Introduction
Colorectal cancer is one of the most frequently
diagnosed malignant diseases and one of the leading causes of
cancer-related death worldwide (1).
In China, the incidence of colorectal cancer increases by
~4.2%/year, and there is a higher incidence among younger adults
(2). Moreover, metastatic cancer
occurs in 40–50% of newly diagnosed patients, which is associated
with high morbidity (3). Recent
therapeutic strategies for metastatic colorectal cancer (mCRC) have
focused on developing molecular-targeted therapies.
It has been reported that increased epidermal growth
factor receptor (EGFR) expression is the hallmark of many human
tumors and an important therapeutic target in mCRC (4). Cetuximab, the first chimeric
monoclonal antibody which has been generated against the EGFR, is
the first-line treatment used as a single agent or in combination
with standard chemotherapy for KRAS wild-type mCRC patients
(5). The mechanism of cetuximab
(C225, Erbitux) involves the specific binding of the extracellular
domain of EGFR, subsequently blocking the downstream signaling of
EGFR that influences cell proliferation, survival, apoptosis,
migration and tumorigenesis. Numerous clinical studies have shown
that cetuximab significantly improves the progression-free and
overall survival of patients with KRAS wild-type mCRC, while
patients with mutant KRAS do not benefit from cetuximab treatment
(6,7). The main reason for this phenomenon is
that downstream signaling of KRAS in the MAPK pathway is not
controlled by EGFR. Moreover, KRAS gene mutations, BRAF, NRAS and
PIK3CA gene mutations can also cause resistance to cetuximab in
mCRC patients (6,8). However, the resistance to cetuximab
still exits in patients with the wild-type genes mentioned above,
which indicates that there are some other resistance mechanisms to
be explored (9).
Several studies have shown that phosphorylation
levels of paxillin (PXN) are related to those of cytokine
receptors; the blocking of cell growth factor receptors leads to a
compensatory increase in PXN phosphorylation levels, and that PXN
plays an important role in mediating signal transduction of the
epidermal growth factor (EGF) (2,10–12).
Therefore, high expression of PXN and increased phosphorylation
levels may be one of the reasons for the poor therapeutic effect of
cetux-imab. Meanwhile, it is necessary and meaningful to explore
the specific mechanism of resistance to cetuximab in mCRC.
PXN, a 68-kDa focal adhesion-associated protein,
contains a number of motifs that mediate protein-protein
interactions, including C-terminal LIM domains resembling a double
zinc-finger domain, N-terminal LD motifs, SH3 and SH2
domain-binding sites, whose motifs serve as docking sites for
cytoskeletal proteins, tyrosine and serine/threonine kinases,
GTPase-activating proteins and other adaptor proteins (13,14).
PXN plays an important role in signal transduction, regulation of
cell morphology, migration, proliferation and apoptosis (2,13,14). A
number of studies have demonstrated that high expression of PXN
also occurs in cancer cells of many other organs, such as the
esophagus, prostate, and lung (2).
Upregulation of PXN phosphorylation levels promotes tumor cell
growth, invasion, migration, recurrence and inhibits cell
apoptosis, which results in the poor prognosis of patients.
However, expression of PXN does not serve as an independent risk
factor for prognosis (15,16). Various studies have mentioned that
PXN mediates the EGF signal transduction process. Sen et al
discovered that after knockdown of the PXN gene in prostate cancer
cells, the phosphorylation level of extracellular regulated-protein
kinase (Erk) significantly decreased in the downstream of
mitogen-activated protein kinase (MAPK) pathway, which proves that
activation of the Erk signaling pathway requires PXN involvement
and PXN is necessary for the proliferation of prostate cancer cells
(11). In a study of non-small cell
lung cancer (NSCLC), Wu et al found that activation of Erk
requires mediation of PXN and further confirms that expression of
the B-cell leukemia/lymphoma-2 (Bcl-2) gene is regulated by PXN
through activation of Erk, while high expression of the Bcl-2 gene
inhibits cancer cell apoptosis, causing resistance to
chemotherapeutic drugs in lung cancer patients (17). Despite these previous studies,
whether PXN overexpression is one of the reasons for resistance to
cetuximab and how PXN affects the MAPK pathway in colorectal cancer
cells remain unknown.
In the present study, immunohistochemical staining
(IHC) in 148 colorectal carcinoma and 126 normal adjacent tissues
was performed to clarify the relationship between TNM stage,
recurrence rate, metastasis, survival and PXN expression. Two human
colon cancer cell lines were chosen to discover the role of PXN
activation in cetuximab resistance: SW480 cells with high
expression of PXN and insensitive to cetuximab, and Caco-2 cells
with the opposite characteristics. It was demonstrated that
inhibition of PXN expression by PXN-siRNA restored sensitivity to
cetuximab in the SW480 cells, and upregulation of PXN expression by
PXN-cDNA (PXN Human cDNA ORF Clone) reduced the sensitivity to
cetuximab in the Caco-2 cells. Notably, the expression of p-Erk and
PXN expression were consistent. We treated these cells with a
selective Erk inhibitor, which restored cetuximab sensitivity in
the SW480 cells and made Caco-2 cells more sensitive to cetuximab.
These results suggest that inhibition of PXN expression can improve
sensitivity to cetuximab by downregulation of p-Erk levels.
Materials and methods
Drugs
Cetuximab, an anti-EGFR human-mouse chimeric
monoclonal antibody (mAb), was purchased from Merck Serono
(Darmstadt, Germany). SCH-772984, a selective inhibitor of
extracellular signal regulated kinase (Erk1/2) that displays
behaviors of both type I and II kinase inhibitors, was purchased
from Selleckchem. SCH-772984 was dissolved in dimethylsulfoxide
(DMSO) and maintained as a concentrated stock at −20°C. Working
concentrations were diluted in culture medium just before each
experiment.
Tumor samples
We received approval for the present study from the
Clinical Research Ethics Committee, and tumor specimens were
collected after obtaining patient informed consent in accordance
with the institutional guidelines. We selected 274 samples from 148
patients (78 men and 70 women) with primary colorectal carcinoma
who underwent surgery at the Department of General Surgery, Peking
University First Hospital from January 2006 to June 2009, including
148 colorectal carcinoma and 126 normal adjacent mucosa (at least 2
cm distant from the tumor). The patients were selected according to
TNM stage; the case number of each stage did not differ greatly.
None of the patients had received chemotherapy and radiotherapy
before surgery. Two pathologists reviewed all of the histological
slides.
Cell lines
The human colon cancer cell lines and a human colon
normal mucosa cell line were obtained from the Colon Cancer
Institute of Peking University First Hospital (Beijing, China),
including SW480, HT29, Rko, Caco-2, Lovo and FHC cell lines. These
cells were routinely cultured in Dulbecco's modified Eagle's medium
[DMEM containing 10% fetal bovine serum (FBS)] (both from Gibco)
and penicillin/streptomycin at 37°C in a 5%
CO2-humidified atmosphere.
Proliferation assay
Cancer cells were seeded into 96-well plates and
treated with different concentrations of SCH-772984 (range, 5–90
mg/ml), cetuximab (range 5–700 mg/ml) alone for 48 h or with
cetuximab (100 mg/ml) for a period of time (6 h-10 days). Cell
proliferation was measured with MTT assay. The IC50
value and proliferation rate were calculated according to optical
density from a microplate reader. Each test was performed in
quadruplicate.
Apoptosis assay
SW480 and Caco-2 cells were seeded in 25
cm2 flasks, treated for 72 h and stained with Annexin
V-fluorescein isothiocyanate (FITC) and 7-amino-actinomycin D
(7-AAD) in the dark. Viable (7-AAD-negative) and dead
(7-AAD-positive) cell populations were quantitated by flow
cytometry using a flow cytometer (BD Influx).
