Introduction
Glioblastoma multiforme (GBM) is the most common
malignant and aggressive primary human brain tumor (1). It is highly proliferative, invasive
and chemoresistant, thus, that even with the combined treatment of
surgery, chemotherapy and radiotherapy, the 5-year survival rate of
WHO grade IV glioblastoma remains at less than 5% (2,3).
Although significant advances have been made in our understanding
of the molecular status of this tumor type, novel efficacious
therapeutic avenues are critically needed.
Natural products have received recent interest in
the discovery of novel anticancer therapeutic agents as have long
been used as alternative remedies for a variety of diseases
including cancer with relatively few side-effects (4,5).
Nitidine chloride (NC), a natural bioactive phytochemical alkaloid,
has displayed anticancer activity in various types of cancer, such
as ovarian, colorectal, hepatic and renal cancers (6–9).
However, whether NC exhibits tumor inhibiting activity on GBM cells
and the molecular mechanisms that underlie its tumoricidal activity
remain largely unknown.
In the present study, GBM cell lines were exposed to
varying concentrations of NC in vitro, and efficacy was
evaluated in functional assays for proliferation, migration and
invasion, and apoptosis. Alterations in metabolism and
mitochondrial function were also investigated as well as changes in
potential molecular targets in order to understand the basis of its
activity. Our results revealed NC as a potential therapeutic agent
in the treatment of human GBM and that the molecular basis for
tumor inhibition may be through targeting of the PI3K/Akt/mTOR
signaling pathway.
Materials and methods
Cell lines and cultures
Human glioma cell lines (U251 and U87) were
purchased from the Chinese Academy of Sciences Cell Bank (Shanghai,
China). Both U251 and U87 have been recently authenticated through
cross species checks, DNA fingerprinting and quarantine. Cells were
grown in Dulbecco's modified Eagle's medium (DMEM; SH30022.01B;
Gibco, GE Healthcare Life Sciences, Pittsburgh, PA, USA)
supplemented with 10% fetal bovine serum (FBS) (10082147; HyClone,
GE Healthcare Life Sciences) in a humidified incubator with 5%
CO2 at 37°C.
Cell viability and proliferation
assays
Cell viability was assessed with the Cell Counting
Kit-8 (CCK-8; CK04-500; Dojindo, Kumamoto, Japan). Tumor cells
(1.0×104 cells/well) were seeded into 96-well,
flat-bottomed plates with DMEM containing 10% FBS and incubated at
37°C overnight. Nitidine chloride (NC) (N117977; Aladdin Biotech,
Shanghai, China) or SC79 (SML0749) was dissolved in dimethyl
sulfoxide (DMSO) (D2650) (both from Sigma-Aldrich, St. Louis, MO,
USA) and diluted to working concentrations in culture medium. After
the desired treatment, the cells were incubated for an additional 4
h at 37°C with 10 µl of CCK-8 in 100 µl of serum-free
DMEM. The absorbance at 450 nm was measured using a microplate
reader (Bio-Rad, Hercules, CA, USA). Proliferation was assessed
using the EdU incorporation assay according to the manufacturer's
protocol (C103103; RiboBio, Guangzhou, China). Briefly, EdU was
incorporated into the proliferating cells and was detected through
a catalyzed reaction with a fluorescently labeled azide. The
percentage of labeled cells (ratio:
EdU+/DAPI+ × 100%) was determined using
images captured in four random fields per sample under
fluorescence.
Cell migration and invasion assays
Cell migration was assessed in wound healing assays.
U251 or U87 glioma cells (1×105/well) were seeded into
6-well plates and incubated overnight. A cell-free gap was
generated by scratching the cell monolayer with a 200 µl
pipette tip. The scratched plates were cultured in DMEM containing
1% FBS, and images used for analysis were captured under a light
microscope at 0 and 24 h. Cell invasion was examined using the
Transwell chamber assay. The bottom of the Transwell membrane was
pretreated with Matrigel (Becton-Dickinson, Bedford, MA, USA) for 4
h. Cells (5×104) were re-suspended in serum-free DMEM
and seeded into the upper chamber of a Transwell apparatus
(8.0-µm pore; Corning, Sigma-Aldrich). DMEM containing 10%
FBS (600 µl) was added to the lower chamber, and chambers
were incubated at 37°C for 24 h. Cells that had migrated to the
bottom of the membrane after 24 h were fixed and stained with
crystal violet for 15 min, while those remaining in the upper
chamber were removed with a cotton swab. Five random views from
images acquired under a light microscope were used to count the
migrated cells.
Measurement of mitochondrial membrane
potential (Δψm) and mitochondrial morphology
Live cells were seeded into 96-well plates and
loaded with tetramethylrhodamine methyl ester (JC-1) for 30 min
according to the manufacturer's protocol. Fluorescence was measured
in a microplate reader, and the fluorescence intensity ratio of
JC-1 aggregates to JC-1 monomers [ratio of 590 nm:530 nm emission
intensities; (10)] was used to
determine the Δψm. For mitochondrial morphology, live cells were
seeded in 6-well plates and fluorescently labeled with 25 nM
MitoTracker Red (Molecular Probes, Thermo Fisher Scientific,
Waltham, MA, USA). In both analyses, cells were examined under an
Olympus BX61 fluorescence microscope, and images were acquired
using a DP71 charge-coupled device (CCD) digital camera (Olympus,
Waltham, MA, USA).
