Introduction
Ovarian cancer is a heterogeneous group of tumors,
where epithelial ovarian cancer accounts for the majority of cases.
The 5-year survival for invasive ovarian cancer strongly depends on
the stage at time of diagnosis and varies from 30 to 90% (1,2). In
the majority of patients late diagnosis and high morbidity result
mainly from asymptomatic manifestation of the disease (3). Although multiple genetic and
epigenetic changes have been studied, and some are characteristic
for ovarian cancer, it is still not clear how these changes affect
tumorigenesis. To date at least 15 oncogenes and 16 potential
tumor-suppressor genes in several signaling pathways have been
associated with ovarian cancer (4).
In the majority of populations, ~5–15% of all
ovarian cancer cases are caused by inheritance of mutations in
genes with an autosomal dominant pattern of transmission (such as
BRCA1/2). Although the actual proportion strongly depends on
the studied population and may be much higher, reaching 30% in the
Ashkenazi Jews (5–8). In Poland, the portion of hereditary
ovarian cancer due to BRCA1/2 mutation ranges from 13.5 to
14.9% (9–11).
Another ~6% of constitutive ovarian cancer cases are
related to alterations in low and moderate-penetrant genes
(12). These genes, products of
which are known to interact with BRCA1/2, are involved in DNA
repair and cell cycle regulation and therefore are good candidates
for possible breast and ovarian susceptibility genes (13,14).
The human BARD1 gene (BRCA1-associated RING
domain 1) is located at the long arm of chromosome 2 (2q34-35) and
encodes a nuclear protein of 777 amino acids that shares many
structural and functional similarities with BRCA1 (15–17).
Like BRCA1, BARD1 has an amino-terminal RING-finger motif and two
carboxy-terminal BRCT domains (15). The RING-finger motif is known to be
essential for BRCA1/BARD1 heterodimer formation, while BRCT domains
are found in proteins maintaining genome integrity (18). Proper interaction between these
proteins is crucial for BRCA1 stability and E3 ubiquitin ligase
activity of the BRCA1/BARD1 complex (16,17,19).
BARD1 has additionally three ankyrin (ANK) repeats
potentially mediating protein-protein interactions and present in
many proteins of various functions (20,21).
Interestingly, no other proteins comprising RING, ANK and BRCT
motifs together are known (16).
Like BRCA1, BARD1 shows highest expression in
actively proliferating cells and those that undergo apoptosis
(17,22). Loss of BARD1, as well as BRCA1,
leads to developmental retardation and early embryonic lethality of
corresponding knockout mice; cells from these mice are
characterized by chromosomal instability (23). In addition, it was observed that
tumors with homozygous deletions of the entire BARD1 gene
displayed a BRCA1 mutation-like expression profile (24).
Germline mutations in the BARD1 gene,
although detected with low frequency and in a limited number of
patients, can be qualified as novel candidates for ovarian cancer
susceptibility in a subset of families negative for BRCA1/2
mutations. The first study linking BARD1 with ovarian cancer
presented a patient with clear cell ovarian carcinoma in who the
missense c.1692G>C (p.Gln564His) mutation was identified
(25). The p.Gln564His reduces
binding of BARD1 to the polyadenylation cleavage specification
complex (CstF-50) and abrogates p53-dependent apoptosis (22,26).
Most recent publications reported three BARD1 mutations: One
affecting splicing, c.1977A>G, p.=, one non-sense, p.Gln715Ter
and one frameshift c.2148delCA; p.Thr716fs*12 (12,27,28).
Finally, germline BARD1 mutations were identified in
patients with a breast cancer (24,27,29–31),
cervical cancer (32), and
neuroblastoma (33).
In the present study, we present four different
possibly pathogenic BARD1 variants identified in a group of
unselected ovarian cancer patients. Three of the identified
alterations result in incorrect splicing of the corresponding
exons, which may prompt expression of specific BARD1 isoforms and
promote carcinogenesis.
Materials and methods
Study population
The study comprised 255 unselected ovarian cancer
patients referred to the Department of Gynaecological Oncology of
Medical University of Gdansk between 1995 and 2009. Within the
study group 162 (63.5%) patients were diagnosed with serous ovarian
cancer. The remaining 36.5% of tumors were classified as
endometroid (n=30/255; 12%); mucinous (n=25/255; 10%); clear cell
(n=17/255; 7%); and non-differentiated (n=17/255; 7%). Four tumors
(n=4/255; 1%) did not have complete histopatological
classification. Average age at diagnosis was 58 (range, 20–88
years). Informed consent was obtained from all of the patients and
the study was approved by the Medical Review Board of Medical
University of Gdansk (NKEB/399/2011-2012). The frequency of
identified BARD1 variants was investigated in an unselected
population-based control group of 1,000 anonymous samples collected
at birth (dried blood spots) and in a group of 200 healthy females
matched by age.
Blood samples
Patient samples: Genomic DNA was extracted from the
whole blood using the Genomic Midi AX kit (A&A Biotechnology,
Poland).
In addition, from selected patients a blood sample
was collected into Tempus™ Blood RNA Tubes and total RNA was
isolated with Tempus™ Spin RNA Isolation kit (Life Technologies,
USA). cDNA was synthesized using the Go Script™ Reverse
Transcriptase according to manufacturer's instructions (Promega,
USA).
Population-based control group: Genomic DNA was
extracted from 1,000 dried blood spots using Kapa Express Extract
kit (Kapa Biosystems, USA).
Tissue samples
Ovarian cancer: DNA from ovarian tumors was
extracted from formalin-fixed, paraffin-embedded (FFPE) tissue
blocks using Kapa Express Extract kit (Kapa Biosystems). Total RNA
from FFPE tissue blocks was isolated using the High Pure FFPE RNA
Micro kit (Roche Diagnostics, Switzerland).