RNA interference
The small inhibitor duplex RNAs (siRNAs) (On-target
paxillin) PXN-siRNAs (human: SR303929) were purchased from OriGene.
OriGene synthesized three siRNA duplexes targeting human paxillin
mRNA (PXN-siRNA). The targeting sequences were siRNA1,
UGACGAAAGAGAAGCCUAAGCGGAA; siRNA2, UGAACGCUGUACAGCAUAACCCGCC; and
siRNA3, GACAAUGCCAGCAUAAAUCCAUCCA. In the present study, siRNA1 was
used since it effectively inhibited PXN expression in our
preliminary experiments. The siCONTROL non-targeting siRNA (human,
SR30004) was used as a negative control (NC). Cells were
transfected with 40 nmol/l siRNAs using Lipofectamine 2000 reagent
(Invitrogen) according to the manufacturer's instructions. The day
before the transfection, the cells were plated in 25 cm2
flasks at 50–70% confluency in medium supplemented with 5% FBS.
Cells were harvested 72 h after the transfection. Efficiency of
gene silencing was detected by western blotting.
PXN-cDNA transient transfection and
co-transfection of PXN-cDNA and PXN-siRNA
The human PXN-cDNA was purchased from OriGene
(RC213811). It was cloned into vector Pcmv6-Entry (cat.
#PS1000-001) and was verified by full sequencing (NM_002859). This
human PXN cDNA was derived from single colony E. coli
cultures and purified through the OriGene ion-exchange plasmid
purification system (PowerPrep. HP Midiprep kits with Prefilters NP
100024). The non-targeting plasmid was used as a negative control.
The cells were transfected with 1.5 mg/ml cDNAs using Lipofectamine
2000 reagent following the manufacturer's protocols. The day before
the transfection, the cells were plated in 25 cm2 flasks
at 50–70% confluency in medium supplemented with 10% FBS. They were
harvested 72 h after the transfection. Efficiency of gene silencing
was detected by western blotting. When transfecting PXN-cDNA and
PXN-siRNA at the same time, Lipofectamine 2000 reagent and 30 pmol
of siRNA/1 mg of DNA was used.
Western blotting
SW480, SW620, HT29, Rko, Caco-2 and Lovo cells were
seeded into 25 cm2 flasks and treated for 72 h. Total
proteins were obtained by a protein extraction kit (KeyGen Biotech,
Nanjing, China), including phosphorylated proteins. The protein
concentration was estimated by a modified Bradford assay (KeyGen
Biotech). Immunoreactive bands were visualized by enhanced
chemiluminescence (ECL; GE Healthcare/Amersham). Antibodies against
p-Erk, cleaved caspase-3 and GAPDH (HRP-conjugated) were purchased
from Cell Signaling Technology. Antibodies against PXN (Tyr-118)
were purchased from Abcam (Hong Kong, China). Secondary antibodies
coupled to horseradish peroxidase were purchased from Signalway
Antibody (USA). The antibodies were diluted according to the
instructions before the experiment. Each experiment was carried out
for several times.
IHC
The immunohistochemical staining was performed on
paraffin sections (4-µm) using a streptavidin-peroxidase
immunostaining kit. After deparaffinization with xylene and
rehydration through a graded ethanol series, the sections were
subjected to microwave antigen retrieval with an
ethylene-diaminetetraacetic acid (EDTA) buffer solution (1 mmol/l,
pH 8.0) at 98°C for 10 min to unmask antigenic epitopes. Endogenous
peroxidase activity was blocked with 0.3% hydrogen peroxide for 15
min at room temperature. After blocking non-specific binding sites
with a blocking serum for 30 min at room temperature, the sections
were incubated with a mouse monoclonal anti-PXN antibody (1:100;
Abcam) at 4°C overnight. Visualization of the antibody-enzyme
complex was achieved with 3,3-diaminobenzidine tetrahydrochloride
(DAB). The sections were counterstained with Mayer's hematoxylin.
Appropriate positive controls were included, and as a negative
control, normal serum and PBS were substituted for the primary
antibody.
Immunohistochemical analysis
To quantify PXN protein expression, both the extent
and intensity of immunoreactivity were assessed and scored. In the
present study, the scores of the extent of immunoreactivity ranged
from 0 to 3 and were determined according to the percentage of
cells that exhibited positive staining in each microscopic field of
view (0, <25%; 1, 25–50%; 2, >50–75%; and 3, >75–100%).
The scores of IHC intensity were as follows: 0, negative staining;
1, weak staining; 2, moderate staining; and 3, strong staining. By
multiplying the scores for extent and intensity, a total score
ranging from 0 to 9 was achieved. The expression level of PXN was
considered high when the total score was ≥4 and low when the total
score was <4. The results were assessed by two independent
observers who did not have any knowledge of the clinicopathological
features of the patients. If the results differed, then they
consulted and reached a final consensus.
Statistical analysis
The statistical analyses of data were carried out
using SPSS 17.0 statistical software (SPSS, Inc., USA). The
relationship between PXN expression and clinicopathological
characteristics was analyzed by the χ2 test. All
P-values represent two-sided tests of statistical significance with
P-value <0.05.
Results
The expression of PXN is higher in tumor
tissues than that in normal mucosa
The expression of PXN was examined in 148 colorectal
adenocarcinoma samples and 126 normal adjacent mucosa samples
through an immunohistochemical staining approach. A total of 101 of
the 148 (68.2%) colorectal adonocarcinoma samples and 56 of the 126
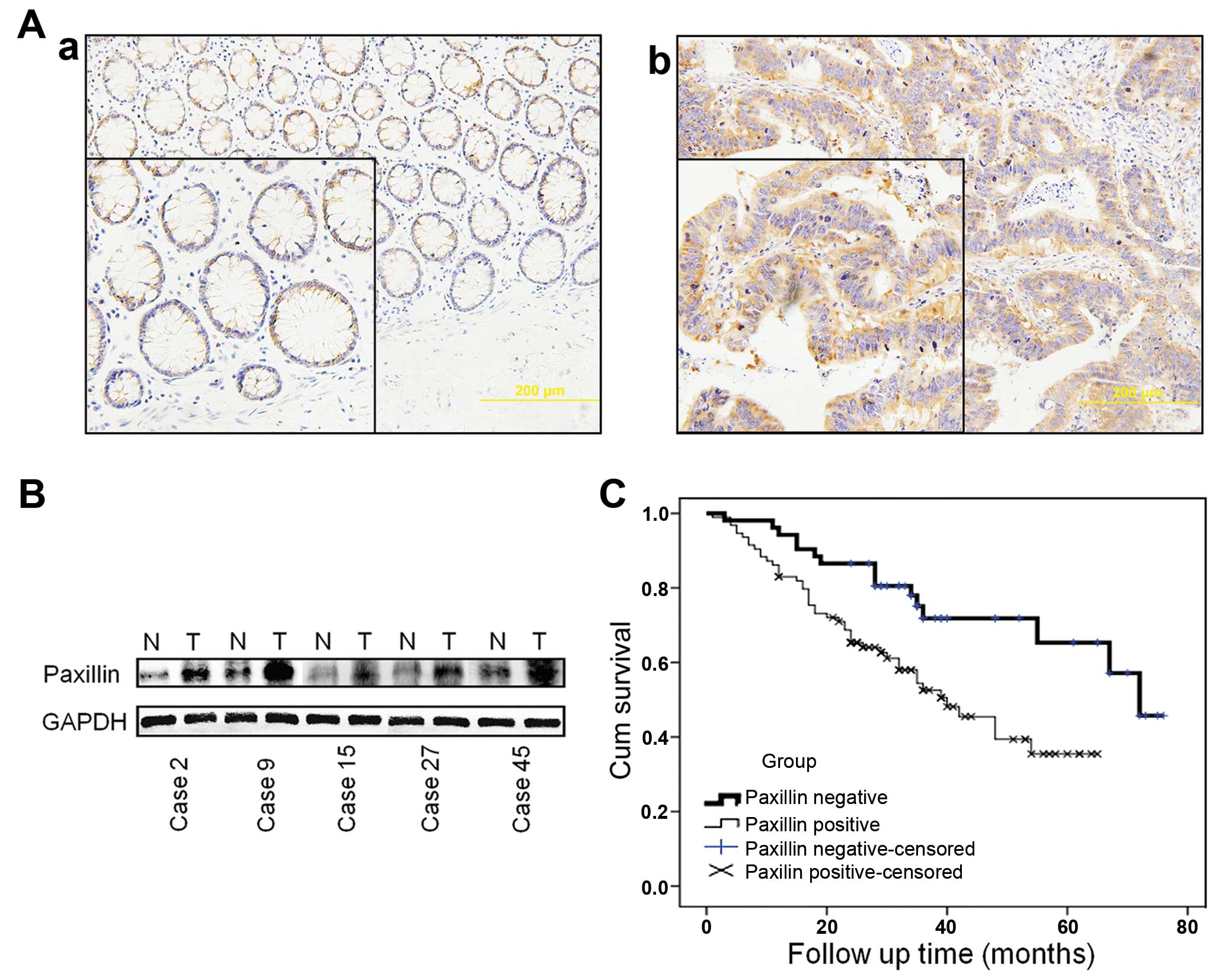
(44.4%) normal tissues were PXN-positive (P<0.001) (Table I). PXN protein was mainly expressed
in the cytoplasm and was found to be overexpressed in the tumors
compared with the normal adjacent tissues (Fig. 1A). This result was further confirmed
in 60 colorectal carcinoma samples with paired normal adjacent
tissues by western blotting. In 43 of 60 (71.7%) cases, PXN
expression levels in the tumor tissues (T) were at least 2-fold
higher than levels in the normal adjacent mucosa (N) (Fig. 1B).