Western blotting
U251 and U87 glioma cells were harvested, rinsed
with phosphate-buffered saline (PBS) and lysed with RIPA buffer
(P0013B; Beyotime, Shanghai, China) containing 1%
phenylmethylsulfonyl fluoride. Protein concentrations were
determined using the BCA method (23225; Beyotime). Proteins (20
µg) were separated on 8–15% SDS-PAGE and transferred onto
polyvinylidene fluoride (PVDF) membranes (ISEQ00010 0.22 µm;
Millipore, Billerica, MA USA). Membranes were blocked for 2 h at
room temperature (RT) with 5% non-fat dry milk in TBST [20 mmol/l
Tris-HCL (pH 8.0), 137 mmol/l NaCl and 0.1% Tween-20 or with 5% BSA
in TBST for phospho-proteins], incubated with primary antibodies
overnight at 4°C, rinsed with TBST, and probed with horseradish
peroxidase (HRP)-conjugated secondary antibody (1:5,000; Santa Cruz
Biotechnology, Dallas, TX, USA) for 1 h at RT. Proteins were
visualized with enhanced chemiluminescence (ECL; Millipore) and the
ChemiDoc Touch detection system (Bio-Rad). The following primary
antibodies were used: rabbit anti-cytochrome c, Bcl-2,
Drp-1, AKT, p-Akt (Ser473), mTOR, p-mTOR (Ser2448), GAPDH (Cell
Signaling Technology, Danvers, MA, USA), rabbit anti-mitofusion-1
and Bax (Abcam, Cambridge, MA, USA).
Measurement of ATP and lactate
Cells (2×105/well) were seeded into
6-well plates. After incubation overnight, the culture medium
supernatants were collected for the determination of the
concentration of lactate. Cells were subsequently trypsinized and
counted for ATP concentrations and normalization. Total ATP and
lactate concentrations were determined using the CellTiter-Glo
Luminescent assay (Promega, Madison, WI, USA) and the L-lactate
colorimetric assay kit (Abcam), respectively, according to the
manufacturer's protocols. Data were normalized to the number of
cells.
Immunofluorescence
U251 and U87 cells were fixed with 4% formaldehyde
in PBS, permeabilized with 0.5% Triton X-100 in PBS, incubated with
rabbit anti-cytochrome c antibody (1:400) in 5% bovine serum
albumin in PBS overnight, and labeled with FITC- and DyLight
594-conjugated anti-rabbit IgG (Santa Cruz Biotechnology). Cells
were incubated in the dark with Rhodamine phalloidin (PHDR1;
Cytoskeleton, Inc., Denver, CO, USA) and DAPI to stain actin and
nuclei, respectively. Slides were examined under an Olympus BX61
fluorescence microscope, and images were acquired using a DP71 CCD
digital camera.
Flow cytometric analysis of
apoptosis
U251 and U87 glioma cells were harvested,
re-suspended in binding buffer, and incubated with Annexin V-FITC
(BD Biosciences, Franklin Lakes, NJ, USA) according to the
manufacturer's instructions. Apoptotic cells were detected by flow
cytometry (NovoCyte Flow Cytometer; ACEA Biosciences, San Diego,
CA, USA), and the results were analyzed using the software FlowJo
(Tree Star, Ashland, OR, USA).
Statistical analysis
Three independent experiments were performed and the
results are expressed as the mean ± standard deviation (SD). Data
were compared using paired t-tests in GraphPad Prism 5 software
(San Diego, CA, USA), and P-values determined from different
comparisons are indicated in the figures as follows:
*P<0.05, **P<0.01 and
***P<0.001.
Results
NC inhibits proliferation of GBM
cells
To determine whether NC may be effective against
GBM, NC treatment was first evaluated in U87 and U251 cells in
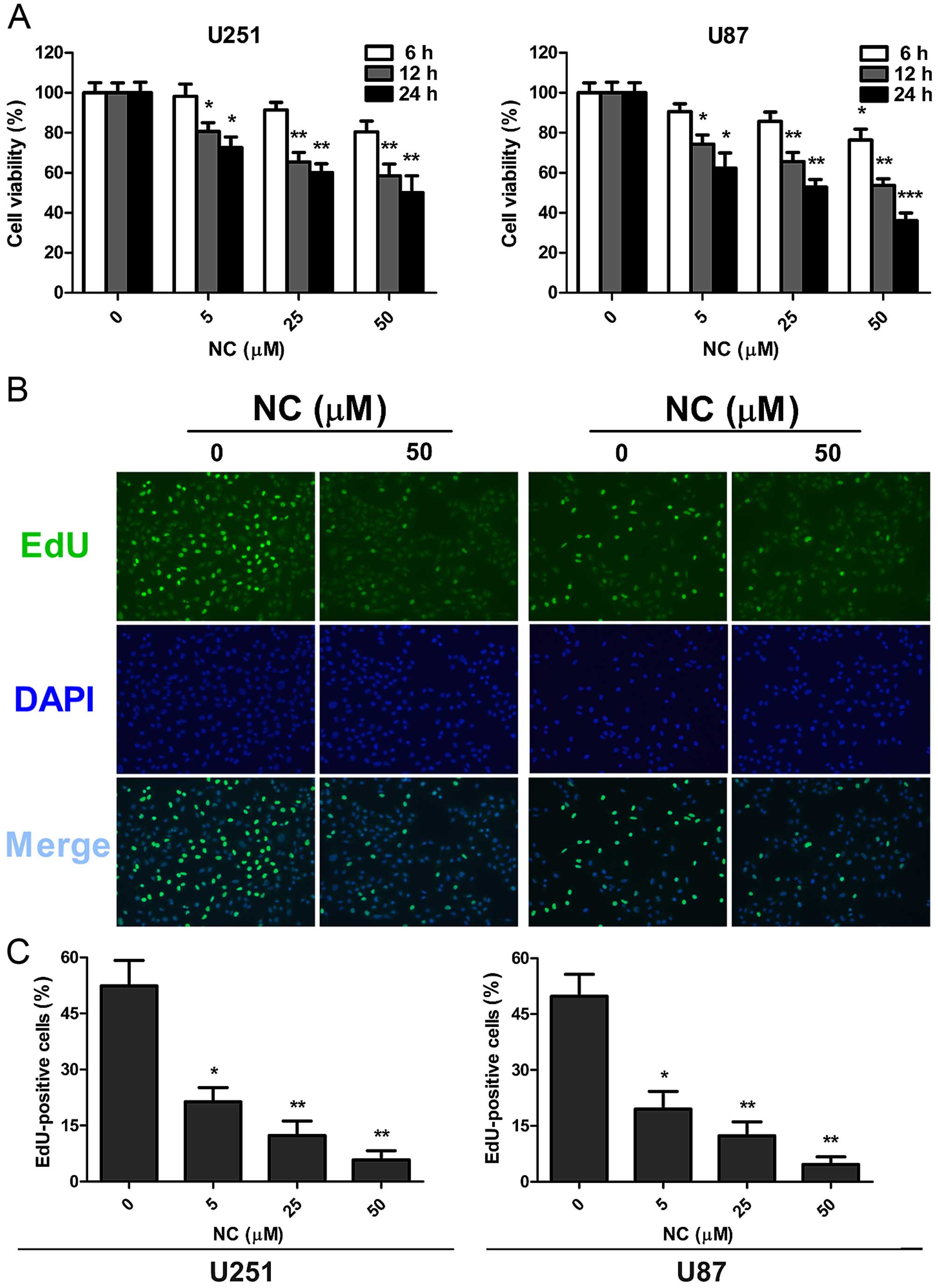
vitro, using the cell viability assay, CCK-8 (Fig. 1A). Cells were treated with differing
concentrations of NC in vitro, and viability was assessed at
6, 12 and 24 h. Decreases in cell viability relative to the
untreated cells were statistically significant at the 12 and 24 h
time points at all concentrations of NC. The most dramatic decrease
in viability was ~50% for both cell lines following treatment with
50 µM NC at 24 h.
