Introduction
High mobility group box-1 (HMGB1) is observed in
most tumor types and its expression is higher in gastric
adenocarcinomas (1–3). HMGB1 overexpression is associated with
hallmarks of cancer, including unlimited replicative potential,
vasculogenesis, evasion of apoptosis and insensitivity to growth
inhibitors (4,5). HMGB1 was first identified as a
non-histone chromosomal protein that participates in DNA
replication, transcription and repair in most eukaryotic cells
(6). Studies of the previous decade
have reported that it also functions as an extracellular
damage-associated molecular patterning molecule, promoting
inflammation, cellular differentiation, survival and migration.
Extracellular localization of HMGB1 may be mediated by passive
release from necrotic cells or active secretion by inflammatory
cells (7,8). Recent data show that certain cytotoxic
chemotherapy agents may induce HMGB1 release that contributes to
autophagy regulation (9,10).
Autophagy is a lysosome-mediated, self-degradation
process that protects normal cells, but also promotes tumor cell
survival under stress (11).
Evidence suggests that endogenous HMGB1 is a critical regulator of
autophagy in tumor cells. Cytoplasmic HMGB1 can directly bind to
autophagy protein Beclin-1, disrupting Beclin-1/Bcl-2 interaction
and sustaining autophagy (12).
Endogenous knockdown of HMGB1 with siRNA or inhibition of its
release with small-molecule inhibitors, abolished the protective
effect of autophagy and increased tumor cell sensitivity to several
clinically useful agents (10,13).
Although autophagy may regulate selective HMGB1 release in some
tumor types, the mechanisms of its extracellular release from
gastric cancer cells undergoing autophagy and activation of surface
receptor-mediated intracellular signaling pathways are not well
characterized.
Multiple receptor types have been implicated in
HMGB1 signaling, including the receptor for advanced glycation end
products (RAGE) and members of the Toll-like family of receptors
(TLRs) (14,15). In the present study, we showed that
HMGB1 was highly expressed in human gastric carcinoma and primarily
located in the cytoplasm of gastric cancer cells. Gefitinib, a
common chemotherapeutic agent, activated autophagy and promoted the
release of HMGB1, which was required to maintain gastric tumor cell
survival. Intriguingly, the interaction between HMGB1 and RAGE
initiated signaling involving ERK1/2 phosphorylation and
contributed to the cell proliferation in gastric tumor cells.
Materials and methods
Patients and specimens
Gastric cancer samples and tissues surrounding the
tumor (>2 cm from the tumor edge) were obtained from 23 patients
that underwent curative gastric cancer resection at the Department
of General Surgery in the First Affiliated Hospital of Fujian
Medical University, China. Isolated tumors were only single gastric
neoplasms; no patient received antitumor treatment before surgery.
Specimens were prepared and maintained anonymously according to
ethical and legal standards.
Cell culture and reagents
We used the subsequent cell lines: BGC-823 (low
differentiated stomach adenocarcinoma cell line), SGC-7901
(moderately differentiated stomach adenocarcinoma cell line) and
gastric epithelial cells (GES-1) (immortalized fetal gastric
mucosal cell line), MKN28 (well-differentiated adenocarcinoma cell
line) and MKN45 (poorly differentiated adenocarcinoma cell line).
These cell lines were kindly provided by Dr Yujuan Dong (Department
of Surgery, The Chinese University of Hong Kong, Shatin, Hong Kong,
China). The cell lines were routinely cultured in RPMI-1640 medium
supplemented with 10% fetal bovine serum (FBS) (both from Gibco,
Carlsbad, CA, USA) and maintained at 37°C in a humidified
environment with 5% CO2. Gefitinib was obtained from
Cayman Chemical (Ann Arbor, MI, USA) and glycyrrhizic acid, MTT,
human recombinant HMGB1, rabbit-derived anti-human HMGB1 antibodies
were obtained from Sigma. Neutralizing anti-RAGE, anti-TLR2 and
anti-TLR4 and isotype-matched control (IgG) were from R&D
Systems (Minneapolis, MN, USA), and the GFP-LC3 expression vector
was kindly provided by Professor George G. Chen (Department of
Surgery, The Chinese University of Hong Kong, Shatin, Hong Kong,
China).
Immunohistochemical analysis of HMGB1
expression in gastric cancer tissues
Human gastric cancer tissue sections were
immunolabeled with rabbit anti-human HMGB1 antibodies (1:1,000)
using a mouse/rabbit specific horseradish peroxidase
(HRP)/diaminobenzidine (DAB) detection IHC kit (Abcam, San
Francisco, CA, USA). Sections were incubated with biotinylated goat
anti-rabbit antibodies and ExtrAvidin-conjugated HRP. Staining was
developed with DAB chromogenic substrate and sections were
counterstained with hematoxylin.
Concentration of HMGB1 in serum and cell
supernatant by ELISA
Serum from patients with gastric cancer and healthy
volunteers was assayed. BGC-823 cells were treated with different
doses of gefitinib for 24 h or with 20 μM gefitinib for designated
time periods and the supernatant was collected for HMGB1 detection.
The HMGB1 levels in serum and cell medium were quantified using an
ELISA kit (Chemicon, Temecula, CA, USA) according to the
manufacturer’s instructions.
Immunofluorescence detection of HMGB1
expression in gastric cancer cells
Cells cultured on coverslips were fixated and
processed with primary antibodies (anti-HMGB1, 1:500 dilution)
followed by Alexa Fluor 488 conjugated anti-rabbit IgG (Invitrogen,
Carlsbad, CA, USA). Nuclei were stained with DAPI (Invitrogen).
Images were captured with a fluorescence microscope (Olympus IX81,
Olympus, Tokyo, Japan).
Western blot analysis
Lysates were isolated from the whole tissue
homogenates or gastric cancer cells using a Total Protein
Extraction kit (Millipore, Billerica, MA, USA) and were cytoplasmic
and nuclear protein extracted using a Cytoplasmic and Nuclear
Protein Extraction kit (Promega, Madison, WI, USA) and were
subjected to western blot analysis. Antibodies against HMGB1
(1:8,000; Sigma), LC3B, ERK1/2, phospho-ERK1/2, p38, p38, AKT,
phospho-AKT, JNK, phospho-JNK (1:1,000; Cell Signaling Technology,
Danvers, MA, USA), RAGE (1:200; R&D Systems), β-tubulin,
albumin and lamin B (1:1,000; Santa Cruz Biotechnology Inc., Santa
Cruz, CA, USA) were used to develop immunoreactive signals.
Densitometry was performed using AlphaImager 2200 system and
Quantity software.
GFP-LC3 analysis
BGC-823 cells were grown on coverslips and
transfected with the GFP-LC3 vector using X-tremeGENE HP DNA (Roche
Applied Science). Twenty hours later, the cells were treated with
the selected agents for 24 h. Autophagic vesicles were monitored by
GFP-LC3 aggregation in stably expressing polyclonal cell lines. A
percentage of the cells with >10 GFP-LC3 puncta/100 cells from
the two experiments were investigated using a Laser Scanning
Confocal Microscope (Leica TCS SP5; Leica, Mannheim, Germany).