Chronic myeloid leukemia (CML) controls: In order to
verify a presence of identified BARD1 isoforms in a
hormone-independent cancer, we examined cDNA from bone marrow cells
from 120 CML patients.
Mutation screening
All samples were tested for the presence of three
previously described BARD1 mutations (c.1315-2A>G in
intron 4; c.1690C>T in exon 8 and c.1977A>G in exon 10) by
using high-resolution melting analysis (LightScanner®
System; BioFire Defense, USA). Primers listed in Table I were designed based on the
BARD1 gene sequence obtained for the Ensemble database
(http://www.ensembl.org; BARD1:
ENSG00000138376). In order to simplify subsequent sequencing
analysis, M13 adaptor sequences (indicated by capital letters) were
added to the 5′-end of each primer. All amplicons demonstrating
melting profiles distinct from those of the wild-type samples were
subsequently sequenced (ABI PRISM 3130; Life Technologies). More
detailed information on screening protocols, can be obtained from
the corresponding author upon request.
 | Table ISequences of the primers used for
amplification of exons 5, 8 and 10 together with PCR and HRM
conditions. |
Table I
Sequences of the primers used for
amplification of exons 5, 8 and 10 together with PCR and HRM
conditions.
| No. | Primer name | Primer
sequence | PCR annealing
temperature (°C) | Product length
(bp) | HRM melting range
(°C) |
|---|
| 1 | BARD1_ex5Frw |
TGTAAAACGACGGCCAGTttttcctttctttccttaatgctt | 64 | 236 | 78–88 |
| 2 | BARD1_ex5Rev |
CAGGAAACAGCTATGACCaagagtatatgtggcagaggatga | | | |
| 3 | BARD1_ex8Frw |
TGTAAAACGACGGCCAGTtcgtctaatgtttttaacactggt | 68 | 210 | 80–90 |
| 4 | BARD1_ex8Rev |
CAGGAAACAGCTATGACCtctaccccacctcccaaaat | | | |
| 5 | BARD1_ex10Frw |
TGTAAAACGACGGCCAGTtcgtctaatgtttttaacactggt | 63 | 293 | 80–90 |
| 6 | BARD1_ex10Rev |
CAGGAAACAGCTATGACCagctgttgaaagggcagaag | | | |
Bioinformatics analysis and sequence
variation nomenclature
BARD1 mutations were numbered according to
the Human Genome Variation Society guidelines (34). The BARD1 sequence was in
accordance with GenBank NM_000465.2 All mutations were analyzed for
potential pathogenic effect using the following in silico
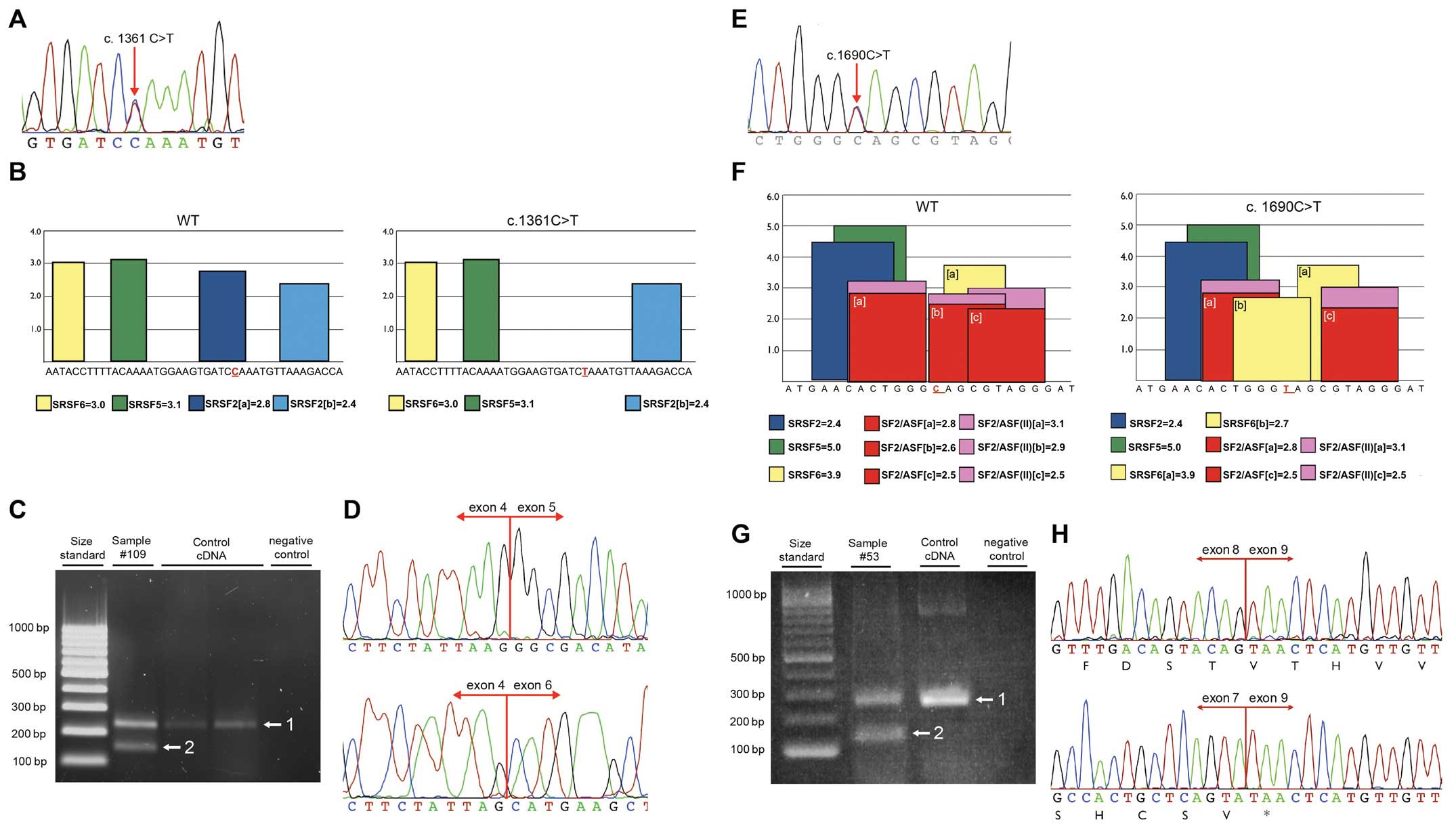
software: Alamut Mutation Interpretation software, ESE
finder, Human Splicing Finder, MutPred Splice,
Mutation Tester, PolyPhen 2, Rescue ESE and
SIFT.