EdU incorporation was used to further evaluate
proliferation of the GBM cell lines in the presence of NC.
Quantification of EdU incorporation revealed a statistically
significant decrease in proliferation for both cell lines at 24 h
after exposure to NC at all concentrations. The NC concentration of
50 µM however was the most effective in both cell lines (~50
vs. 5%; untreated vs. treated cells). These results indicated that
NC potently arrested proliferation in both the U251 and U87 cells
and in a dose-dependent manner (Fig. 1B
and C).
NC attenuates migration and invasion of
GBM cells
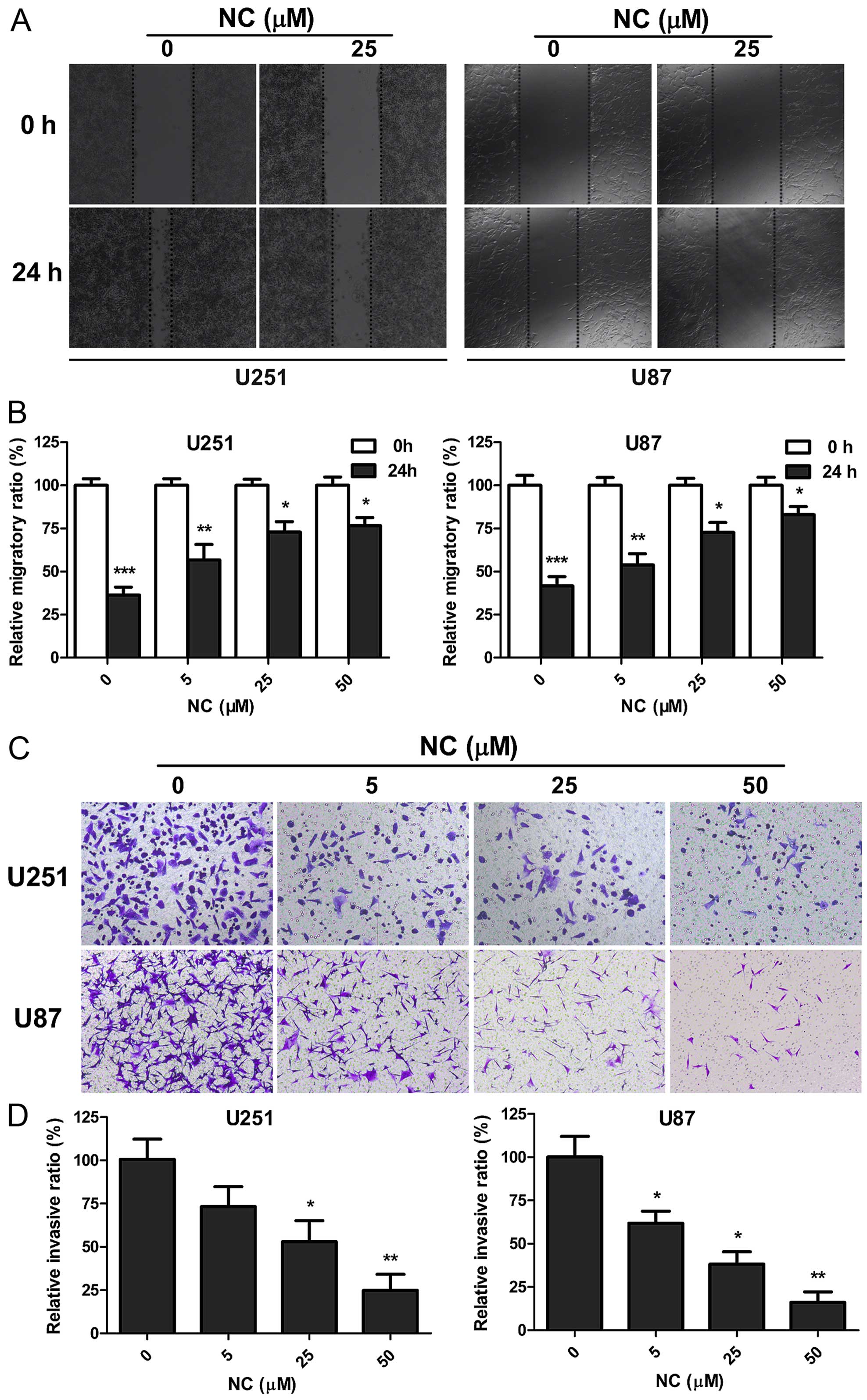
To determine whether NC inhibits the migration and
invasive properties of GBM cells, wound healing and invasion assays
were performed on U251 and U87 cells in the presence of different
concentrations of NC. Wound closure at 24 h in monolayer culture
was inhibited at all NC concentrations for both U251 and U87 cells
(Fig. 2A and B). Increasing NC
concentrations were more effective, with only partial wound closure
at 50 µM after 24 h (~20%; P<0.05). The Transwell assay
was used to further evaluate the effect of NC treatment on the
invasion of GBM cells. Upper chambers of Transwell apparatuses were
coated with Matrigel in order to provide extracellular matrix (ECM)
simulating a normal tumor microenvironment, and seeded with U251 or
U87 cells. Counts of cells that had migrated through the membrane
at 24 h were decreased with increasing concentrations of NC. At 50
µM, cell counts were decreased by ~80% for both cell lines
(Fig. 2C and D). These results
indicate that NC profoundly inhibited migration and invasion of
U251 and U87 cells in vitro.