Culture medium preparation
BGC-823 cells (2×106) were cultured in a
medium containing 20 μM gefitinib or DMSO for 24 h. The culture
media were collected and used in a mixture with fresh media to
treat BGC-823 cells for indicated periods. Cell proliferation was
assessed via MTT assay.
Cell growth assessment
Cell proliferation was analyzed with an MTT assay.
Cells incubated with a medium containing different concentrations
of HMGB1 or a mixture of cultured media were treated with 20 μl MTT
dye (5 mg/ml) at 24, 48, 72 and 96 h. Optical density was
determined at 570 and 630 nm using ELISA (Bio-Tek Instruments,
Inc., Winooski, VT, USA). For the colony formation assay, BGC-823
colonies that contained >50 cells were counted and stained with
0.1% of crystal violet.
RNA interference (siRNA)
RAGE siRNA (Shanghai GenePharma Co., Ltd., Shanghai,
China) was transfected into the cells using X-tremeGENE siRNA
(Roche Applied Science) according to the manufacturer’s
protocol.
Statistical analysis
All the results are expressed as mean ± SEM.
Differences between the two groups were analyzed using a Student’s
t-test and among multiple groups by a one-way analysis of variance
with a Dunnett’s multiple comparison post hoc test. A two-way ANOVA
followed by Dunnett’s test was performed for multiple comparisons.
P<0.05 was considered statistically significant. Statistical
analyses were performed using GraphPad Prism 6.0 (GraphPad
Software, San Diego, CA, USA).
Results
HMGB1 is overexpressed in gastric
cancer
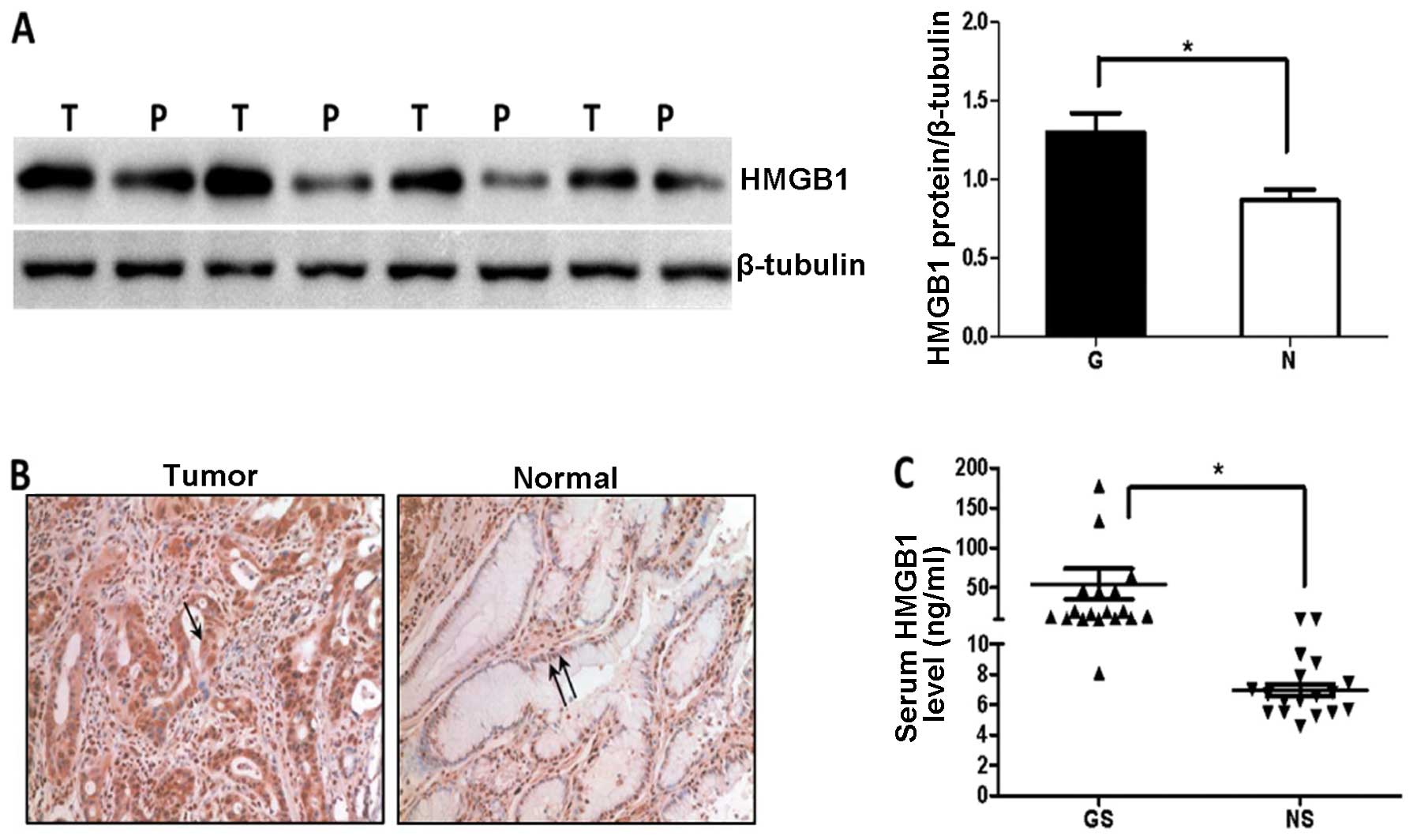
We first examined the amount of HMGB1 in 8 gastric
cancer tissue samples and corresponding non-tumor gastric tissues
by immunoblotting analysis. Expression of HMGB1 protein was
significantly higher in the tumor compared to that in the peritumor
tissues (P=0.0101, Fig. 1A). HMGB1
was predominantly localized in the tumor cell cytoplasm, while low
expression was detected mainly in the peritumor cell nuclei
(Fig. 1B). HMGB1 was actively
released into the circulation of patients with gastric cancer and
serum levels were significantly increased in gastric cancer
patients compared to the level in the healthy volunteers (Fig. 1C).