 | Table IPatient clinicopathological factors
and paxillin expression in normal and colorectal cancer
tissues. |
Table I
Patient clinicopathological factors
and paxillin expression in normal and colorectal cancer
tissues.
| Parameters | Total | Paxillin
| P-value |
|---|
| + | − |
|---|
| Tissues | | | | <0.001 |
| Normal colorectal
mucosa | 126 | 56 | 70 | |
| Colorectal
cancer | 148 | 101 | 47 | |
| Distant
metastasis | | | | 0.014 |
| Absent | 123 | 79 | 44 | |
| Present | 25 | 22 | 3 | |
| Recurrence | | | | 0.032 |
| Absent | 114 | 73 | 41 | |
| Present | 34 | 28 | 6 | |
| TNM stage | | | | 0.023 |
| I | 28 | 14 | 14 | |
| II | 39 | 24 | 15 | |
| III | 59 | 44 | 15 | |
| IV | 22 | 19 | 3 | |
PXN expression is negatively correlated
with the survival of the patients
Follow-up results in a total of 148 patients were
analyzed. The median duration of follow-up was 48.6 months (range
1–75 months), and a total of 63 patients died during the follow-up
period. Univariate analyses revealed that the 3- and 5-year
survival rates of the PXN-negative group were higher (71.8 and
65.3%) than those of the positive group (52.5 and 35.4%, log-rank
testing, P=0.004, Fig. 1C). The
survival rate was correlated with the expression of PXN.
PXN is expressed differently in the colon
cell lines
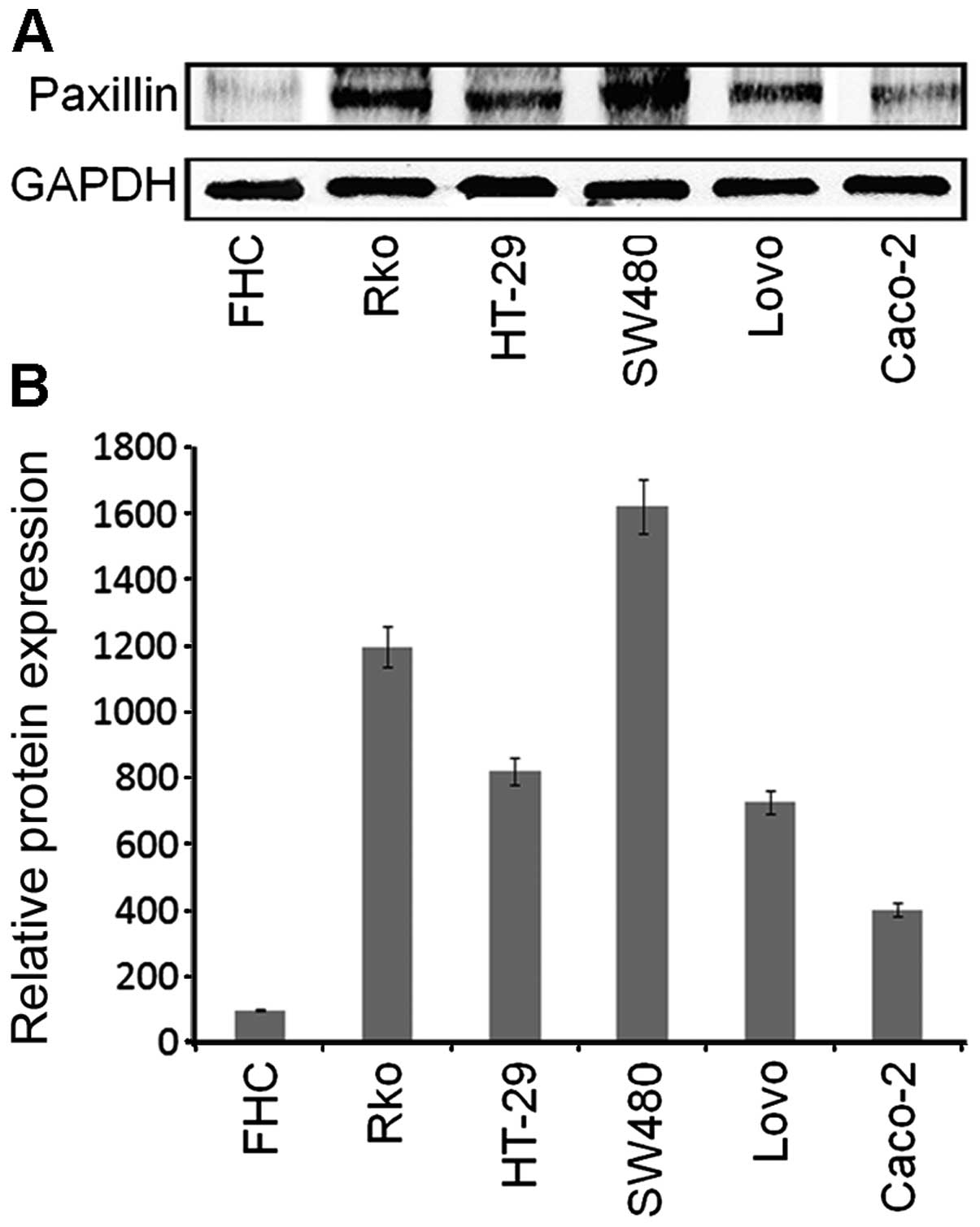
The expression of PXN was assessed in five different
colon cancer cell lines and a normal mucosal cell line by western
blotting. Compared with FHC, a normal human colon cell line, the 5
colon cancer cell lines showed a higher level of PXN. Among the
Caco-2, Lovo, SW480, HT-29 and Rko cells, PXN was expressed at the
highest level in the SW480 cells, secondly in the Lovo, HT-29 and
Rko cells, and least in the Caco-2 cells (Fig. 2). Therefore, we chose SW480 and
Caco-2 cells to perform subsequent experiments.