Results
Here we investigated a role of BARD1 germline
mutations in predisposition to ovarian cancer. Frequency of
BARD1 recurrent mutations was estimated in a group of 255
unselected ovarian cancer patients. Exons 5, 8 and 10 together with
flanking intron sequences were analyzed by HRM technique followed
by bi-directional sequencing. A summary of the identified
BARD1 alterations is presented in Table II.
| Table IIBARD1 gene variants identified
in intron and exon sequences in 255 patients with ovarian
cancer. |
Table II
BARD1 gene variants identified
in intron and exon sequences in 255 patients with ovarian
cancer.
| No. | Intron/exon | Nucleotide
change | Effect | Status | Patients (n=255)
| Unselected controls
(n=1,000)
| Healthy controls
(n=200)
|
|---|
| n | % | n | % | n | % |
|---|
| Sequence variants
identified in intron sequences |
| 1 | 4 |
c.1315-19A>G | NE | rs6704780 | 130 | 51 | NA | NA | NA | NA |
| 2 | 10 |
c.2001+66A>G | NE | De Brakeleer et
al (29) | 7 | 2.75 | NA | NA | NA | NA |
|
| Sequence variants
identified in exon sequences |
| 1 | 5 |
c.1361C>T
r.[=,1315_1395del]
p.Gly439_Leu465del | p.Pro454Leu | Novel | 1 | 0.39 | 0 | 0 | 0 | 0 |
| 2 | 8 |
c.1690C>T
r.[=,1678_1810del]
p.Met560Ter | p.Gln564Ter | Ratajska et
al (27) | 1 | 0.39 | 0 | 0 | 0 | 0 |
| 3 | 10 | c.1972C>T | p.Arg658Cys | rs3738888 | 1 | 0.39 | 3 | 0.3 | 2 | 1 |
| 4 | |
c.1977A>G
r.[=, 159_1903del]
p.Cys53_Trp635delinsfs*12 | p.= | Ratajska et
al (27) | 1 | 0.39 | 1 | 0.1 | 2 | 1 |
BARD1 germline mutations
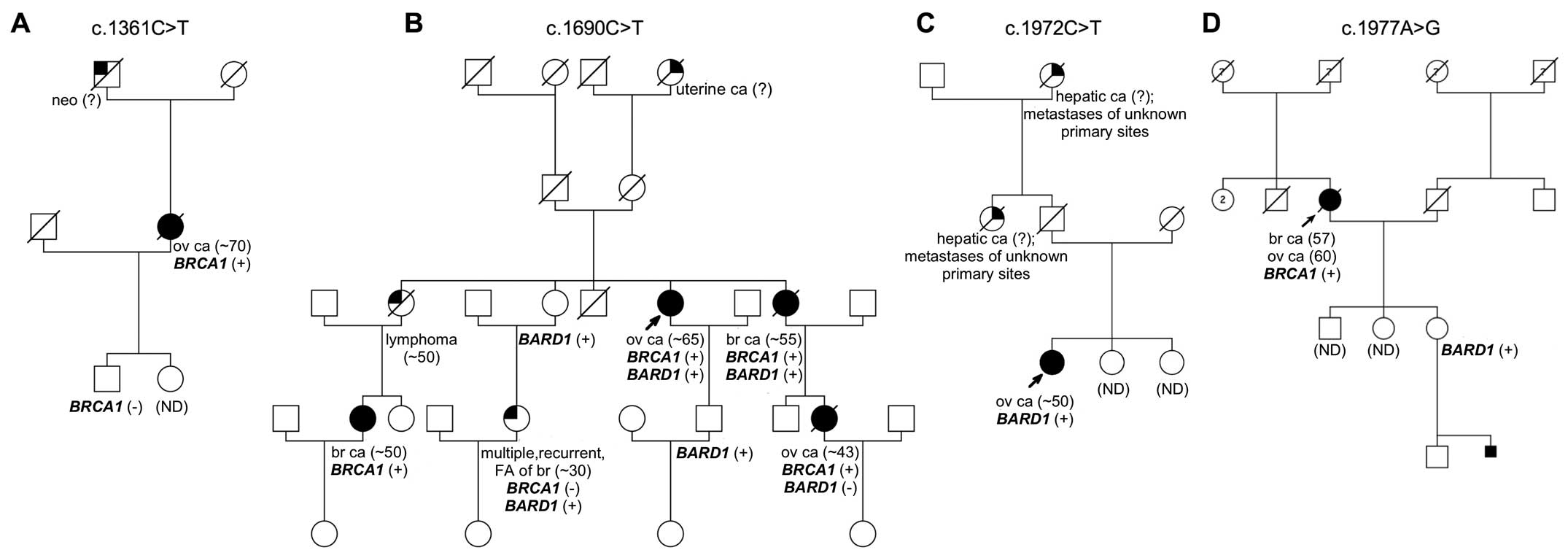
In patient #109, diagnosed with advanced serous
ovarian cancer at the age of 70, a novel substitution c.1361C>T
located in exon 5 was identified. The substitution results in a
change of a highly conserved amino acid (p.Pro454Leu), located
within the ANK repeats of the protein which suggest possible
alteration of the BARD1 structure and/or interactions with other
proteins. The information regarding the patient family was limited
and it was unclear whether additional breast and/or ovarian cases
were present in other family members (Fig. 1A). The germline mutation was found
heterozygous in the tested tumor sample. The c.1361C>T was not
detected in the control groups.