Overexpression and cytoplasmic
localization of HMGB1 in gastric cancer cells
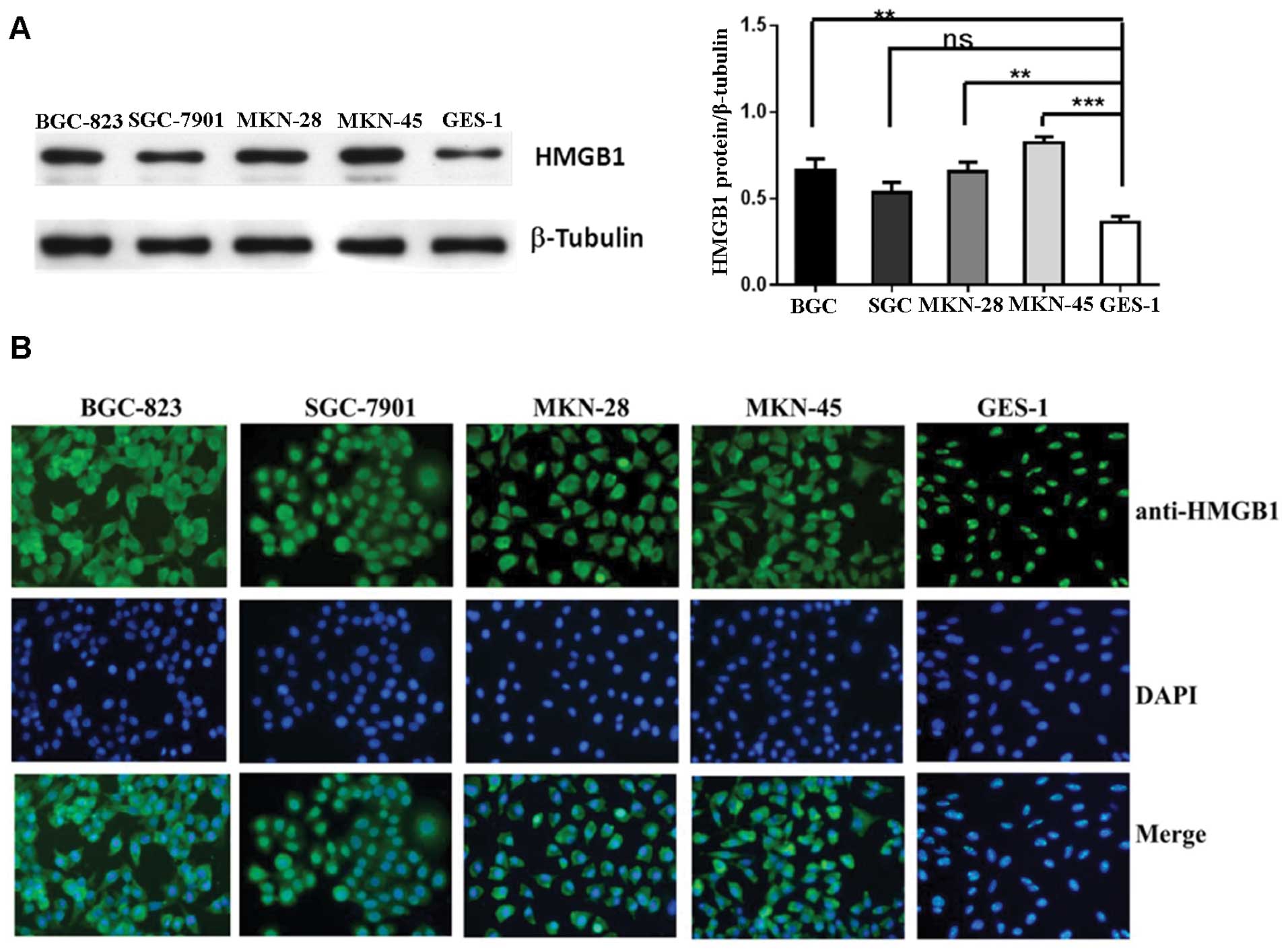
Western blot analysis and immunofluorescence were
performed in four gastric cancer cell lines and non-malignant GES-1
cells and showed that HMGB1 protein expression was much higher in
the cancer cells compared to the level in the GES-1 cells (Fig. 2). HMGB1 was absent in the cytoplasm
of the non-malignant gastric epithelial cells, whereas cytoplasmic
HMGB1 was abundant in all four cancer cell lines. High
extracellular and cytoplasmic levels of HMGB1 suggest that its
detection may occur in the context of active HMGB1 release.
Autophagy induces HMGB1 release from
gastric cancer cells
HMGB1 is released from tumor cells undergoing
classical necrotic cell death. Recent discoveries suggest that
autophagy regulates selective HMGB1 release in tumor cells
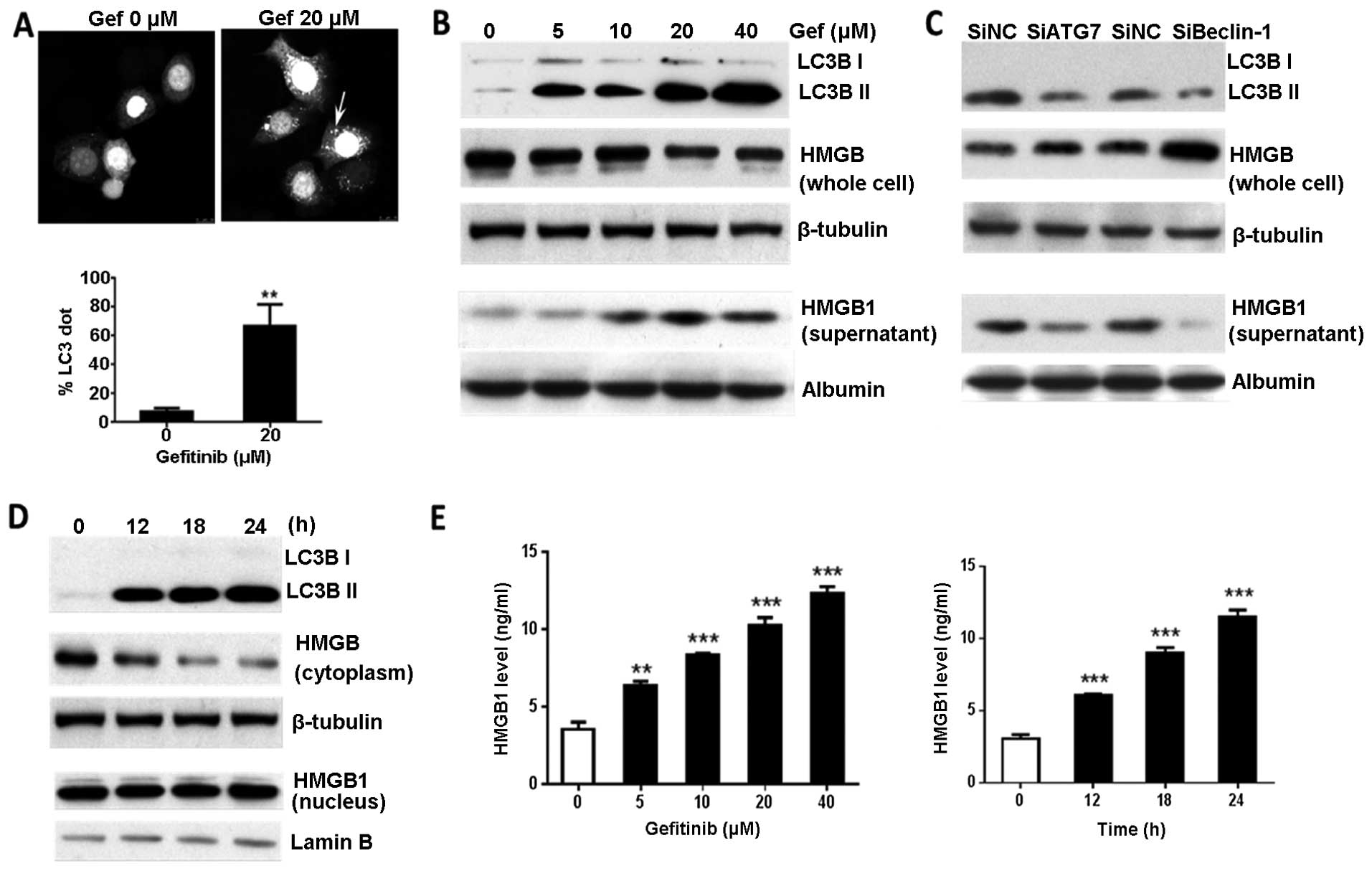
(8,16). In the present study, we observed
that gefitinib, an epidermal growth factor receptor (EGFR)
inhibitor, activated autophagy in the gastric cancer cells, as
indicated by LC3-positive puncta and increased the levels of the
autophagosome-bound form of LC3, LC3 II (Fig. 3A and B). Gefitinib-induced autophagy
triggered a dose-dependent increase in HMGB1 release into the media
of the BGC-823 cells. Western blot analysis showed that HMGB1
protein was reduced in the BGC-823 cells and increased in the
supernatants, after treatment with gefitinib (Fig. 3B). Knockdown of Atg7 or Beclin-1
prevented HMGB1 release from the gefitinib-treated cells suggesting
regulation by autophagy (Fig. 3C).