NC inhibits production of ATP and
L-lactate in GBM cells
Tumor cell invasion is a high energy and nutrient
consuming process (11,12), with increased glycolysis as one of
the most prominent metabolic alterations universally occurring in
cancer cells (13). Molecules that
may uncouple glycolysis have thus been the basis for several
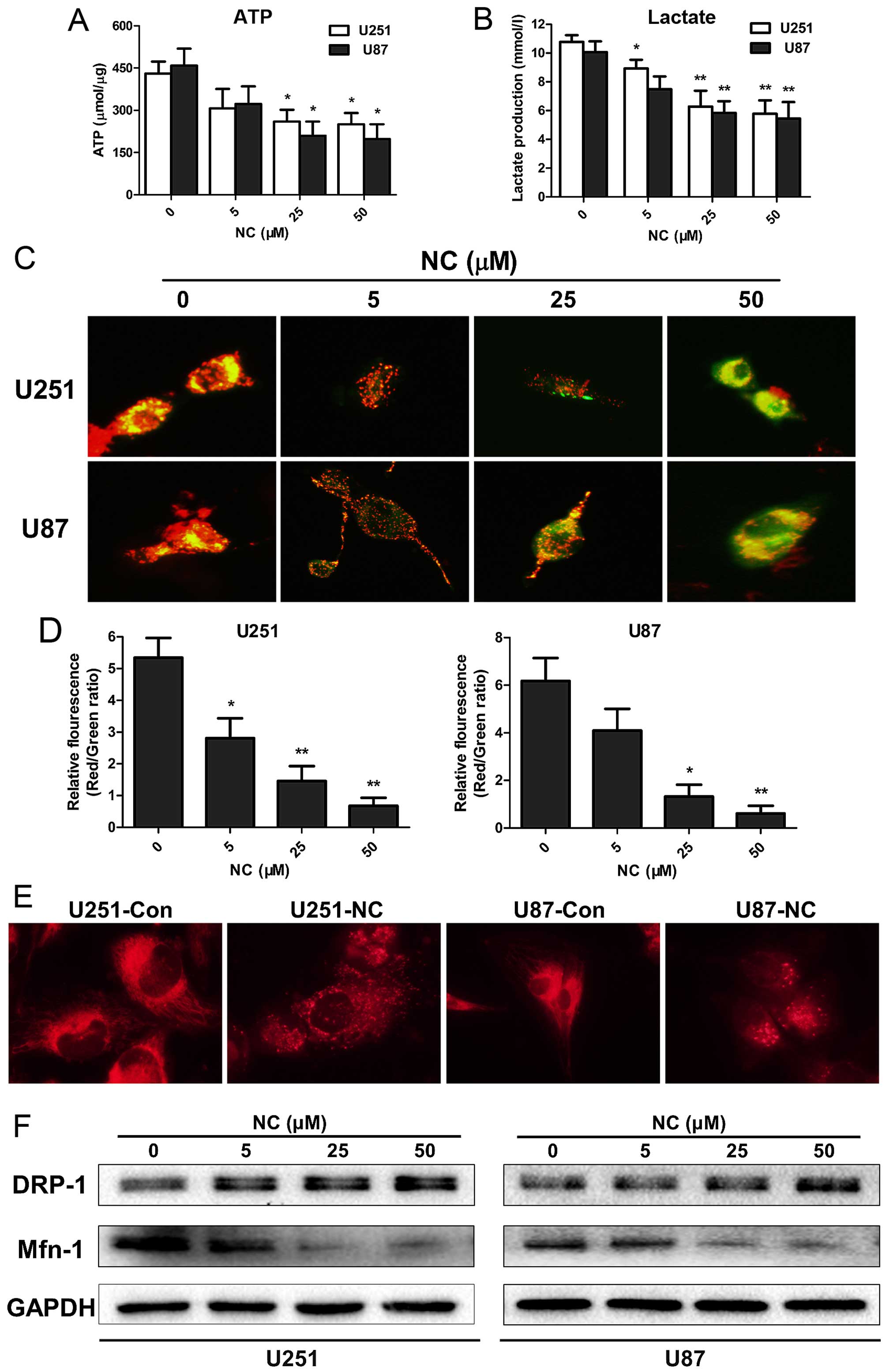
treatment strategies for cancer. To begin to understand whether NC
may interfere with cancer cell metabolism, ATP synthesis levels
were first examined in the NC-treated GBM cells. In both the U251
and U87 cells, ATP levels were moderately decreased after treatment
with NC at 25 and 50 µM concentrations at 24 h (P<0.05;
Fig. 3A). L-lactate is the main
metabolic product generated in glycolysis, and high levels are
typically observed in cell culture media collected from cancer
cells. L-lactate concentrations in cell media from U251 and U87
cells were therefore also evaluated under treatment with NC at 24
h. Maximum decreases in L-lactate concentrations were found in
media from both U251 and U87 cells following NC treatment at 25
µM with no further enhancement at 50 µM (P<0.01).