The second identified genetic variant was a
non-sense mutation in exon 8 (c.1690C>T, p.Gln564Ter) previously
reported by us (27). This mutation
is located between the ANK repeats and the BRCT domains. The
alteration was found in patient #53 diagnosed with serous ovarian
cancer at the age of 65. The patients' family showed a strong
aggregation of the disease (Fig.
1B). The sisters of the proband were affected with breast
cancer and lymphoma. In addition, three nieces were diagnosed with
ovarian cancer at the age of 43 and a breast cancer at the age of
~50. This variant was not found in the studied control groups.
The third, possibly deleterious alteration, located
in exon 10 (c.1972C>T) was identified in patient #150 affected
with ovarian cancer at the age of ~50. The proband's paternal aunt
and grandmother were diagnosed with hepatic metastases of unknown
primary site (Fig. 1C). The
substitution c.1972C>T results in an amino acid change at
position 658 (p.Arg658Cys), which is located between the two BRCT
domains. This genetic variant was also detected in both control
groups (unselected control group: n=3/1,000; 0.3% and healthy
individuals: n=2/200; 1%) suggesting its limited significance.
Analysis of the tumor sample revealed the heterozygous status of
this substitution.
Finally, we identified a carrier (#4321) of a
previously described c.1977A>G mutation in exon 10 (27). The patient was diagnosed with both
breast and ovarian cancer at the age of 57 and 60, respectively.
There was no information on other cancer cases within the proband's
family. Although co-segregation analysis revealed that one of the
proband's daughters was a carrier of this alteration (Fig. 1D). Tumor tissue was not available
for the study. c.1977A>G variant was also detected among control
samples (unselected control group: n=1/1,000; 0.1% and healthy
individuals: n=2/200; 1%).
Analysis of amplicons spanning the flanking intronic
sequence of exons 5, 8 and 10 lead to identification of two
frequent intronic variants c.1315-19A>G (rs6704780) and
c.2001+66A>G (rs75237746), which are well-known polymorphisms
(18). These were present in a
studied group of patients with a frequency of 49 and 2.6%,
respectively. Because of the high frequency, and therefore
presumably neutral character of these SNPs, we did not investigate
their frequency in control groups.
Identification of a BARD1/BRCA1 double
mutation carrier
Interestingly, mutational analysis of the
BRCA1 gene in patient #53, positive for BARD1
alteration (c.1690C>T), revealed the presence of a deleterious
mutation c.5266dupC in exon 20. Further segregation analysis showed
that both BRCA1 and BARD1 mutations were present in
other family members (Fig. 1B).
Investigation of the mutation status in tumor tissue of patient #53
reveled a heterozygotic character of both alterations.
Cancer-associated alterations of the
BARD1 gene affect splicing
In order to evaluate the possible effect of the
identified BARD1 sequence alterations on protein function
and splicing, in silico analysis was performed. The
synonymous substitution c.1977A>G, previously reported to affect
splicing (27), was used as
indicator of accuracy in predicting potential splicing disruption.
Although the employed software packages individually generated
inconsistent results, the combined analyses indicated a possible
influence of the tested mutations on the splicing process (Table III).
| Table IIIPrediction of possible pathogenicity
of identified mutations performed by combining seven different
in silico tools. |
Table III
Prediction of possible pathogenicity
of identified mutations performed by combining seven different
in silico tools.
| Exon | Nucleotide
change | Effect | Predicted effect on
splicing
| Predicted
pathogenicity
|
|---|
| MutPred Splice | Human Splicing
Finder | Alamut | Rescue ESE | PolyPhen 2 | SIFT | Mutation
Tester |
|---|
| Sequence variants
identified in exon sequences |
| 5 | c.1361C>T | p.Pro454Leu | Neutral | Probably affects
splicing | Eliminates binding
motifs for SC35 | Eliminates one
binding motif | Probably
damaging | Deleterious |
Disease-causing |
| 8 | c.1690C>T | p.Gln564Ter | Affecting | Probably affects
splicing | Eliminates binding
motifs for SF2/ASF; creates new SRp55 binding motif | No effect on
existing binding motifs | Not relevant | Not relevant | Not relevant |
| 10 | c.1972C>T | p.Arg658Cys | Neutral | Probably no effect
on splicing | Eliminates binding
motifs for SF2/ASF; affects SC35 and SRp55 binding motifs | No effect on
existing binding motifs | Probably
benign | Deleterious |
Disease-causing |
| 10 | c.1977A>G | p.= | Affecting | Probably affects
splicing | Eliminates binding
motifs for SF2/ASF and SRp55; creates new SRp40 binding motif;
affects second SF2/ASF binding motif | Eliminates five
binding motifs | Not relevant | Not relevant | Not relevant |
In order to confirm the in silico results,
RT-PCR using RNA isolated from patients' peripheral blood cells was
applied. In one case, due to the death of the patient with
BARD1 c.1361C>T substitution, RNA was extracted from the
normal tissue macrodissected from the resected tumor sample. RT-PCR
was performed with primers located within exons 4 and 6. Agarose
gel electrophoresis showed two prominent bands. The upper band had
the expected wild-type size (218 bp) and the lower band of 137 bp
corresponded to a fragment lacking exon 5. Sequencing of the
shorter band confirmed exon 5 skipping, resulting in in-frame
deletion from c.1315 to c.1395 [r.(=, 1315_1395del);
p.Gly439_Leu465del] (Fig. 2A–D). On
the protein level, this deletion results in disruption of the 1st
and 2nd ANK repeat.