At 12 h post-gefitinib treatment, cytoplasmic HMGB1 levels
declined, however, nuclear HMGB1 expression showed no significant
change by 24 h after treatment (Fig.
3D). Furthermore, we utilized ELISA to quantify the HMGB1
released in culture supernatants. Consistent with the western blot
analysis results, the HMGB1 released from the gefitinib-treated
cells was significantly increased in a time-and dose-dependent
manner compared with that released in the untreated cells (Fig. 3E). This demonstrated that HMGB1,
particularly the cytoplasmic HMGB1, was rapidly released and
accumulated in the culture media of the gefitinib-treated gastric
tumor cells.
Effect of extracellular HMGB1 on gastric
cancer cell proliferation
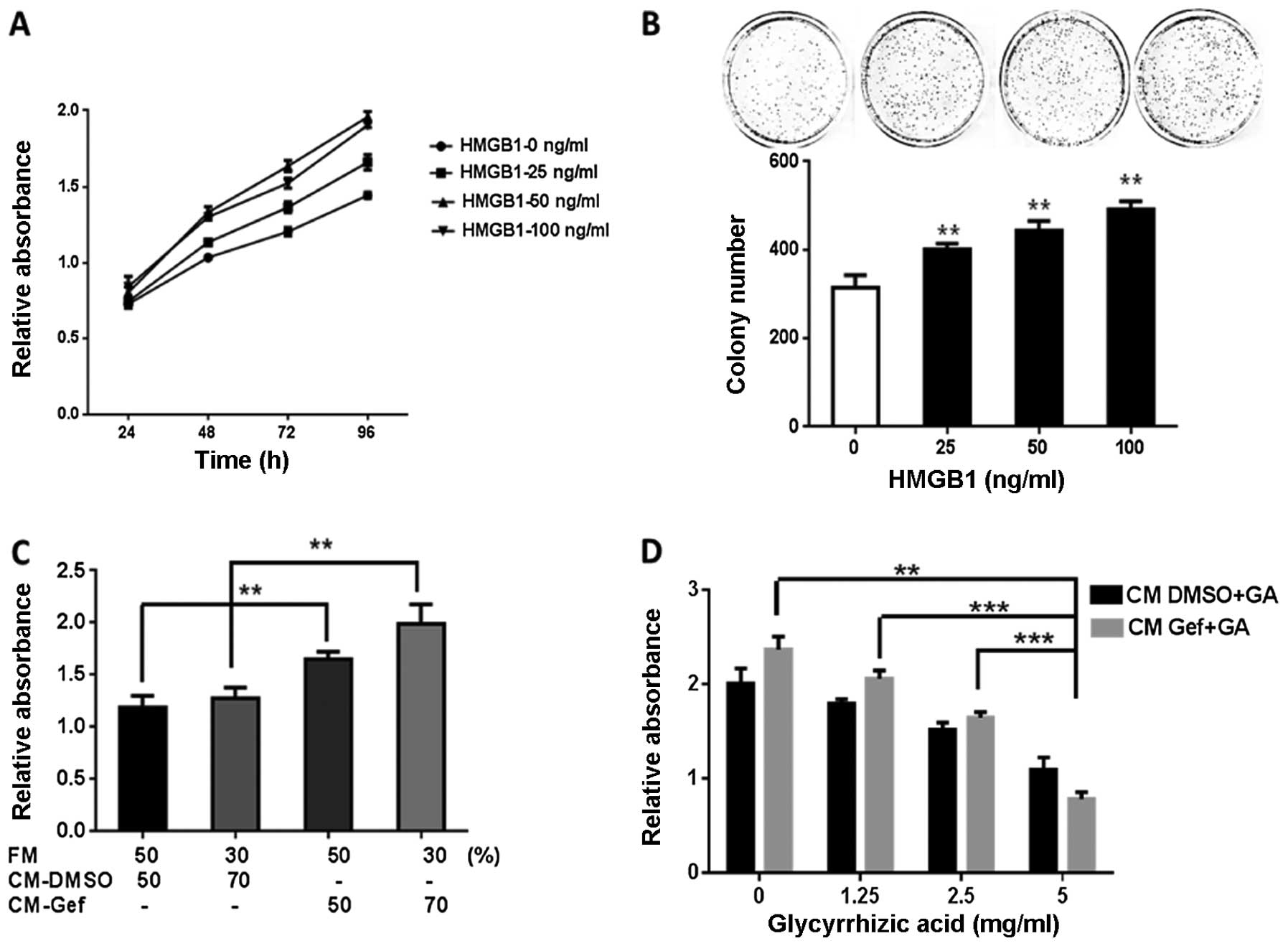
The extracellular HMGB1 effects on cell
proliferation and growth were examined via MTT and colony formation
assays in BGC-823 cells. Human recombinant HMGB1 significantly
enhanced the cell proliferation in a dose- and time-dependent
manner compared to that in the control group (Fig. 4A). The colony formation rate of the
BGC-823 cells was higher in the HMGB1-treated vs. the control group
(Fig. 4B). The proliferative effect
of the natural HMGB1 released by autophagy was assessed in the
BGC-823 cells cultivated with a gefitinib-treated cell medium, with
or without glycyrrhizic acid, a known inhibitor of HMGB1 (17). Cells cultivated with
gefitinib-treated cell medium demonstrated an enhanced growth
compared to the DMSO-treated controls (Fig. 4C and D). Glycyrrhizic acid
noticeably suppressed the growth promoting-effect of gefitinib
(Fig. 4D). These findings indicated
that glycyrrhizic acid bound to HMGB1, released by cells undergoing
autophagy and attenuated HMGB1-induced tumor proliferation.