PXN expression in the tumors is related
with clinicopatho-logical parameters
The positive rate of PXN was higher in the
colorectal adenocarcinoma samples than that in the normal
colorectal mucosa samples (68.2 vs. 44.4%, P<0.001, Table I). The rate of recurrence in the
colorectal patients was higher in the PXN-positive group (27.7%)
than that in the PXN-negative group (12.8%, P=0.032; Table I). PXN positivity was closely
related to TNM stage (86.4, 74.6, 61.5 and 50.0%, P=0.023, Table I) and distant metastasis (88 vs.
64.2%, P=0.014; Table I).
High expression level of PXN confers
resistance to cetuximab in SW480 cells
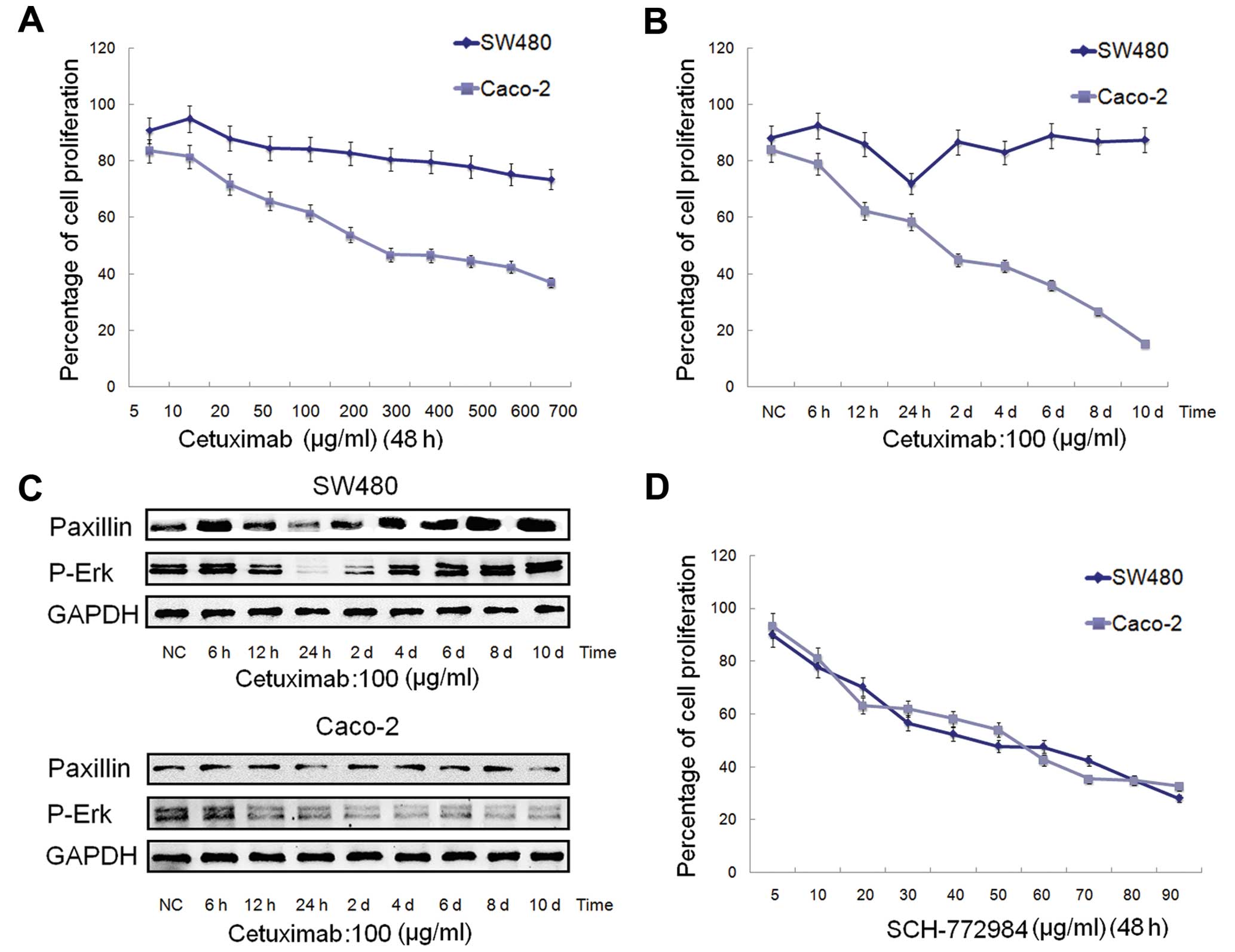
SW480 and Caco-2 cells were treated with increasing
concentrations of cetuximab (5–700 mg/ml). The percentage of
proliferation in the SW480 cells slightly increased at first and
then slightly decreased (Fig. 3A).
This showed that the SW480 cells were not sensitive to cetuximab.
Compared with the SW480 cells, the Caco-2 cells were relatively
sensitive to cetuximab, and the IC50 value was ~189.3
mg/ml (Fig. 3A).
To explore how PXN and p-Erk change with the
treatment of cetuximab in SW480 and Caco-2 cells, the cells were
treated for a period of time (from 6 h to 10 days) at a
concentration of 100 mg/ml (Fig.
3B). As shown in Fig. 3C, the
expression of PXN declined at first, and then increased gradually
and stabilized in the SW480 cells, which was very consistent with
the proliferation rate in Fig. 3B,
the lowest point of which was at ~24 h. In contrast to the SW480
cells, PXN expression in the Caco-2 cells did not significantly
change. These data demonstrated that the high expression level of
PXN may be one of the reasons for the insensitivity to cetuximab of
SW480 cells, which warrants further experiments for confirmation.
Moreover, the expression changes in p-Erk in both the SW480 and
Caco-2 cells (Fig. 3C) were
consistent with the proliferation rates in Fig. 3B.
To clarify the effect of an Erk1/2 inhibitor on
SW480 and Caco-2 cells, the cells were treated with increasing
concentrations of SCH-772984 (5–90 mg/ml) for 48 h. SCH-772984
treatment of the SW480 and Caco-2 cells induced a dose-dependent
inhibition of cell growth with an IC50 value of ~47.5
and 56.4 mg/ml, respectively (Fig.
3D), which demonstrated that both SW480 and Caco-2 cells were
sensitive to SCH-772984.
Inhibition of PXN expression by PXN-siRNA
restores sensitivity to cetuximab in the SW480 cells
To determine whether PXN activation could be one of
the reasons for cetuximab resistance in SW480 cells, a further
investigation was carried out to ascertain whether reduction in PXN
expression restores cetuximab sensitivity. First of all, we
compared the effect of transfection between siRNA1 and siRNA2. As
shown in Fig. 4A, transfection with
siRNA1 for 72 h significantly reduced PXN protein expression in the
SW480 cells, and thus, siRNA1 (PXN-siRNA) was chosen to carry out
the following transfection study.
