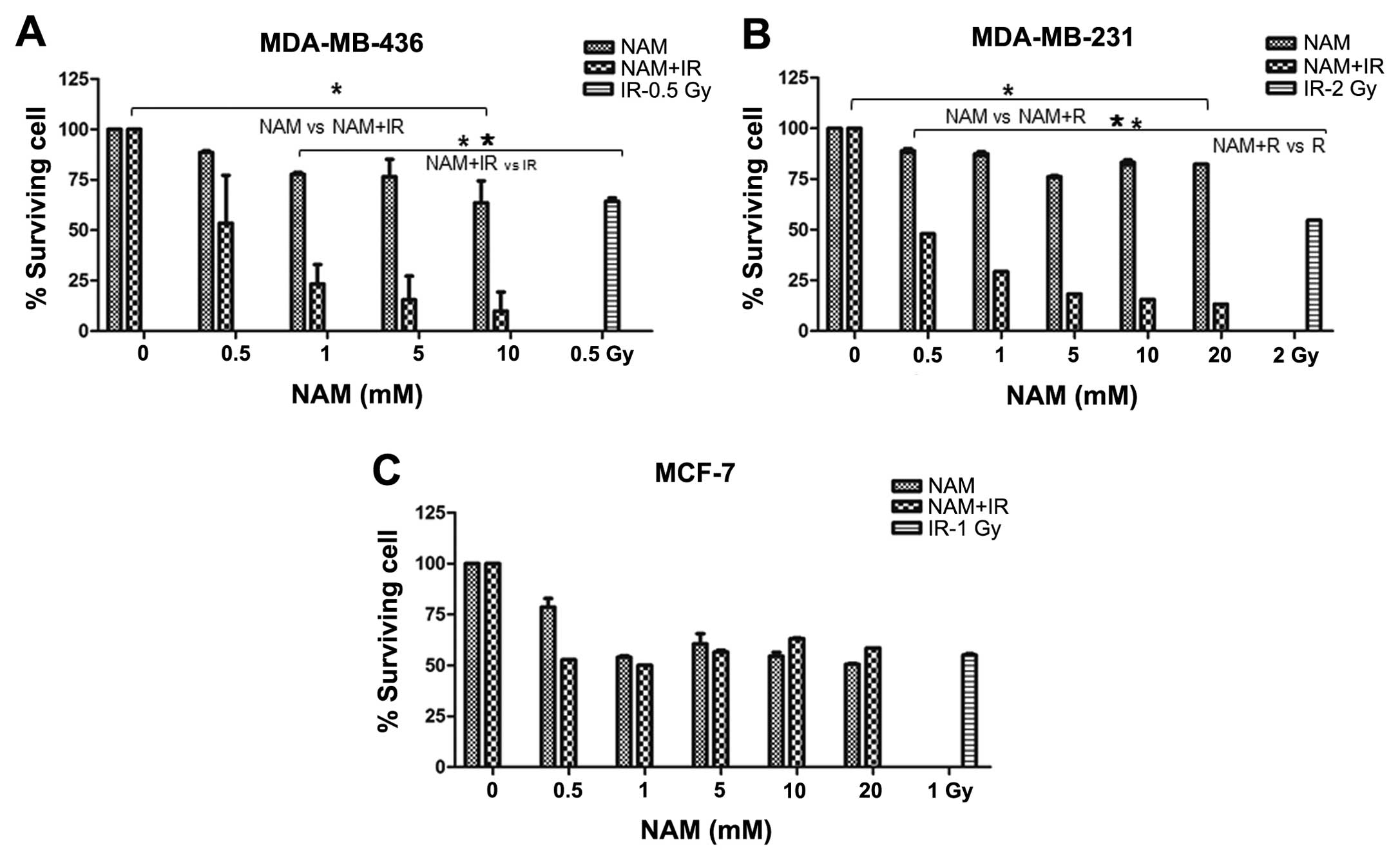
Radiosensitivity in breast cancer cells
by nicotinamide
The effect of NAM in combination with ionizing
radiation (IR) through clonogenic assay was analyzed to determine
the colony-forming ability from a cell after inducing DNA damage.
Survival curves were performed to determine the median lethal doses
(LD50), which were defined as the absorbed dose of ionizing
radiation (Gy) required to induce 50% cell death. The results
demonstrated a different sensitivity to IR in cell lines.