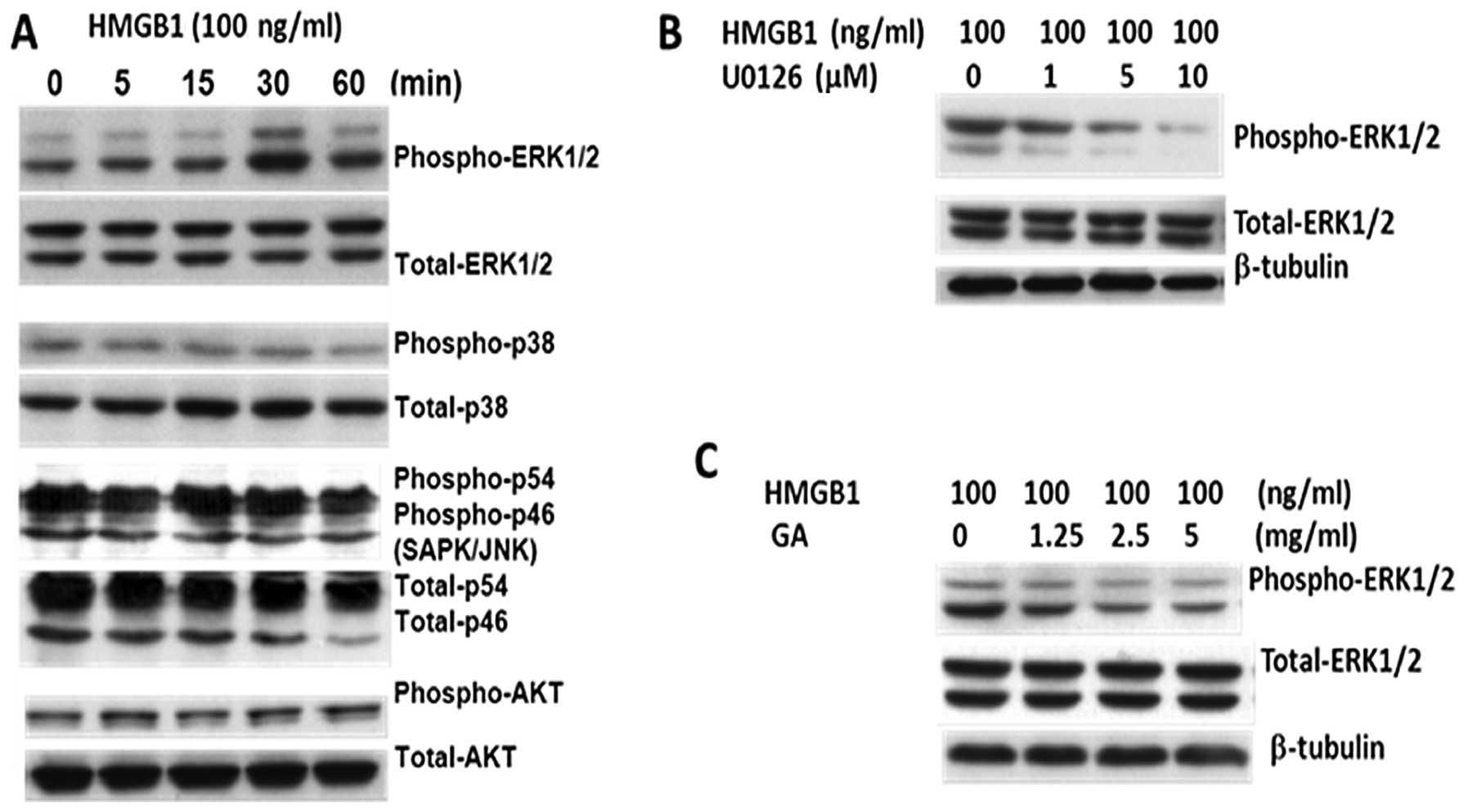
HMGB1 induces ERK activation in gastric
tumor cells
HMGB1 activates multiple signaling pathways involved
in cell proliferation, including mitogen-activated protein kinase
(MAPK), AKT and JNK pathways (14,18,19).
BGC-823 cells were incubated with human recombinant HMGB1 for
different times and harvested for analysis of ERK1/2
phosphorylation via western blot analysis. HMGB1 induced ERK
activation in a time-dependent manner (Fig. 5A); however, p38 MAPK, AKT and JNK
phosphorylation and activation were not detected (Fig. 5A). Treatment with U0126 (MEK1 and
MEK2-specific inhibitor) and glycyrrhizic acid significantly
reduced the ERK1/2 phosphorylation (Fig. 5B and C). Therefore, HMGB1 induced
cell proliferation in gastric cancer cells via activation of the
MEK/ERK signaling pathway.
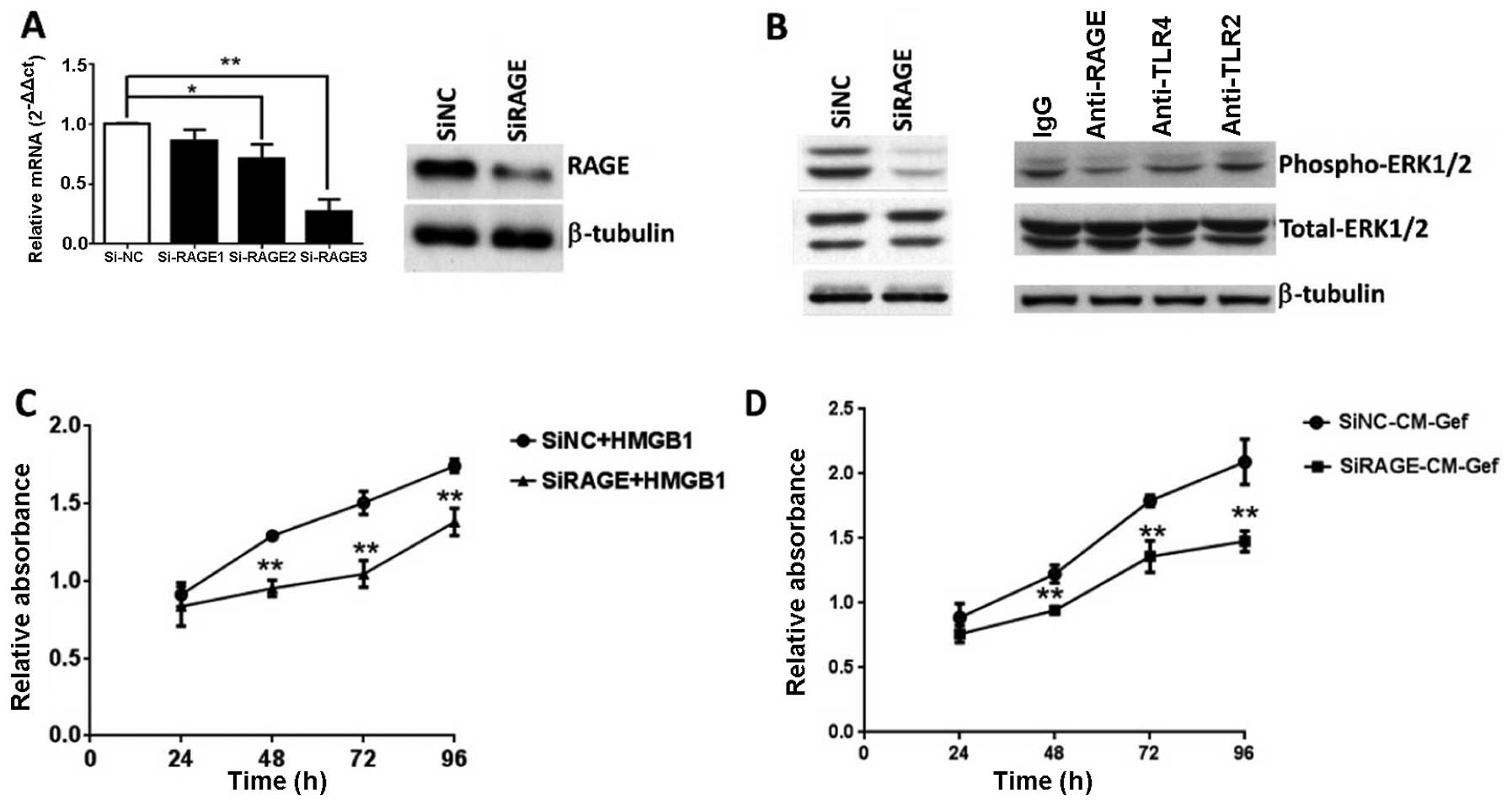
RAGE is required for HMGB1-induced MAPK
activation and gastric cancer cell proliferation
Several receptors have been linked to HMGB1
signaling, including RAGEs and TLRs (14,15).