These results indicated that NC treatment suppressed glycolysis in
the GBM cells (Fig. 3B).
Mitochondrial function is altered in GBM
cells treated with NC
Mitochondria are the key organelles generating ATP
in the cell, and the Δψm is a critical factor in the production of
ATP. Changes in Δψm detected on the basis of fluorescence from the
JC-1 probe can be a signal of reduced mitochondrial function. The
JC-1 probe is an indicator of Δψm, which emits red fluorescence
upon formation of J-aggregates under high Δψm conditions and green
fluorescence from J-monomers under low Δψm conditions. Thus, the
conversion between red and green fluorescence directly reflects
changes in Δψm (14). In the U251
and U87 cells, green fluorescence was increased following treatment
with NC at 25 and 50 µM concentrations (Fig. 3C) as demonstrated by the decrease in
the red/green fluorescence ratios (Fig.
3D). These results indicate that mitochondrial activity was
suppressed in the NC-treated U251 and U87 cells relative to the
untreated cells.
ATP production has been associated with changes in
mitochondrial morphology which largely occur due to a dynamic
switch between fusion and fission states in mitochondria in order
to accommodate diverse cellular scenarios (15). The fusion state, for example, has
been found to be a more favorable form in cell invasion apices
while the fission state was associated more often with cell
trafficking (16). To determine
whether mitochondrial morphology was altered under NC treatment,
live cells were exposed to the fluorescent dye MitoTracker Red
which is selective for the staining of mitochondria. The results
demonstrated that the NC-treated cells favored the fission state,
while in the untreated GBM cells the fusion state predominated
(Fig. 3E).
Mitochondrial fission and fusion status is also
accompanied by changes in proteins specifically associated with
each of these processes. The protein Drp-1, for example, is
essential for mitochondrial fission, while the protein Mfn-1 is
required for mitochondrial fusion (17). To determine whether the levels of
these proteins paralleled the morphological changes observed,
western blot analyses were performed with lysates prepared from the
U251 and U87 cells treated with NC at different concentrations for
24 h. Protein levels of Drp-1 remained unchanged at all
concentrations of NC. Mfn-1 protein levels were however decreased
by ~2-fold at 25 and 50 µM NC concentrations (Fig. 3F). Taken together, these results
indicate that NC treatment resulted in the inhibition of glycolysis
and a decrease in mitochondrial function through changes in Δψm and
mitochondrial morphology in the GBM cells.
NC induces mitochondrial apoptosis in GBM
cells
Studies have shown that mitochondrial fission
renders cells more sensitive to apoptosis (17). Our results above revealed that the
morphology of mitochondria transitioned from fusion to fission
after exposure to NC. We therefore investigated whether NC induces
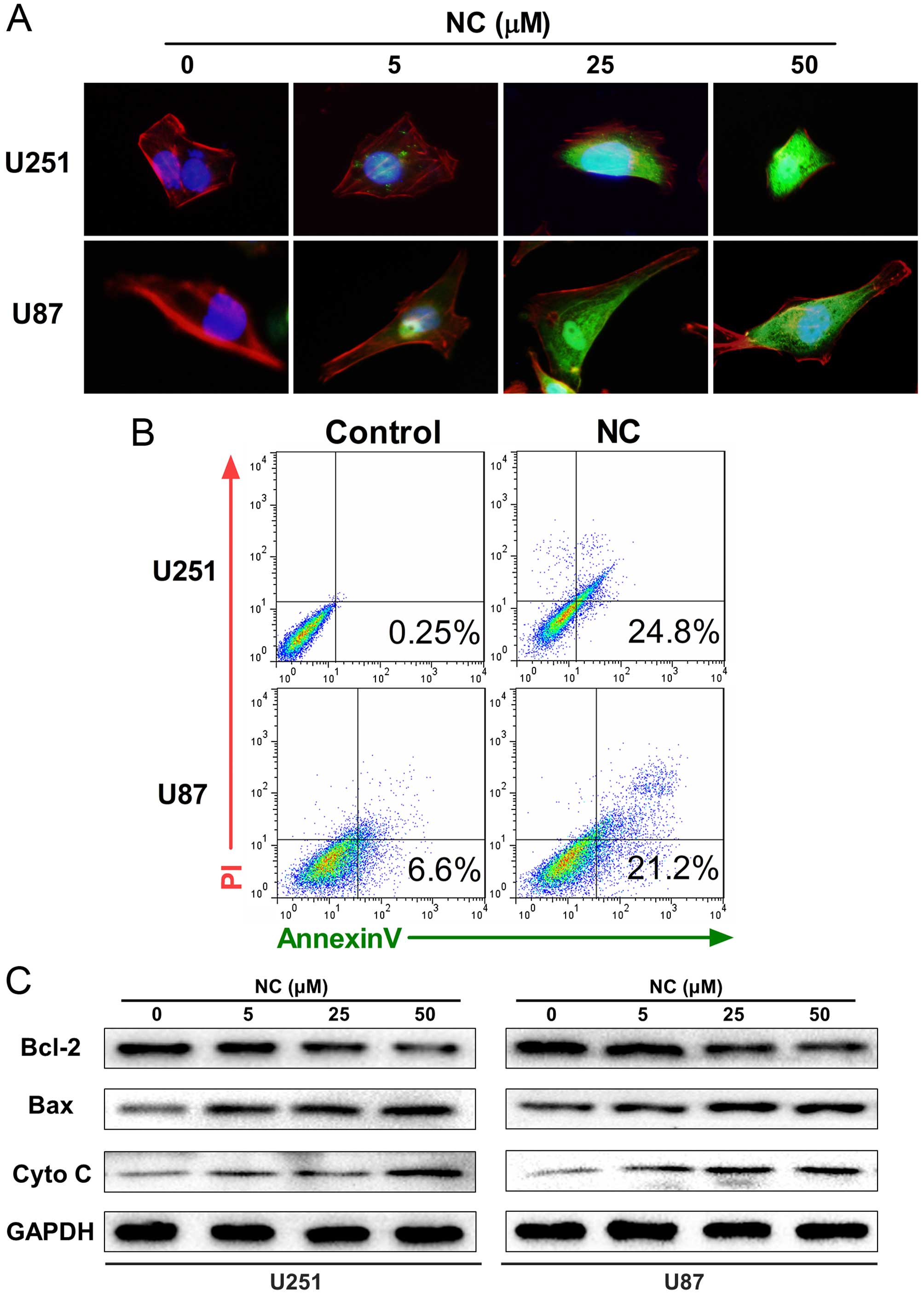
mitochondrial apoptosis in GBM cells. Immunofluorescent imaging
(IFI) was first performed in order to localize cytochrome c
which is released from mitochondria into the cytosol during
apoptosis. Cytochrome c was found to be increased in the
cytosol in the NC-treated U251 and U87 cells, particularly at 25
and 50 µM concentrations after 24 h (Fig. 4A). The percentage of cells
undergoing apoptosis was subsequently determined through FACS
analysis of Annexin V/PI staining. Increases in apoptosis were
observed in both the U251 and U87 cells in early (Annexin
V+/PI−) and late (Annexin
V+/PI+) stages with 50 µM NC at 24 h
(Fig. 4B).