MDA-MB-436 cells were more sensitive to IR than MDA-MB-231 and
MCF-7 cells (data not shown). Subsequently, we analyzed the effect
of NAM combined with IR. A significant decrease in cell survival
levels in MDA-MB-436 and MDA-MB-231 cells exposed to the
combination compared with NAM only was observed (P<0.001,
Fig. 6A and B), while MCF-7 cells
showed no significant differences (P<0.001, Fig. 6C). When comparing the effect of the
combination of NAM and IR versus IR only, we observed a significant
effect on survival (P<0.001, Fig. 6A
and B), while MCF-7 cells showed no significant difference
(P<0.001, Fig. 6C).
Discussion
The results of this study show that NAM inhibits the
growth of breast cancer cell lines in a dose-dependent manner. In
addition, depletion of PARP1 mRNA reduces cell viability and NAM
increases the cytotoxic effects of cisplatin and induces
radiosensitization to various degrees in MDA-MB-436, MDA-MB-231 and
MCF-7 breast cancer cell lines.
A number of PARP inhibitors are being developed in
the clinic as single agents and/or in combination with other drugs
as potential enhancers of DNA-damaging cytotoxic agents, such as
alkylating agents or radiation therapy. The chemistry of most of
these agents is that of reversible NAD+ mimetics,
although they have different bioavailability and molar equivalence
for PARP enzyme inhibition (1,8).
Nevertheless, cancer drug development needs alternative approaches
for drug identification because of increasing failure rates, high
cost, poor safety, limited efficacy, and a lengthy design and
testing process of new entities. In this sense, drug repurposing of
established non-cancer drugs that have anticancer activity provides
an opportunity to rapidly advance therapeutic strategies in
clinical trials (9,10).
Despite the long-established activity of NAM as a
PARP1 inhibitor this drug has not been extensively evaluated as
anticancer agent. However, it has largely been used in the clinic
at pharmacological doses over many years with a low incidence of
side effects and toxicity for diverse conditions including
dermatological, metabolic and psychiatric disorders (19–22).
In cancer, NAM has been used in clinical studies in combination
with accelerated radiotherapy with carbogen and nicotinamide
(ARCON) for radiosensitation (12,16–18).
In the present study, we demonstrate that NAM effectively inhibits
PARP activity in vitro, results that are consistent with
those of previous reports (13). In
addition, we show that NAM inhibits endogenous PARP activity in
extracts from BRCA1-deficient MDA-MB-436 cells. PARP is important
in DNA repair, and our results showed that NAM inhibits
ADP-ribosylation in the presence of DNA damage induced by
H2O2 in a dose-dependent manner in the three
breast carcinoma cell lines examined, suggesting that NAM blocks
DNA repair through PARP-1 inhibition. This effect is observed in
the three breast cancer lines, indicating that its action is
independent of the BRCA1 status. We analyzed whether inhibition of
PARP induced by NAM or knockdown of PARP1 mRNA may have an effect
on cell viability, demonstrating that both experimental conditions
reduce cell viability.
It has been reported that PARP inhibition may
potentiate the effects of antineoplastic DNA-damaging agents such
as temozolomide, cyclophosphamide and platinum in BRCA1-deficient
cells (23). Similarly, AZD2281,
another PARP inhibitor, in combination with cisplatin
synergistically induced cell growth inhibition of breast cancer
cells deficient in BRCA2 (24). In agreement with findings of that
report, our results demonstrate that the combination of NAM with
cisplatin significantly decreased cell viability in the three
breast cancer cell lines regardless of the BRCA1 status. Thus, NAM
induces synthetic lethality in BRCA1-deficient breast cancer cells
as is the case for other PARP inhibitors.
Inhibition of PARP activity reduced the
single-strand breaks (SSBs) repair range and increased sensitivity
to ionizing radiation and antineoplastic agents. As such, PARP
inhibition exerts radiosensitization by facilitating the conversion
of an unrepaired SSB to double-strand breaks (DSBs) during the S
phase of the cell cycle (25). Our
results demonstrate a radiosensitizing effect of NAM (0.5 Gy) in
cells deficient of BRCA1 (MDA-MB-436) and p53
(MDA-MB-231), compared to MCF-7 cells. In agreement with our
results, it was previously reported that MCF-7 cells exhibit
resistance to cell death induced by ionizing radiation caused by
lack of caspase-3 activity (26).