We analyzed TLR2, TLR4 and RAGE gene expression in human gastric
tumor cells using quantitative real-time PCR. The three receptors
were expressed in multiple gastric tumor cell lines in
vitro. Interestingly, RAGE expression was greater in the
BGC-823 cells, where HMGB1 was relatively highly expressed (data
not shown). To assess the contribution of RAGE to ERK activation
and tumor growth, siRNA was designed to silence RAGE. RAGE
expression was significantly reduced in the siRAGE group as long as
96 h post-transfection, which covered the maximal duration of the
cell proliferation assay (Fig. 6A).
RAGE knockdown almost completely abrogated ERK phosphorylation in
response to HMGB1 (Fig. 6B). To
confirm the interaction of extracellular HMGB1 and its receptor,
anti-TLR2, anti-TLR4 and anti-RAGE antibodies were used to block
the respective receptors in BGC-823 cells. Consistent with the RAGE
knockdown results, anti-RAGE antibodies significantly lowered the
ERK response to HMGB1, although anti-TLR2 and anti-TLR4 antibodies
demonstrated no inhibition of ERK response (Fig. 6B). Thus, HMGB1-induced ERK
activation is dependent on RAGE. Cells subjected to different
treatments were analyzed via MTT assay to investigate the RAGE
involvement in HMGB1-induced cell proliferation. Compared to the
siRNA-control, the RAGE-reduced BGC-823 cells displayed markedly
decreased proliferation in response to recombinant HMGB1 or mixed
medium (a mixture of gefitinib-treated cell medium and fresh medium
in a ratio of 7:3) (Fig. 6C and D).
Extracellular HMGB1 interacted with RAGE and activated ERK
signaling responsible for gastric cancer cell proliferation.
Discussion
Increased HMGB1 is observed in many cancer types,
including prostate cancers, leukemia, colorectal and hepatocellular
cancer and is related to occurrence, progression, and metastasis
(10,20–22).
HMGB1 expression has also been described in gastric epithelial
cancer (1–3). Nearly all gastric adenocarcinomas show
HMGB1-positive labeling, primarily in the nucleus and HMGB1 in
gastric cancer cells may be significantly increased compared to
that in the epithelial and stromal cells in normal tissues
(1). Chung et al (2) reported that the serum HMGB1 levels
were higher than normal in patients with gastric cancer, while a
positive correlation was observed between serum levels and the
depth of invasion, lymph node metastasis, tumor size and poor
prognosis. Similar results were obtained in the present study. We
observed that the HMGB1 expression in gastric cancer tissues was
increased compared to the non-cancerous tissues, while the serum
HMGB1 levels in cancer patients were higher than that in the
healthy volunteers (Fig. 1).
Gastric carcinoma cell lines (BGC-823, SGC-7901, MKN-28 and MKN-45)
exhibited high HMGB1 levels in both the nuclei and cytoplasm,
whereas gastric epithelial cells showed a reduced HMGB1 level,
primarily localized to the nucleus (Fig. 2). High serum HMGB1 levels in cancer
patients and predominant cytoplasmic localization indicate that
HMGB1 can be actively released into the circulation.
HMGB1 is actively secreted from activated innate
immune cells or passively from cells undergoing classical necrotic
cell death (4). Recently, it was
observed that HMGB1 was selectively released from tumor cells
undergoing autophagy (8,16). Evidence suggests that HMGB1 may
induce autophagy in cancers associated with increased sensitivity
to cytotoxic anticancer agents (10). Contrarily, HMGB1-mediated autophagy
may protect gastric cancer cells from the chemotherapeutic vinca
alkaloid, vincristine (23). In the
present study, data indicated that the protective effects of HMGB1
occurred through its upregulation of the protein myeloid cell
leukemia-1 (Mcl-1). Other studies suggest that vincristine may
reduce Mcl-1 expression and promote the death of cancer cells
(24), complicating the
interpretation of our findings. Autophagy, a process in which
subcellular membranes undergo dynamic morphological changes
resulting in intracellular degradation of proteins, cytoplasmic
organelles and pathogens, is a mechanism exploited by tumor cells
for survival and used in determining tumor response to anticancer
therapy. Increasing evidence suggests that autophagy represents a
resistant mechanism to chemotherapy in many malignancies and our
findings support this notion. Here we observed that BGC-823 cells
(an EGFR-rich human gastric carcinoma cell line) were resistant to
the EGFR tyrosine kinase inhibitor, gefitinib. The IC50
value of gefitinib for growth inhibition of BGC-823 cells was
92.83±1.92 μM (data not shown).
To investigate the effect of gefitinib on autophagy,
we employed transiently expressed GFP-LC3 in BGC-823 cells and
quantified puncta formation. Gefitinib (20 μM) increased autophagic
flux (Fig. 3A) and increased
autophagosome-bound LC3II in the BGC-823 cells in a dose- and
time-dependent manner (Fig. 3B and
D). We then investigated the HMGB1 levels in the BGC-823 cell
supernatants and homogenates after treatment with low doses of
gefitinib (10, 20 and 40 μM), which did not affect the cell
viability. HMGB1 accumulated rapidly in the culture medium and was
slightly reduced in the homogenates after adding gefitinib. The
increased levels of extracellular HMGB1 in the gefitinib-treated
supernatants were confirmed by ELISA. Unlike dying cells, no
significant release of lactate dehydrogenase (LDH) was detected in
the autophagic cells induced by gefitinib (data not shown).