Finally, western blot analysis was used to determine
the expression levels of mitochondrial proteins executing
apoptosis, including Bax and cytochrome c, as well as Bcl-2,
which inhibits apoptosis, in the NC-treated cells. Treatment with
NC led to increases in Bax and cytochrome c and simultaneous
decreases in Bcl-2 compared to the untreated U251 and U87 cells
(Fig. 4C).
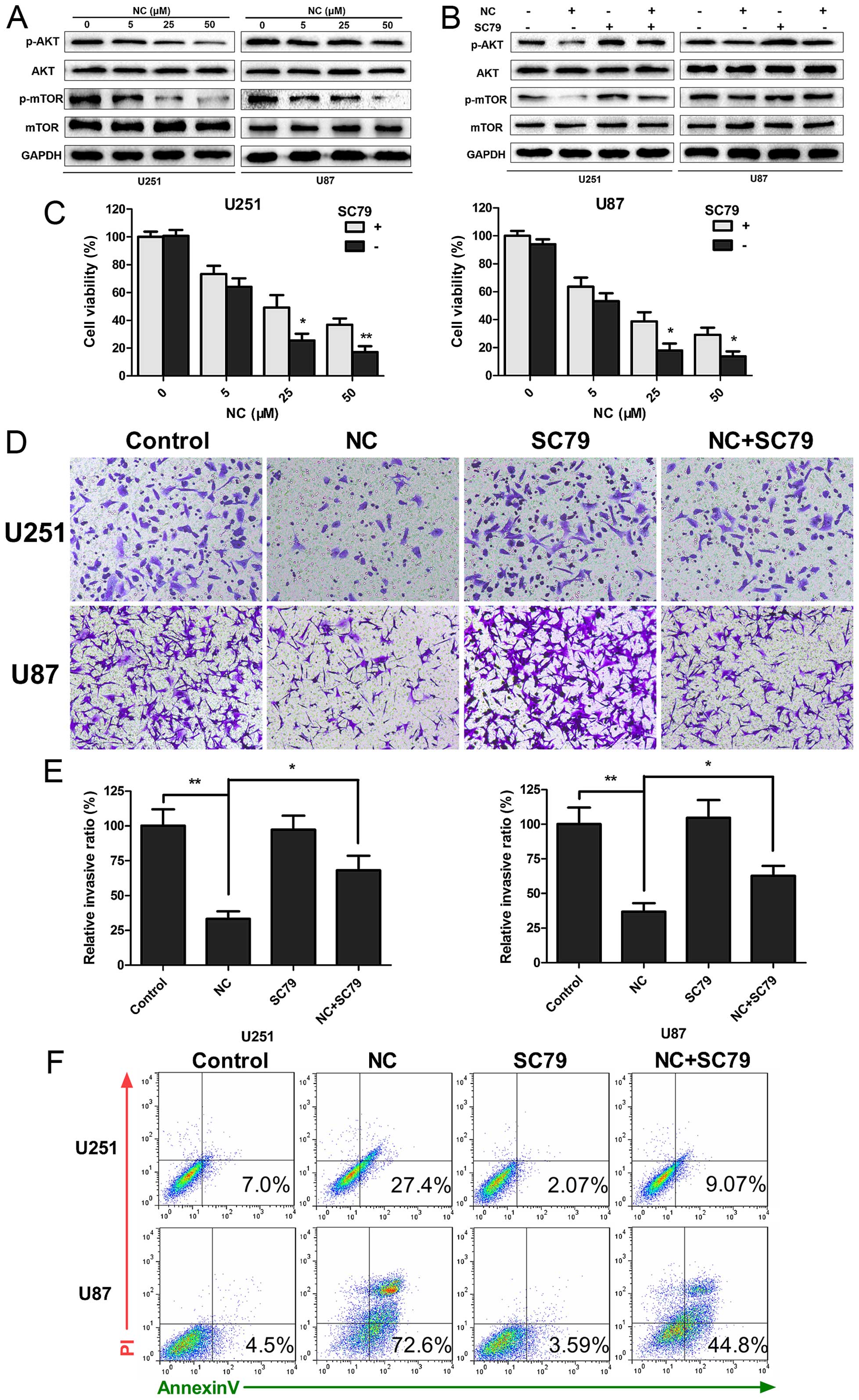
NC inhibits PI3K/Akt/mTOR signaling
Aberrant activation of the PI3K/Akt/mTOR signaling
pathway is known to promote tumorigenesis, and mutations in the
pathway frequently occur in human GBM. The phosphorylation status
of Akt and mTOR proteins in the presence of NC was therefore
examined by western blotting to determine whether the observed
responses could be due to decreased PI3K/Akt/mTOR signaling.