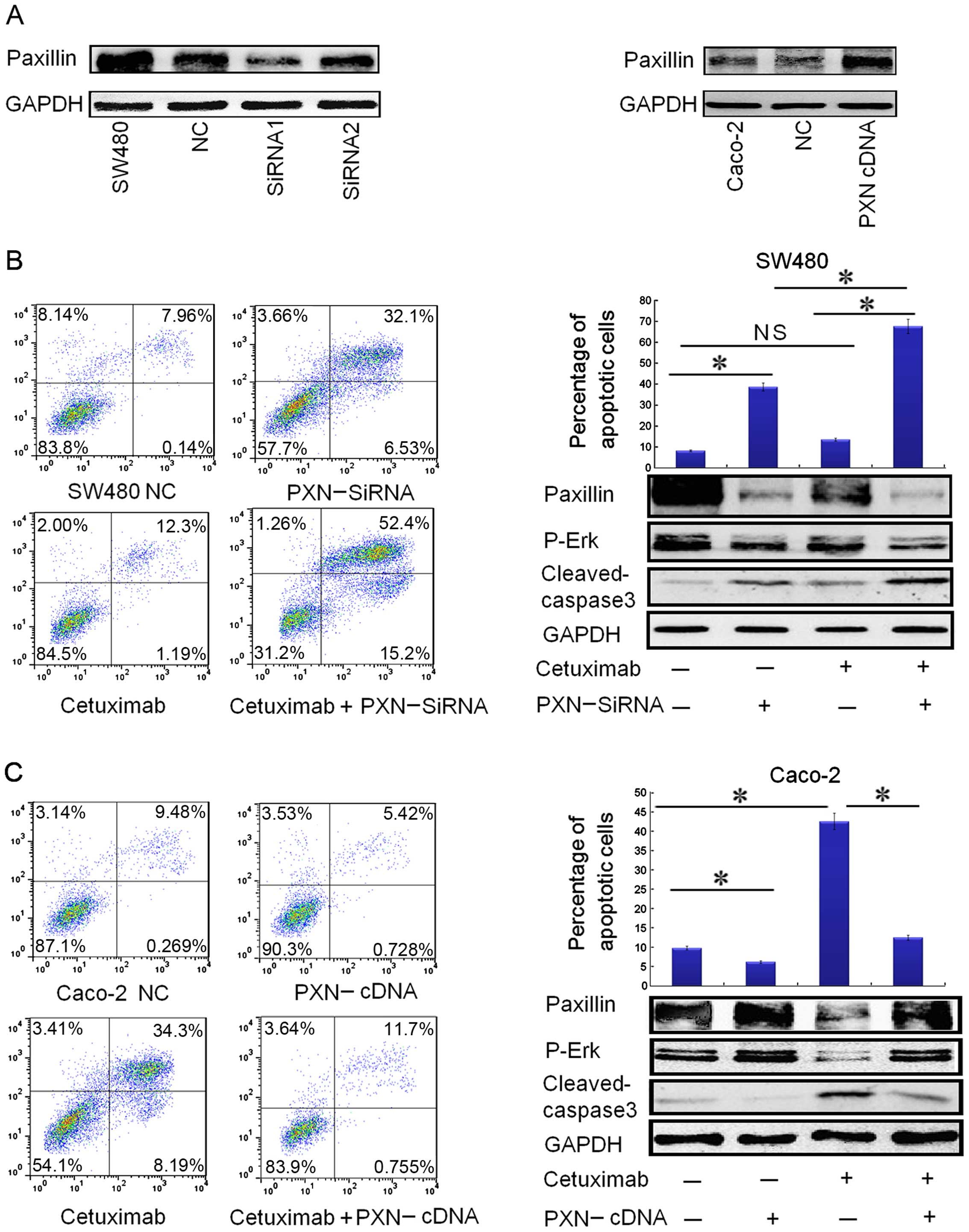 | Figure 4Downregulation of paxillin by
PXN-siRNA significantly restores the sensitivity to cetuximab in
SW480 cells and upregulation of paxillin expression by PXN-cDNA
(PXN Human cDNA ORF Clone) reduces the sensitivity to cetuximab in
the Caco-2 cells. (A) Transfection with siRNA1 (PXN-siRNA) for 72 h
significantly reduced paxillin protein expression in the SW480
cells, and the expression level of paxillin increased significantly
in the Caco-2 cells after transfection with PXN-cDNA for 72 h. (B)
Downregulation of paxillin by PXN-siRNA in combination with
cetuximab obviously increased the apoptotic rate in the SW480
cells, illustrated by flowgram and histogram. Columns, means of
three independent experiments. PXN-siRNA vs. negative control (NC)
(*P<0.001), cetuximab plus PXN-siRNA vs. PXN-siRNA
(*P<0.001), cetuximab plus PXN-siRNA vs. cetuximab
(*P<0.001), cetuximab vs. negative control (NC) (not
significant NS, P>0.05). The expression of apoptotic protein
cleaved caspase-3 was consistent with the data in the flowgram and
histogram. Paxillin silencing also restored the ability of
cetuximab to inhibit p-Erk activation in the SW480 cells, as shown
by downregulation of p-Erk levels. (C) Upregulation of paxillin by
PXN-cDNA clearly reduced the apoptotic rate in Caco-2 cells,
illustrated by the flowgram and histogram. Columns: means of three
independent experiments. Cetuximab vs. negative control (NC)
(*P<0.001), PXN-cDNA vs. negative control (NC)
(*P<0.05), PXN-cDNA plus cetuximab vs. cetuximab
(*P<0.005). Corresponding to the data, the expression
of apoptotic protein cleaved caspase-3 was downregulated as the
apoptotic rate declined. Paxillin overexpression also promoted the
activation of p-Erk in Caco-2 cells, as shown by upregulation of
p-Erk levels. |
Although single-agent cetuximab did not
significantly affect SW480 apoptosis, cetuximab treatment in
combination with PXN silencing showed a statistically significant
apoptotic effect on SW480 cells (Fig.
4B). The expression of apoptotic protein cleaved caspase-3 was
consistent with the apoptotic rate as shown in Fig. 4B. PXN silencing also inhibited p-Erk
activation in the SW480 cells as shown by downregulation of p-Erk
levels (Fig. 4B), which may be the
mechanism through which SW480 cells regained sensitivity to
cetuximab.
Activation of PXN by PXN-cDNA (PXN Human
cDNA ORF Clone) transfection reduces the sensitivity to cetuximab
in the Caco-2 cells
To further evaluate whether PXN activation confers
resistance to cetuximab in colorectal cancer cells, the Caco-2 cell
line, confirmed to have low expression of PXN, was used. After
transfection with PXN-cDNA for 72 h, PXN protein expression was
significantly increased in the Caco-2 cells (Fig. 4A). As illustrated in Fig. 4C, the overexpression of PXN
effectively suppressed apoptosis in the Caco-2 cells with or
without cetuximab treatment. Corresponding to the apoptotic data,
the expression of apoptotic protein cleaved caspase-3 was
downregulated with the decrease in the apoptotic rate (Fig. 4C). PXN overexpression also promoted
the activation of p-Erk in the Caco-2 cells as shown by
upregulation of p-Erk levels (Fig.
4C). These data further demonstrated that PXN confers
resistance to cetuximab and knockdown of PXN improves sensitivity
to cetuximab in colorectal cancers by downregulating the expression
of p-Erk.
PXN overexpression regulates resistance
to cetuximab in a p-Erk-dependent pattern in the SW480 and Caco-2
cells
To further evaluate the role of PXN activation in
cetuximab resistance, co-transfection with PXN-siRNA and PXN-cDNA
in SW480 and Caco-2 cells was performed. Compared with transfection
with PXN-siRNA alone, the apoptotic rate was greatly reduced after
co-transfection with PXN-siRNA and PXN-cDNA for 72 h in the SW480
cells (Fig. 5A). The expression of
apoptotic protein cleaved caspase-3 increased corresponding to the
apoptotic rate (Fig. 5A). Moreover,
the expression of PXN increased as expected after co-transfection,
compared with transfection with PXN-siRNA alone and activation of
PXN also upregulated p-Erk levels (Fig.