Moreover, gefitinib-induced HMGB1 release was prevented by
knockdown of Atg7 or Beclin-1 in the BGC-823 cells (Fig. 3C), which confirmed HMGB1 release due
to increased autophagy. HMGB1 levels in the cytoplasm and nucleus
were assessed at different time-points following gefitinib
treatment and cytoplasmic levels were decreased at 12 h
post-treatment. However, HMGB1 levels did not change after the DMSO
treatment (Fig. 3D). These data
show that HMGB1, particularly the cytoplasmic HMGB1, is rapidly
released and accumulated in the culture media after
gefitinib-induced autophagy in gastric cancer cells.
Autophagy is a cell survival mechanism in many types
of malignant tumors (25,26). Kang et al (27) found that intracellular HMGB1
regulates autophagy through interaction with Beclin-1, in
competition with Bcl-2, indicating its functional importance in
cross-regulating apoptosis and survival. However, detailed
functions of extracellular HMGB1 remain largely undefined. Our data
showed that exogenous HMGB1 induced a dose-dependent upregulation
of BGC-823 cell proliferation (Fig. 4A
and B). Treatment with a medium containing HMGB1 released from
the autophagic cells resulted in enhanced cell growth. Therefore,
HMGB1 released by autophagic cells serves as a pro-survival signal
for residual cells.
HMGB1 acts as a growth factor for cancer cells, it
activates MAPK or PI3K/AKT signaling and enhances proliferation via
RAGE receptor activation (18,28,29).
Our findings confirmed that exogenous HMGB1 increased ERK1/2
phosphorylation, with no effect on the phosphorylation of p38, JNK
and PI3K/AKT pathways in BGC-823 cells (Fig. 5A). HMGB1-induced ERK1/2 activation
was blocked by pretreatment with either U0126, an MEK1/2 inhibitor,
or glycyrrhizic acid, an HMGB1 inhibitor. Therefore, we propose
that extracellular HMGB1 regulates cell proliferation through
MEK-ERK signaling. Recently, numerous studies indicated that RAGE,
a multi-ligand receptor for certain stress-associated factors,
affected the proliferation of various types of cancer cells
(14,29,30).
Consequently, we employed siRNA to silence RAGE and investigate its
function in our experimental model (Fig. 6A). ERK activation was partially
reversed in the RAGE-reduced BGC-823 cells (Fig. 6B). Anti-RAGE antibody, but not
anti-TLR2 and anti-TLR4 antibodies, significantly inhibited the ERK
response to HMGB1 (Fig. 6B). RAGE
knockdown also suppressed HMGB1-induced cell proliferation
validating our presumption that HMGB1/RAGE interaction modulates
gastric cancer cell proliferation. Therefore, we deduce that
exogenous HMGB1 binds to RAGE and initiates MEK/ERK signal
transduction, a process that may play a crucial role in cancer cell
survival and resistance to chemotherapy.
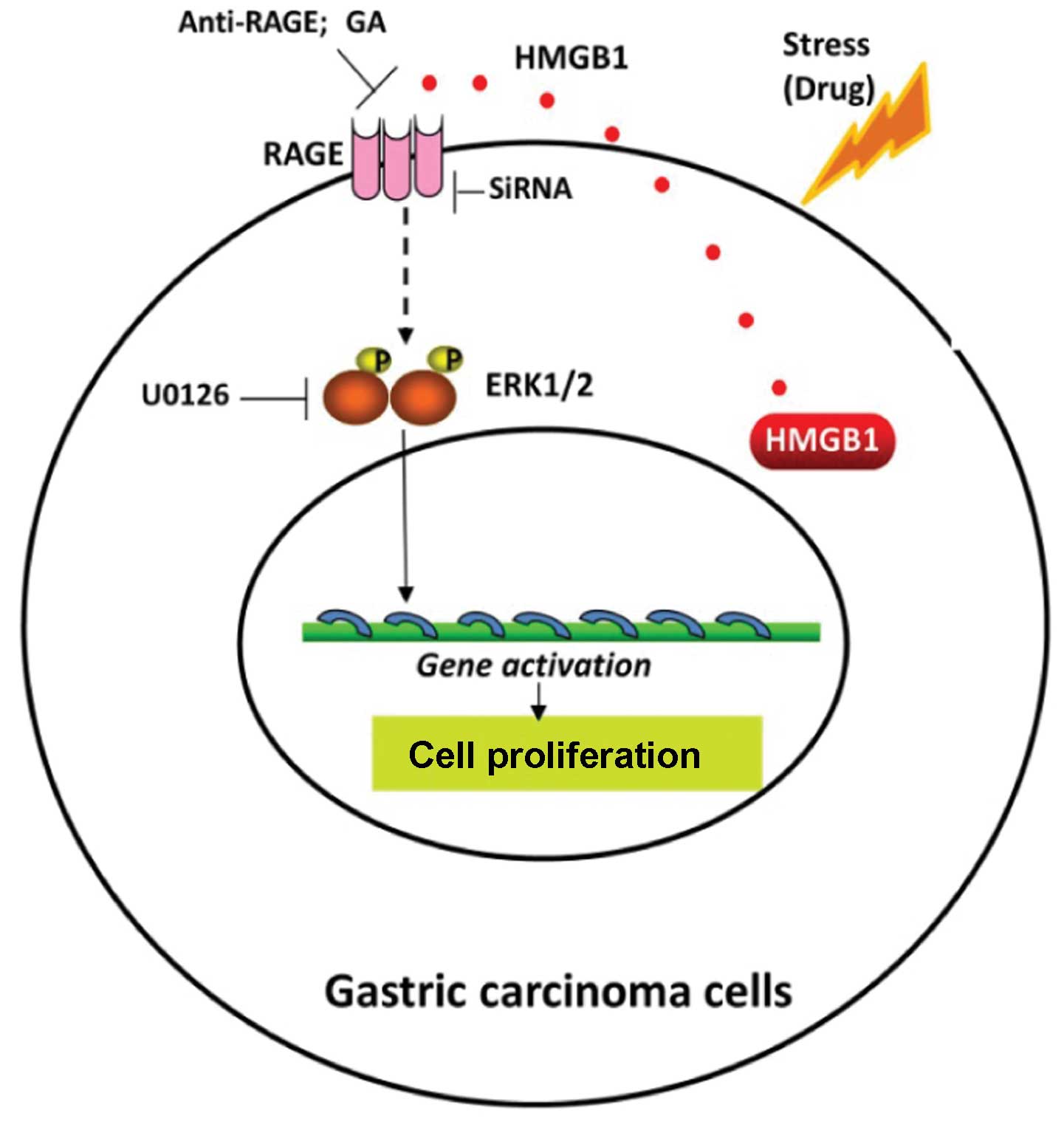
In conclusion, the present study demonstrated
(Fig. 7) that HMGB1 was highly
expressed, particularly within the cytoplasm of human gastric
carcinoma cells. As a regulator of cell death and survival, HMGB1
was released from the tumor cells undergoing gefitinib-induced
autophagy, bound with RAGE and initiated signaling involving
phosphorylation of ERK1/2, which contributed to gastric tumor cell
proliferation. Thus, we propose HMGB1 release as a pro-survival
signal for residual cells following various cytotoxic cancer
treatments. HMGB1 inhibitors or RAGE suppressants may be effective
in prohibiting cancer regrowth, supported by HMGB1-related
autophagy during chemotherapy. Such methods may be considered for
future chemotherapy protocols to increase their efficacy in human
gastric adenocarcinoma and other epithelial neoplasms.