Similarly, the c.1690C>T mutation in exon 8 was
examined. The RT-PCR experiment was performed using forward and
reverse primers in exons 7 and 9, respectively. Once again agarose
gel electrophoresis exhibited two bands: One band corresponding to
the wild-type fragment (264 bp) and a lower band. Sequencing of the
smaller band confirmed the presence of frame-shift deletion of exon
8 [r.(=,1678_1810del)], resulting in formation of premature stop
codon at position p.Met560 (Fig.
2E–H).
Finally, c.1972C>T substitution located in exon
10 was analyzed. RNA was extracted and RT-PCR using previously
described primers was applied (27). Alteration c.1972C>T does not
affect the splicing process.
Ovarian cancer-associated BARD1
alterations might be cancer-specific
Ultimately, we analyzed 120 samples of patients with
CML to assess the frequency of the newly identified BARD1 isoforms
(lacking exon 5 and 8, respectively) in other, hormone-independent
types of cancer. None of the analyzed sample showed the presence of
BARD1 alterations resulting in formation of the isoforms
lacking exons 5 or 8 (data not shown).
Discussion
All initial BARD1 studies described a limited
number of sequence variants, including several missense mutations
and one in-frame deletion of 21 bp with unknown consequence for the
protein function. However, more recent studies reported several
truncating mutations, which were segregating with the disease.
Interestingly, BARD1 germline variants were identified not
only in patients with breast and ovarian cancer (12,24,27–29,31),
but also among individuals with familial neuroblastoma (33).
In the present study, in a cohort of 255 unselected
ovarian cancer cases, one novel and three previously reported
genomic BARD1 alterations in exons 5, 8 and 10
(c.1361C>T; c.1690C>T, c.1972C>T, c.1977A>G) were
identified. All four genetic variants were absent or infrequent in
the control group (0.1–0.3%), indicating their pathogenic
potential.
c.1361C>T mutation disrupts the binding motif for
splicing factor SC35 and results in an in-frame deletion of exon 5
and disruption of two presumably important ANK repeats. Based on
previously published data, we assume that lack of 27 amino acids at
positions p.Gly439_Leu465 may diminish the ability of BARD1 to
induce apoptosis (35,36). BARD1 mRNAs lacking exon 5 could be a
full length (FL) BARD1 or mRNA isoforms lacking also other exons,
namely BARD1β, BARD1α, BARD1κ, BARD1ϕ, BARD1γ (Fig. 3B), which have been described before
for breast, ovarian, lung, colon cancer and neuroblastoma (37–40).
 | Figure 3The schematic structure of protein
coding FL BARD1 mRNA and splice isoforms is presented (A). Thick
line, non-coding sequences; dashed bar, exon skipping; gray bars,
protein coding sequence corresponding the main BARD1 reading frame.
White bars, alternative, in-frame ORFs. The positions of RING
domain, ANK repeats and BRCT domains are shown on the top. The
expected length of the polypeptides encoded by the isoforms is
shown on the right. (B) The structure of the BARD1 splice isoforms
with the exon 5 skipping. (C) The structure of the BARD1 splice
isoforms with exon 8 skipping. BARD1, BRCA1-associated RING domain
1; FL, full length; ORF, open reading frame; ANK, ankyrin. |
c.1361C>T is the second BARD1 mutation
resulting in exon 5 skipping. In a previous study, we identified an
intronic variant located in the donor site of intron 4
(c.1315-2A>G) that also resulted in in-frame deletion of exon 5
(27). Exon 5 deletion was first
observed in the ovarian cancer cell line NuTu-19 (41), derived from spontaneous mutation of
rat ovarian cancer cells that recapitulates human ovarian cancer
when injected intraperitoneally into mice. This cell line expressed
no FL BARD1, but BARD1β which had an additional deletion of exon 5
(41). The NuTu-19 cells were
resistant to apoptosis induction, but became sensitive when
expressing exogenous FL BARD1 suggesting that loss of exon 5 leads
to isoforms that have lost tumor-suppressor functions affecting the
apoptosis pathway (41). BARD1β
with an additional exon 5 deletion was also observed in mouse
spermatogenesis (42). However,
impact of exon 5 deletion on the function of FL BARD1 or BARD1β has
not been determined.
The second identified alteration, c.1690C>T, was
located in exon 8. In silico analysis showed that this
alteration significantly changed the binding motif for splicing
factors. Splicing alteration was confirmed by RT-PCR
[r.(=,1678_1810del), p.Met560*]. Translation of the
c.1690C>T mutation results in a truncated protein lacking the
BRCT domains, which play an integral role in the DNA damage
response (43). BRCA1
mutations located in BRCT domains are correlated with an increased
risk for both breast and ovarian cancer (44,45).
It was demonstrated that BRCA1 truncated only by 10 amino acids is
less stable, does not accumulate in the nucleus and fails to
colocalize with BARD1 and BRIP1 (44). It is therefore likely that lack of
BARD1 BRCT domains have a similar impact on protein function.
In addition, this mutation could not only produce a
truncated product of FL BARD1 mRNA, but also truncated proteins
from BARD1 isoform mRNAs (Fig. 3C).
Previously we reported the BARD1 c.1690C>T alteration as
a non-sense mutation (27), but
additional studies showed its impact on splicing alteration.
Moreover, the carrier of c.1690C>T mutation in
BARD1 was also diagnosed with mutation c.5266dupC in the
BRCA1 gene. The duplication of the cytosine at position
c.5266 is the second most frequent BRCA1 mutation all over
the world and the most common in Poland (10,11,18).