5A). Similarly, after co-transfection with PXN-siRNA and
PXN-cDNA for 72 h in the Caco-2 cells, the apoptotic rate was
greatly reduced, compared with transfection with PXN-cDNA alone
(Fig. 5B). Not surprisingly, the
expression of apoptotic protein cleaved caspase-3 increased, and
activation of PXN and p-Erk was inhibited as shown by
downregulation of p-Erk levels (Fig.
5B).
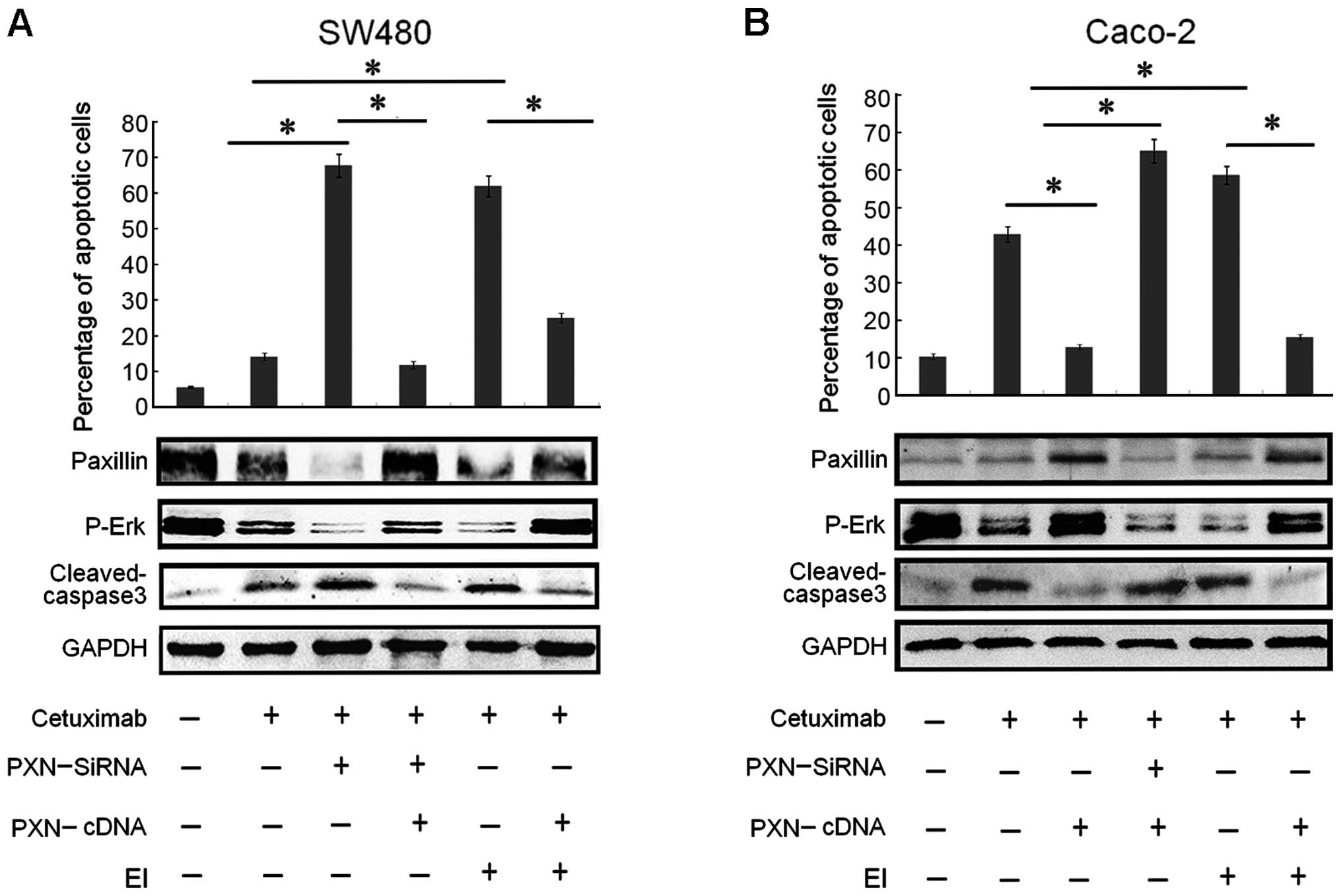 | Figure 5Paxillin overexpression regulates the
resistance to cetuximab in a p-Erk-dependent pattern in SW480 and
Caco-2 cells. (A) Both downregulation of paxillin and treatment
with a selective Erk inhibitor (EI) drastically increased the
apoptotic rate in SW480 cells, which were illustrated by
histograms. Columns: means of three independent experiments.
Cetuximab plus PXN-siRNA vs. cetuximab (*P<0.001),
PXN-siRNA plus cetuximab vs. PXN-cDNA plus PXN-siRNA plus cetuximab
(*P<0.001), cetuximab plus EI vs. PXN-cDNA plus EI
plus cetuximab (*P<0.005), cetuximab plus EI vs.
cetuximab (*P<0.001). The expression of apoptotic
protein cleaved caspase-3 in the SW480 cells changed corresponding
to its apoptotic rate. The expression of paxillin increased as
expected after co-transfection, compared with transfection with
PXN-siRNA alone. Activitation of paxillin also upregulated p-Erk
levels. (B) Although Caco-2 cells were relatively sensitive to
cetuximab, downregulation of paxillin and treatment with a
selective Erk inhibitor (EI) increased the apoptotic rate in the
Caco-2 cells, which was illustrated by histograms. Columns, means
of three independent experiments. Cetuximab plus PXN-cDNA vs.
cetuximab (*P<0.005), PXN-cDNA plus cetuximab vs.
PXN-cDNA plus PXN-siRNA plus cetuximab (*P<0.001),
cetuximab plus EI vs. cetuximab (*P<0.001), cetuximab
plus EI vs. PXN-cDNA plus EI plus cetuximab
(*P<0.001). The expression of apoptotic protein
cleaved caspase-3 corresponded to the apoptotic rate. The paxillin
expression decreased as expected after co-transfection, compared
with transfection with PXN-cDNA alone. Inhibition of paxillin
expression also downregulated p-Erk levels. |
From the experiments above, we found that the
expression levels of protein PXN and p-Erk were very synchronous.
To explore the relationship between PXN and p-Erk and further
evaluate the role of PXN activation, SW480 and Caco-2 cells were
treated with cetuximab and SCH-772984, a selective Erk1/2
inhibitor. SCH-772984 treatment of SW480 and Caco-2 cells induced a
dose-dependent inhibition of cell growth with an IC50
value of ~47.5 and 56.4 mg/ml, respectively (Fig. 3D). Although cetuximab treatment had
little effect on cell growth in the SW480 cells, the combined
treatment with SCH-772984 restored the sensitivity of SW480 cells
to cetuximab (Fig. 5A). Moreover,
as illustrated in Fig. 5A, the
percentage of apoptotic cells also obviously increased, compared
with the group treated with cetuximab or SCH-772984 alone.
Meanwhile, the expression levels of PXN and p-Erk were suppressed
significantly in both cell lines (Fig.