The results of the siRNA demonstrate that depletion
of PARP1 has only a modest effect on reducing cell viability.
However, it is known that NAM exerts a number of biological actions
including inhibition of SIRT1 (silent mating-type information
regulation 2, homolog 1), which is a NAD+-dependent
deacetylase that regulates the processes of stress response and
cell survival (23). Studies have
shown that SIRT1 inhibition by NAM decreases the viability of MCF-7
cells by inducing apoptosis through the activation of caspases
(27) as well as growth inhibition
and chemosensitization to gemcitabine in pancreatic cells (28).
Our findings and those of other studies on the
antitumor effects of NAM in a number of cancer models suggest that
this drug that can be clinically tested as a repositioned cancer
drug. A major drawback for its potential application is that the
antitumor effects of NAM require drug concentrations in millimolar
ranges (>10 mM) when used as single agent. However, when used
for radiosensitization or in combination with cytotoxic drugs, the
effects are seen at lower molar concentrations. Pharmacokinetic
studies in cancer patients receiving oral high-dose NAM show that
at doses of 6 g daily, peak plasma concentrations can be as high as
>200 μg/ml, which corresponds to molar concentrations >2 mM.
No clinical significant toxicity other than easily controlled
nausea and vomiting were observed (29–31).
In summary, further investigation on NAM is required to evaluate
its activity in other tumor models as well as to demonstrate
whether it increases the efficacy of combined chemoradiation.
Acknowledgements
The authors would like to thank Dr Patricia García
López for valuable comments concerning this study as well as M.C.
Roberto Lazzarini for technical assistance. This study was
supported by CONACYT Mexico grant (203457).
References
|
1
|
Davar D, Beumer JH, Hamieh L and Tawbi H:
Role of PARP inhibitors in cancer biology and therapy. Curr Med
Chem. 19:3907–3921. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirai T, Shirai H, Fujimori H, Okayasu R,
Sasai K and Masutani M: Radiosensitization effect of
poly(ADP-ribose) polymerase inhibition in cells exposed to low and
high liner energy transfer radiation. Cancer Sci. 103:1045–1050.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Michels J, Vitale I, Senovilla L, et al:
Synergistic interaction between cisplatin and PARP inhibitors in
non-small cell lung cancer. Cell Cycle. 12:877–883. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
De Soto JA, Wang X, Tominaga Y, et al: The
inhibition and treatment of breast cancer with poly (ADP-ribose)
polymerase (PARP-1) inhibitors. Int J Biol Sci. 2:179–185. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bryant HE, Schultz N, Thomas HD, et al:
Specific killing of BRCA2-deficient tumours with inhibitors of
poly(ADP-ribose) polymerase. Nature. 434:913–917. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Farmer H, McCabe N, Lord CJ, et al:
Targeting the DNA repair defect in BRCA mutant cells as a
therapeutic strategy. Nature. 434:917–921. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Murphy CG and Moynahan ME: BRCA gene
structure and function in tumor suppression: a repair-centric
perspective. Cancer J. 16:39–47. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hilton JF, Hadfield MJ, Tran MT and
Shapiro GI: Poly(ADP-ribose) polymerase inhibitors as cancer
therapy. Front Biosci. 1:1392–1406. 2013. View Article : Google Scholar
|
|
9
|
Gupta SC, Sung B, Prasad S, Webb LJ and
Aggarwal BB: Cancer drug discovery by repurposing: teaching new
tricks to old dogs. Trends Pharmacol Sci. 34:508–157. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dueñas-González A, García-López P, Herrera
LA, Medina-Franco JL, González-Fierro A and Candelaria M: The
prince and the pauper. A tale of anticancer targeted agents. Mol
Cancer. 7:822008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Surjana D, Halliday GM and Damian DL: Role
of nicotinamide in DNA damage, mutagenesis, and DNA repair. J
Nucleic Acids. 2010:pii: 157591. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Knip M, Douek IF, Moore WP, et al:
European Nicotinamide Diabetes Intervention Trial Group Safety of
high-dose nicotinamide: a review. Diabetologia. 43:1337–1345. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Virag L and Szabo C: The therapeutic
potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev.