The c.1690C>T mutation in BARD1 was previously described
in a family with an aggregation of breast, colon, and uterine
cancer and was further identified in four different families with
aggregation of breast and ovarian cancer (27, and Ratajska
unpublished data). Co-existence of BARD1 and BRCA1
could have either an aggravating or a mitigating affect. However,
co-segregation of both mutations may indicate the latter. Another
case of co-occurrence of germline BARD1 (p.Gln715Ter) and
BRCA1 (c.3600del11) mutations was described in a patient
with serous carcinoma (28).
Moreover, co-occurrence of a BARD1 (p.Cys557Ser) germline
alteration with a BRCA2 (c.771_775del) truncating mutation
was reported as a risk amplifying factor to carriers of both
mutations (46). As BRCA1, BRCA2
and BARD1 act together in several tumor suppressor pathways, it is
likely that double mutations increase the risk of cancer.
Within this study group, we identified a carrier of
recurrent BARD1 missense mutation (c.1972C>T;
p.Arg658Cys) located in exon 10. Several studies have classified
c.1972C>T substitution as possibly deleterious (29) or with unknown significance (25,30,47).
Rudd et al suggested its correlation with a risk of lung
cancer with an odds ratio 1.55 (47). In our study, in silico
analysis performed by using Mutation Tester,
PolyPhen, and SIFT indicated its potentially damaging
character. However, RT-PCR showed that this variant does not alter
the splicing of BARD1 gene.
Finally, we described another case of synonymous
change (c.1977A>G) that results in aberrant splicing, leading to
a transcript lacking exons 2–9. This mRNA is identical to BARD1η
(Fig. 3A), which can either be
translated from the first methionine and end with a premature stop
codon in exon 10 (p.Cys53_Trp635delinsfs*12), or
translation could start with a first methionine in an alternative
open reading frame (ORF) (39,48).
This c.1977A>G alteration was previously identified and
characterized in a patient with clear cell ovarian cancer with
familial aggregation of the disease (27). Additionally, BARD1η isoform was
found by Li et al in human cytotrophoblast invasion and in
gynecological cancers, and authors showed the presence of a 21-kDa
protein, consistence with the predicted molecular weight of BARD1η
(39,48).
Importantly, our results underscore the necessity of
multilevel BARD1 mutation screening, as the molecular
analysis limited to DNA may result in high-level misclassification
of the detected genetic variants. This phenomenon was widely
studied in NF1 and ATM, where authors demonstrated
that ~13% of patients may be incorrectly diagnosed (49–51).
Several studies demonstrated that BARD1 isoforms are
widely expressed in different types of cancer and that spliced
isoforms are often more abundant than FL BARD1 (37–40).
Moreover, RNA interference experiments suggested that BARD1 splice
variants have functional roles and are the driving force of
tumorigenesis (37,39,40,52–54).
It was shown that the expression of alternatively spliced BARD1
isoforms was associated with poor outcome and short survival of
breast and lung cancer patients. The link between sequence
alterations and alternative splicing causing tumorigenesis was
convincingly demonstrated for neuroblastoma (40). SNPs that are significantly
associated with aggressive neuroblastoma were identified in
intronic sequences of BARD1. Increased expression of BARD1β
was linked to the disease-associated SNP and to functions in
malignant transformation (40).
Thus, we suggest that germline BARD1
mutations are responsible for a portion of hereditary ovarian
cancers and BARD1 should be included in gene panels that are
used for molecular diagnosis of breast and ovarian patients. The
actual pathogenic role of BARD1 sequence variants is rather
due to activation of alternative splicing and enhanced expression
of certain isoforms than mutations per se. Even apparently
harmless variants may lead to incorrect splicing process and
expression of oncogenic dominant negative forms of BARD1.
Acknowledgments
The authors wish to thank all the patients and their
families who participated in this study for their invaluable
contribution. This study was supported by the Polish National
Science Centre projects: N407 627740, 2011/02/A/NZ2/00017,
2011/01/B/NZ5/02773.
References
|
1
|
Jemal A, Murray T, Ward E, Samuels A,
Tiwari RC, Ghafoor A, Feuer EJ and Thun MJ: Cancer statistics,
2005. CA Cancer J Clin. 55:10–30. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Urban N and Drescher C: Potential and
limitations in early diagnosis of ovarian cancer. Adv Exp Med Biol.
622:3–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ruddon RW: Cancer Biology. Oxford
University Press; USA: 2007
|
|
4
|
Bast RC Jr, Hennessy B and Mills GB: The
biology of ovarian cancer: New opportunities for translation. Nat
Rev Cancer. 9:415–428. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berchuck A, Heron KA, Carney ME, Lancaster
JM, Fraser EG, Vinson VL, Deffenbaugh AM, Miron A, Marks JR,
Futreal PA, et al: Frequency of germline and somatic BRCA1
mutations in ovarian cancer. Clin Cancer Res. 4:2433–2437.
1998.PubMed/NCBI
|
|
6
|
Malander S, Ridderheim M, Måsbäck A, Loman
N, Kristoffersson U, Olsson H, Nilbert M and Borg A: One in 10
ovarian cancer patients carry germ line BRCA1 or BRCA2 mutations:
Results of a prospective study in Southern Sweden. Eur J Cancer.
40:422–428. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Risch HA, McLaughlin JR, Cole DE, Rosen B,
Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson
JL, et al: Prevalence and penetrance of germline BRCA1 and BRCA2
mutations in a population series of 649 women with ovarian cancer.