Increasing concentrations of NC suppressed phosphorylation of both
Akt and mTOR, indicating that PI3K/Akt/mTOR signaling was inhibited
by NC (Fig. 5A). Activation of the
pathway was partially restored when cells were treated
simultaneously with a novel Akt activator, SC79 (18). SC79 increased phosphorylation of Akt
and mTOR in the NC-treated cells, and thus further established
PI3K/Akt/mTOR signaling as a molecular target for NC suppression of
cell growth (Fig. 5B). Functional
assays, including CCK-8, Transwell migration/invasion and Annexin
V/PI staining, were repeated in order to determine whether SC79 may
restore cell growth and migration of GBM cells under NC treatment.
SC79 led to increased viability/proliferation (U251 and U87, ~15
vs. 40%; NC vs. NC + SC79; P<0.05) and invasion (U251 and U87,
~30 vs. 60%; NC vs. NC + SC79; P<0.05) in the NC-treated GBM
cells (Fig. 5C–E). In contrast,
apoptosis was decreased (U251, 27.4 vs. 9.04%; U87, 72.6 vs. 44.8%,
NC vs. NC + SC79; Fig. 5F). Taken
together, our data indicated that changes in proliferation,
invasion and apoptosis induced by NC were in part due to inhibition
of PI3K/Akt/mTOR signaling in the GBM cells.
Discussion
Therapeutic compounds are desperately needed to
improve the dismal survival outcome for glioblastoma multiforme
(GBM) patients. Multiple studies have provided compelling evidence
that nitidine chloride (NC) plays an anticancer role in various
aggressive malignancies (7–9,19–21).
In the present study, we demonstrated that NC has anticancer
properties affecting many critical cellular processes by targeting
the PI3K/Akt/mTOR pathway in GBM cells.
Our focus was to identify new potential therapies
for GBM. NC however has also been reported to inhibit proliferation
of colorectal and hepatic cancer cells (7,9), and
to attenuate migration and invasion of breast and ovarian cancer
cells (8,22). We therefore chose to examine the
functional and molecular pathways that are commonly altered among
diverse cancers in order to understand the mechanistic basis of NC
inhibition. Metabolism has emerged as a potential therapeutic
target due to the fact that increased glycolysis is one of the most
prominent metabolic alterations common among cancer types (13). Lactate, the main product of
glycolysis, also promotes metabolism in cancer cells (23). NC attenuated both ATP and lactate
generation. Furthermore, mitochondria, which are the key organelles
for the generation of ATP, exhibited changes upon exposure to NC,
including lower Δψm and unbalanced mitochondrial fusion and
fission. Such decreases in mitochondrial output and function in
response to NC may ultimately lead to the inhibition of tumor cell
migration and invasion which are high-energy consuming
processes.
Disruptions in mitochondrial fusion and fission
processes have also been shown to sensitize cells to apoptosis
(17). The mitochondrial morphology
in the U251 and U87 cells transitioned from mitochondrial fusion to
fission after exposure to NC. Annexin V/PI staining was consistent
with GBM cells undergoing apoptosis upon NC treatment. Recent
research has shown that the release of cytochrome c is
initiated by the rupture of the outer mitochondrial membrane after
an increase in Bax protein levels, which initiates apoptosis
(24). Thus, the release of
cytochrome c and the increase in Bax protein are considered
indicators of mitochondrial apoptosis. Cytochrome c release
and increases in Bax protein levels were observed in the NC-treated
GBM cells. These results corroborate previous studies, that NC
induces apoptosis in various cancer cells (7,9,20,21,25,26).
Apoptosis is thus an additional attribute that may contribute to
the efficacy of NC in the treatment of human cancer.
Inhibition of tumor growth through these different
cellular functions led us to examine the PI3K/Akt/mTOR signaling
pathway as a potential target of NC. The PI3K/Akt/mTOR signaling
cascades regulate a wide variety of cellular processes, such as
cell proliferation, differentiation, survival, cell transformation
and metastasis of tumor cells (27), and mutations in the PTEN
tumor-suppressor gene, a critical regulator of PI3K/Akt/mTOR, lead
to unregulated activity of the pathway in diverse cancers. In our
in vitro system, p-Akt and p-mTOR levels were decreased in
the NC-treated GBM cells (Fig. 5A).
p-Akt and p-mTOR levels were however partially restored in the
presence of SC79, a novel Akt activator, as were functional
activities including proliferation, invasion and apoptosis. Taken
together, PI3K/Akt/mTOR signaling provides a molecular basis for
the inhibition of tumor growth by NC.
In summary, our data indicate that NC inhibits
malignant behavior of GBM cells by targeting PI3K/Akt/mTOR
signaling. NC thus warrants further investigation as a natural
bioactive molecule with cancer-killing potential.
Acknowledgments
The present study was supported by the Natural
Science Foundation of China grant (nos. 81402060 and 8157248), the
Special Foundation for Taishan Scholars (nos. ts20110814 and
tshw201502056), the Shandong Provincial Science and Technology
Major Project (emerging industry) (2015ZDXX0801A01), the
Fundamental Research Funds of Shandong University (2015QY001), the
Key Research and Development Program of Shandong Province (nos.
2015GSF118074 and 2015GGE27101), the Department of Science and
Technology of Shandong Province (nos. 2015GGE27101 and
2015ZDXX0801A01), the University of Bergen, the Helse Bergen,
Norway and the Norwegian Centre for International Cooperation in
Education (SIU) (UTF-2014/10047).
References
|
1
|
Ricard D, Idbaih A, Ducray F, Lahutte M,
Hoang-Xuan K and Delattre JY: Primary brain tumours in adults.