54:375–342. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen G and Zeller WJ: Reversal of acquired
cisplatin resistance by nicotinamide in vitro and in vivo. Cancer
Chemother Pharmacol. 3:157–162. 1993. View Article : Google Scholar
|
|
15
|
Kaanders JH, Bussink J and Van der Kogel
AJ: ARCON: a novel biology-based approach in radiotherapy. Lancet
Oncol. 3:728–737. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Janssens GO, Rademakers SE, Terhaard CH,
et al: Accelerated radiotherapy with carbogen and nicotinamide for
laryngeal cancer: results of a phase III randomized trial. J Clin
Oncol. 30:1777–1783. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bernier J, Denekamp J, Rojas A, et al:
ARCON: accelerated radiotherapy with carbogen and nicotinamide in
head and neck squamous cell carcinomas. The experience of the
Co-operative group of radiotherapy of the european organization for
research and treatment of cancer (EORTC). Radiother Oncol.
55:111–119. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bernier J, Denekamp J, Rojas A, et al:
ARCON: accelerated radiotherapy with carbogen and nicotinamide in
non-small cell lung cancer: a phase I/II study by the EORTC.
Radiother Oncol. 52:149–56. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Handfield-Jones S, Jones SK and Peachey
RD: Nicotinamide treatment in diabetes. Br J Dermatol. 116:2771987.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Niren NM: Pharmacologic doses of
nicotinamide in the treatment of inflammatory skin conditions: a
review. Cutis. 77(1 Suppl): 11–16. 2006.PubMed/NCBI
|
|
21
|
Kolb H and Burkart V: Nicotinamide in type
1 diabetes. Mechanism of action revisited. Diabetes Care. 22(Suppl
2): B16–B20. 1999.PubMed/NCBI
|
|
22
|
Vallely JF, Lovegrove TD and Hobbs GE:
Nicotinic acid and nicotinamide in the treatment of chronic
schizophrenia. Can Psychiatr Assoc J. 16:433–435. 1971.PubMed/NCBI
|
|
23
|
Jung-Hynes B, Nihal M, Zhong W and Ahmad
N: Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A
target for prostate cancer management via its inhibition? J Biol
Chem. 284:3823–3832. 2009. View Article : Google Scholar :
|
|
24
|
Audrito V, Vaisitti T, Rossi D, et al:
Nicotinamide blocks proliferation and induces apoptosis of chronic
lymphocytic leukemia cells through activation of the
p53/miR-34a/SIRT1 tumor suppressor network. Cancer Res.
71:4473–4483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Evers B, Drost R, Schut E, et al:
Selective inhibition of BRCA2-deficient mammary tumor cell growth
by AZD2281 and cisplatin. Clin Cancer Res. 14:3916–3925. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Löser DA, Shibata A, Shibata AK, Woodbine
LJ, Jeggo PA and Chalmers AJ: Sensitization to radiation and
alkylating agents by inhibitors of poly(ADP-ribose) polymerase is
enhanced in cells deficient in DNA double-strand break repair. Mol
Cancer Ther. 9:1775–1787. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang T, Cui H, Ma N and Jiang Y:
Nicotinamide-mediated inhibition of SIRT1 deacetylase is associated
with the viability of cancer cells exposed to antitumor agents and
apoptosis. Oncol Lett. 6:600–604. 2013.PubMed/NCBI
|
|
28
|
Gong DJ, Zhang JM, Yu M, Zhuang B and Guo
QQ: Inhibition of SIRT1 combined with gemcitabine therapy for
pancreatic carcinoma. Clin Interv Aging. 8:889–897. 2013.PubMed/NCBI
|
|
29
|
Dragovic J, Kim SH, Brown SL and Kim JH:
Nicotinamide pharmacokinetics in patients. Radiother Oncol.
36:225–228. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bernier J, Stratford MR, Denekamp J, et
al: Pharmacokinetics of nicotinamide in cancer patients treated
with accelerated radiotherapy: the experience of the Co-operative
Group of Radiotherapy of the European Organization for Research and
Treatment of Cancer. Radiother Oncol. 48:123–133. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Horsman MR, Hoyer M, Honess DJ, Dennis IF
and Overgaard J: Nicotinamide pharmacokinetics in humans and mice:
a comparative assessment and the implications for radiotherapy.
Radiother Oncol. 27:131–139. 1993. View Article : Google Scholar : PubMed/NCBI
|