Am J Hum Genet. 68:700–710. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hirsh-Yechezkel G, Chetrit A, Lubin F,
Friedman E, Peretz T, Gershoni R, Rizel S, Struewing JP and Modan
B: Population attributes affecting the prevalence of BRCA mutation
carriers in epithelial ovarian cancer cases in Israel. Gynecol
Oncol. 89:494–498. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Menkiszak J, Gronwald J, Górski B,
Jakubowska A, Huzarski T, Byrski T, Foszczyńska-Kłoda M, Haus O,
Janiszewska H, Perkowska M, et al: Hereditary ovarian cancer in
Poland. Int J Cancer. 106:942–945. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brozek I, Ochman K, Debniak J, Morzuch L,
Ratajska M, Stepnowska M, Stukan M, Emerich J and Limon J: High
frequency of BRCA1/2 germline mutations in consecutive ovarian
cancer patients in Poland. Gynecol Oncol. 108:433–437. 2008.
View Article : Google Scholar
|
|
11
|
Ratajska M, Krygier M, Stukan M, Kuźniacka
A, Koczkowska M, Dudziak M, Śniadecki M, Dębniak J, Wydra D, Brozek
I, et al: Mutational analysis of BRCA1/2 in a group of 134
consecutive ovarian cancer patients. Novel and recurrent BRCA1/2
alterations detected by next generation sequencing. J Appl Genet.
56:193–198. 2015. View Article : Google Scholar :
|
|
12
|
Walsh T, Casadei S, Lee MK, Pennil CC,
Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A,
et al: Mutations in 12 genes for inherited ovarian, fallopian tube,
and peritoneal carcinoma identified by massively parallel
sequencing. Proc Natl Acad Sci USA. 108:18032–18037. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Apostolou P and Fostira F: Hereditary
breast cancer: The era of new susceptibility genes. Biomed Res Int.
2013:7473182013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walsh T and King MC: Ten genes for
inherited breast cancer. Cancer Cell. 11:103–105. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wu LC, Wang ZW, Tsan JT, Spillman MA,
Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM and Baer R:
Identification of a RING protein that can interact in vivo with the
BRCA1 gene product. Nat Genet. 14:430–440. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Irminger-Finger I and Jefford CE: Is there
more to BARD1 than BRCA1? Nat Rev Cancer. 6:382–391. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Irminger-Finger I, Soriano JV, Vaudan G,
Montesano R and Sappino AP: In vitro repression of Brca1-associated
RING domain gene, Bard1, induces phenotypic changes in mammary
epithelial cells. J Cell Biol. 143:1329–1339. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Williams RS, Bernstein N, Lee MS,
Rakovszky ML, Cui D, Green R, Weinfeld M and Glover JN: Structural
basis for phosphorylation-dependent signaling in the DNA-damage
response. Biochem Cell Biol. 83:721–727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Baer R and Ludwig T: BRCA1/BARD1
heterodimer, a tumor suppressor complex with ubiquitin E3 ligase
activity. Curr Opin Genet Dev. 12:86–91. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sedgwick SG and Smerdon SJ: The ankyrin
repeat: A diversity of interactions on a common structural
framework. Trends Biochem Sci. 24:311–316. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Mahajan A and Tsai MD: Ankyrin
repeat: A unique motif mediating protein-protein interactions.
Biochemistry. 45:15168–15178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Irminger-Finger I, Leung WC, Li J,
Dubois-Dauphin M, Harb J, Feki A, Jefford CE, Soriano JV, Jaconi M,
Montesano R, et al: Identification of BARD1 as mediator between
proapoptotic stress and p53-dependent apoptosis. Mol Cell.
8:1255–1266. 2001. View Article : Google Scholar
|
|
23
|
McCarthy EE, Celebi JT, Baer R and Ludwig
T: Loss of Bard1, the heterodimeric partner of the Brca1 tumor
suppressor, results in early embryonic lethality and chromosomal
instability. Mol Cell Biol. 23:5056–5063. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sabatier R, Adélaïde J, Finetti P, Ferrari
A, Huiart L, Sobol H, Chaffanet M, Birnbaum D and Bertucci F: BARD1
homozygous deletion, a possible alternative to BRCA1 mutation in
basal breast cancer. Genes Chromosomes Cancer. 49:1143–1151. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thai TH, Du F, Tsan JT, Jin Y, Phung A,
Spillman MA, Massa HF, Muller CY, Ashfaq R, Mathis JM, et al:
Mutations in the BRCA1-associated RING domain (BARD1) gene in
primary breast, ovarian and uterine cancers. Hum Mol Genet.
7:195–202. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kleiman FE and Manley JL: The
BARD1-CstF-50 interaction links mRNA 3′ end formation to DNA damage
and tumor suppression. Cell. 104:743–753. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ratajska M, Antoszewska E, Piskorz A,
Brozek I, Borg Å, Kusmierek H, Biernat W and Limon J: Cancer
predisposing BARD1 mutations in breast-ovarian cancer families.
Breast Cancer Res Treat. 131:89–97. 2012. View Article : Google Scholar
|
|
28
|
Pennington KP, Walsh T, Harrell MI, Lee
MK, Pennil CC, Rendi MH, Thornton A, Norquist BM, Casadei S, Nord
AS, et al: Germline and somatic mutations in homologous
recombination genes predict platinum response and survival in
ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer
Res. 20:764–775. 2014. View Article : Google Scholar :
|
|
29
|
De Brakeleer S, De Grève J, Loris R, Janin
N, Lissens W, Sermijn E and Teugels E: Cancer predisposing missense
and protein truncating BARD1 mutations in non-BRCA1 or BRCA2 breast
cancer families. Hum Mutat. 31:E1175–E1185. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karppinen SM, Heikkinen K, Rapakko K and
Winqvist R: Mutation screening of the BARD1 gene: Evidence for
involvement of the Cys557Ser allele in hereditary susceptibility to
breast cancer. J Med Genet. 41:e1142004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Castéra L, Krieger S, Rousselin A, Legros
A, Baumann JJ, Bruet O, Brault B, Fouillet R, Goardon N, Letac O,
et al: Next-generation sequencing for the diagnosis of hereditary
breast and ovarian cancer using genomic capture targeting multiple
candidate genes. Eur J Hum Genet. 22:1305–1313. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Narayan G, Pulido HA, Koul S, Lu XY,
Harris CP, Yeh YA, Vargas H, Posso H, Terry MB, Gissmann L, et al:
Genetic analysis identifies putative tumor suppressor sites at
2q35-q36.1 and 2q36.3-q37.1 involved in cervical cancer
progression. Oncogene. 22:3489–3499. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pugh TJ, Morozova O, Attiyeh EF,
Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M,
Kiezun A, et al: The genetic landscape of high-risk neuroblastoma.