5A and B). Based on the outcomes above, another test was
performed to make the entire experiment more rigorous. After
transfection with PXN-cDNA in the SW480 and Caco-2 cells for 24 h,
the cells were treated with cetuximab and SCH-772984 for another 48
h. The results in Fig. 5A and B
showed that the percentage of apoptotic cells decreased
significantly, compared with the group that was treated only with
cetuximab or SCH-772984. Correspondingly, the downregulation of
cleaved caspase-3 and upregulation of both PXN and p-Erk expression
levels are illustrated in Fig. 5A and
B. In summary, knockdown of PXN restored or improved
sensitivity to cetuximab by downregu-lating the expression level of
p-Erk.
Discussion
Clarifying the mechanisms of colorectal cancer cell
resistance to cetuximab is critical for the development of
effective targeted therapies. In the past few years, extensive
effort has been made to elucidate the mechanisms of cetuximab
resistance. At present, it is generally acknowledged that specific
gene mutations are responsible for resistance to anti-EGFR
therapies in patients with colorectal cancer, including the KRAS,
BRAF, NRAS, PI3KCA (exon 20) genes or inactivation of the PTEN
phosphatase (3,8,18).
However, ~25% of colorectal cancer patients with wild-type KRAS,
BRAF, NRAS, PI3KCA and PTEN genes do not respond to EGFR inhibitors
(3), which suggests there exists
some other mechanism of resistance.
By analyzing the clinical data, it was demonstrated
that the positive rate of paxillin (PXN) was much higher in the
adenocarcinoma samples than in the normal mucosa samples (68.2 vs.
44.4%, P<0.001). The rate of recurrence in the colorectal cancer
patients was higher in the PXN-positive group than this rate in the
PXN-negative group (27.7 vs. 12.8%, P=0.032). PXN presence was
closely related with TNM stage (86.4, 74.6, 61.5 and 50.0%,
P=0.023) and distant metastasis (88 vs. 64.2%, P=0.014). Univariate
analyses revealed that the 3- and 5-year survival rates of the
PXN-negative group were higher (71.8 and 65.3%) than those of the
positive group (52.5 and 35.4%, log-rank testing, P=0.004). Above
all, PXN could be a risk factor for distant metastasis, recurrence
and reduced survival time. However, PXN was not found to be an
independent predictor due to the small number of samples and
follow-up time was not long enough. Several studies have shown that
PXN plays an important role in regulating tumor cell proliferation
and mediating signal transduction of epidermal growth factor (EGF)
(2,10–12).
Various scholars have also demonstrated that PXN leads to
resistance to drugs by inhibiting the apoptosis of cancer cells in
non-small cell lung cancer (17).
In the present study, following treatment of cetuximab in SW480 and
Caco-2 cells for a period of time, the expression of PXN in SW480
cells declined at first, and then gradually increased and finally
stabilized, which was consistent with the proliferation rate.
Different from SW480 cells, PXN expression in Caco-2 cells
decreased gradually and the downward trend also corresponded to the
proliferation curve. These data demonstrated that a high expression
level of PXN may be one of the reasons for the insensitivity to
cetuximab of SW480 cells in addition to the fact that the KRAS gene
in these cells is mutant. It was hypothesized that high expression
of PXN could be one reason for cetuximab resistance in metastatic
colorectal cancer (mCRC) patients.
The SW480 cell line was chosen to conduct the first
part of the experiment. It was reported that the expression of PXN
in SW480 cells is very high and its KRAS gene is in a mutational
status (2,4). In the present study, we confirmed the
high expression of PXN and that SW480 cells were not sensitive to
cetuximab. The main reason for the resistance to cetuximab may be
that the signaling pathway in the downstream of KRAS was not
blocked although EGFR was inhibited by cetuximab treatment
(19,20). The high expression of PXN may also
be one of the reasons. Our results showed that knockdown of PXN by
PXN-siRNA transfection restored the sensitivity to cetuximab in the
SW480 cells. As for the mechanisms, it is assumed that PXN plays a
role in regulating the signaling pathway in the downstream of KRAS.
As described in the Introduction, activation of the ERK signaling
pathway requires the participation and mediation of PXN in prostate
cancer and non-small cell lung cancer (10,17).
Moreover, Ishibe et al demonstrated that PXN serves as a
scaffold for the organization and activation of the MAPK proteins
Raf, Mek and Erk at focal adhesions (21). Ishibe et al also pointed out
that Erk can either associate directly with PXN or interact via an
intermediate protein (11). Several
studies have demonstrated that Erk always gives a positive feedback
on PXN and plays an important part in the activitation of PXN and
interaction between FAK and PXN (11,22,23).
Based on these facts, we decided to examine the expression of p-Erk
during this experiment. Following different treatments, the
expression levels of p-Erk and PXN were always consistent.
Moreover, the cells were treated with a selective Erk inhibitor,
which resulted in downregulation of PXN and p-Erk, upregulation of
apoptotic protein cleaved caspase-3 and an increase in the
apoptotic rate. These results suggest that low expression of p-Erk
could significantly increase the percentage of apoptotic cells and
PXN may play a relevant role in regulating the expression of p-Erk.
To further evaluate the role of PXN, upregulation of PXN expression
was carried out in the Caco-2 cell line since the expression of PXN
in Caco-2 cells is low and its KRAS gene is wild-type (4). Thus, PXN-cDNA transfection was more
effective. Upregulation of PXN by PXN-cDNA decreased the percentage
of apoptotic cells and increased the expression levels of p-Erk and
paxilllin in the Caco-2 cells. Treatment of Erk inhibitor was also
carried out in Caco-2 cells, the results of which were similar with
that in the SW480 cells.
The results of the present study suggest that PXN
plays a relevant role in cetuximab resistance, and inhibition of
PXN expression restores the sensitivity to cetuximab in colorectal
cancer cells by downregulating p-Erk levels. In order to find more
evidence to support the conclusion above, the co-transfection of
PXN-cDNA and PXN-siRNA was performed in SW480 and Caco-2 cells.
Compared with transfection with PXN-siRNA or PXN-cDNA alone, the
same results were obtained following co-transfection in both the
SW480 and Caco-2 cells, showing that overexpression of PXN could be
one of the reasons for cetuximab resistance and downregulation of
PXN plays an important role in improving the sensitivity to
cetuximab by suppressing the activation of p-Erk in SW480 and
Caco-2 cells. Moreover, the MAPK pathway was completely blocked
when activation of p-Erk was inhibited. Knockdown of PXN in
combination with cetuximab blocked the EGF pathway more completely,
particularly when the genes located upstream of Erk were mutant.
Besides the overexpression of PXN, there must exist other reasons
for cetuximab resistance. In the past few years, researchers have
focused on the relationship among apoptosis, drug resistance and
autophagy in cancer cells. Various studies have demonstrated that
increased induction of autophagy can become a mechanism of allowing
tumor cells to survive the conditions of hypoxia, acidosis or
chemotherapy, which results in a decrease in apoptosis and drug
resistance (24,25). There are also other studies
indicating out that autophagy inhibitors in combination with
chemotherapeutic agents may provide a premise for the treatment of
colorectal cancer (26–28). Taken together, these results suggest
that overexpression of PXN could be one of the reasons for
cetuximab resistance, and knockdown of PXN plays an important role
in improving sensitivity to cetuximab in colorectal cancer cells.
Downregulation of the p-Erk level that results from the decreased
expression of PXN blocks the EGF/MAPK pathway more completely.
Therefore, knockdown of PXN represents a rational therapeutic
strategy for increasing the sensitivity or overcoming cetuximab
resistance in patients with colorectal cancer.