Acknowledgements
The present study was supported in part by a grant
from the National Natural Science Foundation of China (no.
81101558), the Fujian Provincial Committee of Natural Science and
Technology (no. 2012J01364) and the fund for the Doctoral Program
of Higher Education of China (no. 20113518120002). The project was
also supported by the Training Program of Fujian Excellent Talents
in the University (FETU).
References
|
1
|
Bao G, Qiao Q, Zhao H and He X: Prognostic
value of HMGB1 overexpression in resectable gastric
adenocarcinomas. World J Surg Oncol. 8:522010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chung HW, Lee SG, Kim H, et al: Serum high
mobility group box-1 (HMGB1) is closely associated with the
clinical and pathologic features of gastric cancer. J Transl Med.
7:382009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Akaike H, Kono K, Sugai H, et al:
Expression of high mobility group box chromosomal protein-1
(HMGB-1) in gastric cancer. Anticancer Res. 27:449–457.
2007.PubMed/NCBI
|
|
4
|
Sims GP, Rowe DC, Rietdijk ST, Herbst R
and Coyle AJ: HMGB1 and RAGE in inflammation and cancer. Annu Rev
Immunol. 28:367–388. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kang R, Zhang Q, Zeh HJ III, Lotze MT and
Tang D: HMGB1 in cancer: good, bad, or both? Clin Cancer Res.
19:4046–4057. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bianchi ME and Beltrame M: Upwardly mobile
proteins. Workshop: the role of HMG proteins in chromatin
structure, gene expression and neoplasia. EMBO Rep. 1:109–114.
2000. View Article : Google Scholar
|
|
7
|
Wang H, Bloom O, Zhang M, et al: HMG-1 as
a late mediator of endotoxin lethality in mice. Science.
285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Scaffidi P, Misteli T and Bianchi ME:
Release of chromatin protein HMGB1 by necrotic cells triggers
inflammation. Nature. 418:191–195. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tang D, Kang R, Cheh CW, et al: HMGB1
release and redox regulates autophagy and apoptosis in cancer
cells. Oncogene. 29:5299–5310. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu L, Yang M, Kang R, et al:
HMGB1-induced autophagy promotes chemotherapy resistance in
leukemia cells. Leukemia. 25:23–31. 2011. View Article : Google Scholar
|
|
11
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tang D, Kang R, Livesey KM, et al:
Endogenous HMGB1 regulates autophagy. J Cell Biol. 190:881–892.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu K, Huang J, Xie M, et al: MIR34A
regulates autophagy and apoptosis by targeting HMGB1 in the
retinoblastoma cell. Autophagy. 10:442–452. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pusterla T, Nèmeth J, Stein I, et al:
Receptor for advanced glycation endproducts (RAGE) is a key
regulator of oval cell activation and inflammation-associated liver
carcinogenesis in mice. Hepatology. 58:363–373. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Davalos AR, Kawahara M, Malhotra GK, et
al: p53-dependent release of Alarmin HMGB1 is a central mediator of
senescent phenotypes. J Cell Biol. 201:613–629. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Thorburn J, Horita H, Redzic J, Hansen K,
Frankel AE and Thorburn A: Autophagy regulates selective HMGB1
release in tumor cells that are destined to die. Cell Death Differ.
16:175–183. 2009. View Article : Google Scholar :
|
|
17
|
Smolarczyk R, Cichoń T, Matuszczak S, et
al: The role of Glycyrrhizin, an inhibitor of HMGB1 protein, in
anticancer therapy. Arch Immunol Ther Exp (Warsz). 60:391–399.
2012. View Article : Google Scholar
|
|
18
|
Taguchi A, Blood DC, del Toro G, et al:
Blockade of RAGE-amphoterin signalling suppresses tumour growth and
metastases. Nature. 405:354–360. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao M, Yang M, Yang L, et al: HMGB1
regulates autophagy through increasing transcriptional activities
of JNK and ERK in human myeloid leukemia cells. BMB Rep.
44:601–606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
van Beijnum JR, Nowak-Sliwinska P, van den
Boezem E, Hautvast P, Buurman WA and Griffioen AW: Tumor
angiogenesis is enforced by autocrine regulation of high-mobility
group box 1. Oncogene. 32:363–374. 2013. View Article : Google Scholar
|
|
21
|
Volp K, Brezniceanu ML, Bosser S, et al:
Increased expression of high mobility group box 1 (HMGB1) is
associated with an elevated level of the antiapoptotic c-IAP2
protein in human colon carcinomas. Gut. 55:234–242. 2006.
View Article : Google Scholar
|
|
22
|
Cheng BQ, Jia CQ, Liu CT, et al: Serum
high mobility group box chromosomal protein 1 is associated with
clinicopathologic features in patients with hepatocellular
carcinoma. Dig Liver Dis. 40:446–452. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhan Z, Li Q, Wu P, et al:
Autophagy-mediated HMGB1 release antagonizes apoptosis of gastric
cancer cells induced by vincristine via transcriptional regulation
of Mcl-1. Autophagy. 8:109–121. 2012. View Article : Google Scholar
|
|
24
|
Brunelle JK, Ryan J, Yecies D, Opferman JT
and Letai A: MCL-1-dependent leukemia cells are more sensitive to
chemotherapy than BCL-2-dependent counterparts. J Cell Biol.
187:429–442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kang R, Tang D, Lotze MT and Zeh HJ III:
AGER/RAGE-mediated autophagy promotes pancreatic tumorigenesis and
bioenergetics through the IL6-pSTAT3 pathway. Autophagy. 8:989–991.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Degenhardt K, Mathew R, Beaudoin B, et al:
Autophagy promotes tumor cell survival and restricts necrosis,
inflammation, and tumorigenesis. Cancer Cell. 10:51–64. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kang R, Livesey KM, Zeh HJ, Loze MT and
Tang D: HMGB1: a novel Beclin 1-binding protein active in
autophagy. Autophagy. 6:1209–1211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang J, Zhu JS, Zhou Z, Chen WX and Chen
NW: Inhibitory effects of ethyl pyruvate administration on human
gastric cancer growth via regulation of the HMGB1-RAGE and Akt
pathways in vitro and in vivo. Oncol Rep. 27:1511–1519.
2012.PubMed/NCBI
|
|
29
|
Kang R, Tang D, Schapiro NE, et al: The
HMGB1/RAGE inflammatory pathway promotes pancreatic tumor growth by
regulating mitochondrial bioenergetics. Oncogene. 33:567–577. 2014.
View Article : Google Scholar
|
|
30
|
Lin L, Zhong K, Sun Z, Wu G and Ding G:
Receptor for advanced glycation end products (RAGE) partially
mediates HMGB1-ERKs activation in clear cell renal cell carcinoma.
J Cancer Res Clin Oncol. 138:11–22. 2012. View Article : Google Scholar
|