Nat Genet. 45:279–284. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
den Dunnen JT and Antonarakis SE:
Nomenclature for the description of human sequence variations. Hum
Genet. 109:121–124. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jefford CE, Feki A, Harb J, Krause KH and
Irminger-Finger I: Nuclear-cytoplasmic translocation of BARD1 is
linked to its apoptotic activity. Oncogene. 23:3509–3520. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fox D III, Le Trong I, Rajagopal P,
Brzovic PS, Stenkamp RE and Klevit RE: Crystal structure of the
BARD1 ankyrin repeat domain and its functional consequences. J Biol
Chem. 283:21179–21186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang YQ, Pilyugin M, Kuester D, Leoni VP,
Li L, Casula G, Zorcolo L, Schneider-Stock R, Atzori L and
Irminger-Finger I: Expression of oncogenic BARD1 isoforms affects
colon cancer progression and correlates with clinical outcome. Br J
Cancer. 107:675–683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang YQ, Bianco A, Malkinson AM, Leoni
VP, Frau G, De Rosa N, André PA, Versace R, Boulvain M, Laurent GJ,
et al: BARD1: An independent predictor of survival in non-small
cell lung cancer. Int J Cancer. 131:83–94. 2012. View Article : Google Scholar
|
|
39
|
Li L, Ryser S, Dizin E, Pils D, Krainer M,
Jefford CE, Bertoni F, Zeillinger R and Irminger-Finger I:
Oncogenic BARD1 isoforms expressed in gynecological cancers. Cancer
Res. 67:11876–11885. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bosse KR, Diskin SJ, Cole KA, Wood AC,
Schnepp RW, Norris G, Nguyen le B, Jagannathan J, Laquaglia M,
Winter C, et al: Common variation at BARD1 results in the
expression of an oncogenic isoform that influences neuroblastoma
susceptibility and oncogenicity. Cancer Res. 72:2068–2078. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Feki A, Jefford CE, Berardi P, Wu JY,
Cartier L, Krause KH and Irminger-Finger I: BARD1 induces apoptosis
by catalysing phosphorylation of p53 by DNA-damage response kinase.
Oncogene. 24:3726–3736. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Feki A, Jefford CE, Durand P, Harb J,
Lucas H, Krause KH and Irminger-Finger I: BARD1 expression during
spermatogenesis is associated with apoptosis and hormonally
regulated. Biol Reprod. 71:1614–1624. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Leung CC and Glover JN: BRCT domains: Easy
as one, two, three. Cell Cycle. 10:2461–2470. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Nelson AC and Holt JT: Impact of RING and
BRCT domain mutations on BRCA1 protein stability, localization and
recruitment to DNA damage. Radiat Res. 174:1–13. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
di Masi A, Gullotta F, Cappadonna V,
Leboffe L and Ascenzi P: Cancer predisposing mutations in BRCT
domains. IUBMB Life. 63:503–512. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Stacey SN, Sulem P, Johannsson OT,
Helgason A, Gudmundsson J, Kostic JP, Kristjansson K, Jonsdottir T,
Sigurdsson H, Hrafnkelsson J, et al: The BARD1 Cys557Ser variant
and breast cancer risk in Iceland. PLoS Med. 3:e2172006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rudd MF, Webb EL, Matakidou A, Sellick GS,
Williams RD, Bridle H, Eisen T and Houlston RS; GELCAPS Consortium:
Variants in the GH-IGF axis confer susceptibility to lung cancer.
Genome Res. 16:693–701. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li L, Cohen M, Wu J, Sow MH, Nikolic B,
Bischof P and Irminger-Finger I: Identification of BARD1
splice-isoforms involved in human trophoblast invasion. Int J
Biochem Cell Biol. 39:1659–1672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ars E, Kruyer H, Gaona A, Serra E, Lazaro
C and Estivill X: Prenatal diagnosis of sporadic neurofibromatosis
type 1 (NF1) by RNA and DNA analysis of a splicing mutation. Prenat
Diagn. 19:739–742. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Baralle D and Baralle M: Splicing in
action: Assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Teraoka SN, Telatar M, Becker-Catania S,
Liang T, Onengüt S, Tolun A, Chessa L, Sanal O, Bernatowska E,
Gatti RA, et al: Splicing defects in the ataxia-telangiectasia
gene, ATM: Underlying mutations and consequences. Am J Hum Genet.
64:1617–1631. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ryser S, Dizin E, Jefford CE, Delaval B,
Gagos S, Christodoulidou A, Krause KH, Birnbaum D and
Irminger-Finger I: Distinct roles of BARD1 isoforms in mitosis:
Full-length BARD1 mediates Aurora B degradation, cancer-associated
BARD1beta scaffolds Aurora B and BRCA2. Cancer Res. 69:1125–1134.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Woditschka S, Evans L, Duchnowska R, Reed
LT, Palmieri D, Qian Y, Badve S, Sledge G Jr, Gril B, Aladjem MI,
et al: DNA double-strand break repair genes and oxidative damage in
brain metastasis of breast cancer. J Natl Cancer Inst. 106:pii:
dju1452014. View Article : Google Scholar
|
|
54
|
Chen J and Weiss WA: Alternative splicing
in cancer: Implications for biology and therapy. Oncogene. 34:1–14.
2015. View Article : Google Scholar
|