References
|
1
|
Troiani T, Martinelli E, Napolitano S,
Vitagliano D, Ciuffreda LP, Costantino S, Morgillo F, Capasso A,
Sforza V, Nappi A, et al: Increased TGF-α as a mechanism of
acquired resistance to the anti-EGFR inhibitor cetuximab through
EGFR-MET interaction and activation of MET signaling in colon
cancer cells. Clin Cancer Res. 19:6751–6765. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yin H, Zhang Q, Wang X, Li T, Wan Y, Liu Y
and Zhu J: Role of paxillin in colorectal carcinoma and its
relationship to clinico-pathological features. Chin Med J.
127:423–429. 2014.
|
|
3
|
Bardelli A and Siena S: Molecular
mechanisms of resistance to cetuximab and panitumumab in colorectal
cancer. J Clin Oncol. 28:1254–1261. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shigeta K, Hayashida T, Hoshino Y,
Okabayashi K, Endo T, Ishii Y, Hasegawa H and Kitagawa Y:
Expression of epidermal growth factor receptor detected by
cetuximab indicates its efficacy to inhibit in vitro and in vivo
proliferation of colorectal cancer cells. PLoS One. 8:e663022013.
View Article : Google Scholar :
|
|
5
|
Galizia G, Lieto E, De Vita F, Orditura M,
Castellano P, Troiani T, Imperatore V and Ciardiello F: Cetuximab,
a chimeric human mouse anti-epidermal growth factor receptor
monoclonal antibody, in the treatment of human colorectal cancer.
Oncogene. 26:3654–3660. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Van Cutsem E, Köhne CH, Láng I, Folprecht
G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D,
Tejpar S, et al: Cetuximab plus irinotecan, fluorouracil, and
leucovorin as first-line treatment for metastatic colorectal
cancer: Updated analysis of overall survival according to tumor
KRAS and BRAF mutation status. J Clin Oncol. 29:2011–2019. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen MC, Chiang FF and Wang HM: Cetuximab
plus chemotherapy as first-line treatment for metastatic colorectal
cancer: Effect of KRAS mutation on treatment efficacy in Taiwanese
patients. Neoplasma. 60:561–567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
De Roock W, Claes B, Bernasconi D, De
Schutter J, Biesmans B, Fountzilas G, Kalogeras KT, Kotoula V,
Papamichael D, Laurent-Puig P, et al: Effects of KRAS, BRAF, NRAS,
and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy
in chemotherapy-refractory metastatic colorectal cancer: A
retrospective consortium analysis. Lancet Oncol. 11:753–762. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Misale S, Yaeger R, Hobor S, Scala E,
Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M,
Siravegna G, et al: Emergence of KRAS mutations and acquired
resistance to anti-EGFR therapy in colorectal cancer. Nature.
486:532–536. 2012.PubMed/NCBI
|
|
10
|
Munshi N, Groopman JE, Gill PS and Ganju
RK: c-Src mediates mitogenic signals and associates with
cytoskeletal proteins upon vascular endothelial growth factor
stimulation in Kaposi's sarcoma cells. J Immunol. 164:1169–1174.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishibe S, Joly D, Zhu X and Cantley LG:
Phosphorylation-dependent paxillin-ERK association mediates
hepatocyte growth factor-stimulated epithelial morphogenesis. Mol
Cell. 12:1275–1285. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sen A, O'Malley K, Wang Z, Raj GV,
Defranco DB and Hammes SR: Paxillin regulates androgen-and
epidermal growth factor-induced MAPK signaling and cell
proliferation in prostate cancer cells. J Biol Chem.
285:28787–28795. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Turner CE: Paxillin interactions. J Cell
Sci. 113:4139–4140. 2000.PubMed/NCBI
|
|
14
|
Xiao LJ, Zhao EH, Zhao S, Zheng X, Zheng
HC, Takano Y and Song HR: Paxillin expression is closely linked to
the pathogenesis, progression and prognosis of gastric carcinomas.
Oncol Lett. 7:189–194. 2014.
|
|
15
|
Jun Q, Zhiwei W, Lilin M, Jing K and
Qichao N: Effects of paxillin on HCT-8 human colorectal cancer
cells. Hepatogastroenterology. 58:1951–1955. 2011.PubMed/NCBI
|
|
16
|
Chen DL, Wang DS, Wu WJ, Zeng ZL, Luo HY,
Qiu MZ, Ren C, Zhang DS, Wang ZQ, Wang FH, et al: Overexpression of
paxillin induced by miR-137 suppression promotes tumor progression
and metastasis in colorectal cancer. Carcinogenesis. 34:803–811.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu DW, Wu TC, Wu JY, Cheng YW, Chen YC,
Lee MC, Chen CY and Lee H: Phosphorylation of paxillin confers
cisplatin resistance in non-small cell lung cancer via activating
ERK-mediated Bcl-2 expression. Oncogene. 33:4385–4395. 2014.
View Article : Google Scholar
|
|
18
|
Therkildsen C, Bergmann TK,
Henrichsen-Schnack T, Ladelund S and Nilbert M: The predictive
value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment
in metastatic colorectal cancer: A systematic review and
meta-analysis. Acta Oncol. 53:852–864. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brand TM, Iida M and Wheeler DL: Molecular
mechanisms of resistance to the EGFR monoclonal antibody cetuximab.
Cancer Biol Ther. 11:777–792. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kumar SS, Price TJ, Mohyieldin O, Borg M,
Townsend A and Hardingham JE: KRAS G13D mutation and sensitivity to
cetuximab or panitumumab in a colorectal cancer cell line model.
Gastrointest Cancer Res. 7:23–26. 2014.PubMed/NCBI
|
|
21
|
Ishibe S, Joly D, Liu ZX and Cantley LG:
Paxillin serves as an ERK-regulated scaffold for coordinating FAK
and Rac activation in epithelial morphogenesis. Mol Cell.
16:257–267. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Güller MC, André J, Legrand A, Setterblad
N, Mauviel A, Verrecchia F, Daniel F and Bernuau D: c-Fos
accelerates hepatocyte conversion to a fibroblastoid phenotype
through ERK-mediated upregulation of paxillin-Serine178
phosphorylation. Mol Carcinog. 48:532–544. 2009. View Article : Google Scholar
|
|
23
|
Teranishi S, Kimura K and Nishida T: Role
of formation of an ERK-FAK-paxillin complex in migration of human
corneal epithelial cells during wound closure in vitro. Invest
Ophthalmol Vis Sci. 50:5646–5652. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Çoker-Gürkan A, Arisan ED, Obakan P and
Palavan-Unsal N: Lack of functional p53 renders DENSpm-induced
autophagy and apoptosis in time dependent manner in colon cancer
cells. Amino Acids. 47:87–100. 2015. View Article : Google Scholar
|
|
25
|
Sui X, Kong N, Wang X, Fang Y, Hu X, Xu Y,
Chen W, Wang K, Li D, Jin W, et al: JNK confers 5-fluorouracil
resistance in p53-deficient and mutant p53-expressing colon cancer
cells by inducing survival autophagy. Sci Rep. 4:46942014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li J, Hou N, Faried A, Tsutsumi S,
Takeuchi T and Kuwano H: Inhibition of autophagy by 3-MA enhances
the effect of 5-FU-induced apoptosis in colon cancer cells. Ann
Surg Oncol. 16:761–771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishikawa T, Tsuno NH, Okaji Y, Shuno Y,
Sasaki K, Hongo K, Sunami E, Kitayama J, Takahashi K and Nagawa H:
Inhibition of autophagy potentiates sulforaphane-induced apoptosis
in human colon cancer cells. Ann Surg Oncol. 17:592–602. 2010.
View Article : Google Scholar
|
|
28
|
Zhai H, Song B, Xu X, Zhu W and Ju J:
Inhibition of autophagy and tumor growth in colon cancer by
miR-502. Oncogene. 32:1570–1579. 2013. View Article : Google Scholar
|