Lancet. 379:1984–1996. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Stupp R, Brada M, van den Bent MJ, Tonn JC
and Pentheroudakis G; ESMO Guidelines Working Group: High-grade
glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment
and follow-up. Ann Oncol. 25(Suppl 3): iii93–iii101. iii2014.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu HK, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: The avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gordaliza M: Natural products as leads to
anticancer drugs. Clin Transl Oncol. 9:767–776. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Newman DJ, Cragg GM and Snader KM: The
influence of natural products upon drug discovery. Nat Prod Rep.
17:215–234. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fang Z, Tang Y, Jiao W, Xing Z, Guo Z,
Wang W, Shi B, Xu Z and Liu Z: Nitidine chloride inhibits renal
cancer cell metastasis via suppressing AKT signaling pathway. Food
Chem Toxicol. 60:246–251. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lin J, Shen A, Chen H, Liao J, Xu T, Liu
L, Lin J and Peng J: Nitidine chloride inhibits hepatic cancer
growth via modulation of multiple signaling pathways. BMC Cancer.
14:7292014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sun X, Lin L, Chen Y, Liu T, Liu R, Wang
Z, Mou K, Xu J, Li B and Song H: Nitidine chloride inhibits ovarian
cancer cell migration and invasion by suppressing MMP-2/9
production via the ERK signaling pathway. Mol Med Rep.
13:3161–3168. 2016.PubMed/NCBI
|
|
9
|
Zhai H, Hu S, Liu T, Wang F, Wang X, Wu G,
Zhang Y, Sui M, Liu H and Jiang L: Nitidine chloride inhibits
proliferation and induces apoptosis in colorectal cancer cells by
suppressing the ERK signaling pathway. Mol Med Rep. 13:2536–2542.
2016.PubMed/NCBI
|
|
10
|
Javadov S, Baetz D, Rajapurohitam V,
Zeidan A, Kirshenbaum LA and Karmazyn M: Antihypertrophic effect of
Na+/H+ exchanger isoform 1 inhibition is
mediated by reduced mitogen-activated protein kinase activation
secondary to improved mitochondrial integrity and decreased
generation of mitochondrial-derived reactive oxygen species. J
Pharmacol Exp Ther. 317:1036–1043. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bettum IJ, Gorad SS, Barkovskaya A,
Pettersen S, Moestue SA, Vasiliauskaite K, Tenstad E, Øyjord T,
Risa Ø, Nygaard V, et al: Metabolic reprogramming supports the
invasive phenotype in malignant melanoma. Cancer Lett. 366:71–83.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Keunen O, Johansson M, Oudin A, Sanzey M,
Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, et al:
Anti-VEGF treatment reduces blood supply and increases tumor cell
invasion in glioblastoma. Proc Natl Acad Sci USA. 108:3749–3754.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Warburg O: On the origin of cancer cells.
Science. 123:309–314. 1956. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gao Y, Su Y, Qu L, Xu S, Meng L, Cai SQ
and Shou C: Mitochondrial apoptosis contributes to the anti-cancer
effect of Smilax glabra Roxb. Toxicol Lett. 207:112–120. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasahara A and Scorrano L: Mitochondria:
From cell death executioners to regulators of cell differentiation.
Trends Cell Biol. 24:761–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhao J, Lendahl U and Nistér M: Regulation
of mitochondrial dynamics: Convergences and divergences between
yeast and vertebrates. Cell Mol Life Sci. 70:951–976. 2013.
View Article : Google Scholar :
|
|
18
|
Jo H, Mondal S, Tan D, Nagata E, Takizawa
S, Sharma AK, Hou Q, Shanmugasundaram K, Prasad A, Tung JK, et al:
Small molecule-induced cytosolic activation of protein kinase Akt
rescues ischemia-elicited neuronal death. Proc Natl Acad Sci USA.
109:10581–10586. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu N, Li P, Zang S, Liu Q, Ma D, Sun X
and Ji C: Novel agent nitidine chloride induces erythroid
differentiation and apoptosis in CML cells through c-Myc-miRNAs
axis. PLoS One. 10:e01168802015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ou X, Lu Y, Liao L, Li D, Liu L, Liu H and
Xu H: Nitidine chloride induces apoptosis in human hepatocellular
carcinoma cells through a pathway involving p53, p21, Bax and
Bcl-2. Oncol Rep. 33:1264–1274. 2015.
|
|
21
|
Sun M, Zhang N, Wang X, Cai C, Cun J, Li
Y, Lv S and Yang Q: Nitidine chloride induces apoptosis, cell cycle
arrest, and synergistic cytotoxicity with doxorubicin in breast
cancer cells. Tumour Biol. 35:10201–10212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan X, Han H, Wang L, Yang L, Li R, Li Z,
Liu J, Zhao Q, Qian M, Liu M, et al: Nitidine Chloride inhibits
breast cancer cells migration and invasion by suppressing c-Src/FAK
associated signaling pathway. Cancer Lett. 313:181–191. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pérez-Escuredo J, Dadhich RK, Dhup S,
Cacace A, Van Hée VF, De Saedeleer CJ, Sboarina M, Rodriguez F,
Fontenille MJ, Brisson L, et al: Lactate promotes glutamine uptake
and metabolism in oxidative cancer cells. Cell Cycle. 15:72–83.
2016. View Article : Google Scholar
|
|
24
|
Knudson CM and Brown NM: Mitochondria
potential, bax 'activation,' and programmed cell death. Methods Mol
Biol. 414:95–108. 2008.
|
|
25
|
Kang M, Ou H, Wang R, Liu W and Tang A:
The effect of nitidine chloride on the proliferation and apoptosis
of nasopharyngeal carcinoma cells. J BUON. 19:130–136.
2014.PubMed/NCBI
|
|
26
|
Wang J, Wu J, Wu H, Liu X, Chen Y, Wu J,
Hu C and Zou D: Liraglutide protects pancreatic β-cells against
free fatty acids in vitro and affects glucolipid metabolism in
apolipoprotein E−/− mice by activating autophagy. Mol
Med Rep. 12:4210–4218. 2015.PubMed/NCBI
|
|
27
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|