Introduction
Hepatocellular carcinoma (HCC) is one of the most
common malignancies worldwide, and 50% of all newly diagnosed HCC
cases and deaths worlwide are estimated to occur in China every
year (1,2). In recent years, the incidence rate of
HCC in the US has increased, and the 5-year survival rate after
cancer diagnosis is only 18% (3).
Compared with other lethal cancers, the genomic alterations of HCC
are poorly understood due to its complexity and heterogeneity
(4). In depth studies on the
genomic alterations may not only help to better understand the
pathogenesis of HCC, but may also facilitate the development of new
treatment strategies.
Chromosomal instability (CIN), a major cause of
genomic instability that is thought to be critical in the
development of human cancer, arises from inaccurate separation of
sister chromatids in mitosis (5).
Kinetochores that consist of more than 15 centromere proteins
(CENPs) are vital for accurate chromosome segregation by mediating
microtubule attachment and controlling the movement of chromosomes
during mitosis and meiosis (6).
Inappropriate expression of CENPs may be a major cause of CIN and
tumorigenesis (7). Centromere
protein-H (CENP-H), a fundamental component of the human active
centromere complex (8), has been
found to be upregulated in human colorectal cancer, and the ectopic
expression of CENP-H induces aneuploidy (9). Recently, accumulating evidence
revealed that the abnormal expression of CENP-H is closely
associated with several cancers including esophageal (10) and gastric carcinoma (11,12),
breast (13) and uterine cervical
cancer (14).
Our previous research demonstrated that CENP-H was
not only overexpressed in HCC samples, but was also closely related
to tumor size, histological grade and clinical stage. Moreover, it
may be a novel prognostic biomarker of HCC progression (15). In the present study, we investigated
the role of CENP-H in HCC cell survival. The results confirmed that
CENP-H knockdown in Hep3B cells induced growth arrest and apoptosis
in vitro and in vivo, and the mitochondrial apoptotic
pathway may play a key role in this process.
Materials and methods
Ethics statement
All animal protocols complied with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals with the approval of the Institutional Animal Care and Use
Committee of Xi'an Jiaotong University.
Cell lines and cell culture
HCC cell lines Hep3B, HepG2, HHCC, SMMC7721 and
MHHC97H and immortalized human liver cells L02 were purchased from
the Institute of Cellular Research of the Chinese Academy of
Sciences (Shanghai, China). All of the cell lines were cultured in
Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY,
USA) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan,
UT, USA) and 1% penicillin/streptomycin, and incubated at 37°C in a
humidified 5% CO2 atmosphere.
RNA isolation and real-time qPCR
Total RNA was extracted with TRIzol reagent
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer's
instructions. First-strand cDNA was synthesized from total RNA with
PrimeScript® RT Master Mix (Takara Biotechnology Co.,
Ltd., Dalian, China). Real-time quantitative qPCR was performed
(Bio-Rad Laboratories, Hercules, CA, USA) with SYBR®
Premix Ex Taq™ II (Takara Biotechnology). The cycling conditions
were as follows: initial denaturation at 95°C for 30 sec, and then
40 cycles of denaturation at 95°C for 5 sec and annealing and
elongation at 60°C for 30 sec. Bio-Rad CFX Manager 2.1 software was
used for qPCR analysis. The GAPDH housekeeping gene was used as an
internal control.
The PCR primers sets were as follows: i) CENP-H
forward, 5′-CAGTCTAGTGTGCTCATGGAT-3′ and reverse,
5′-TCCATCTGTAGGTTTTGTCG-3′; ii) Bax forward,
5′-GGTTGTCGCCCTTTTCTACTTT-3′ and reverse,
5′-GTGAGGAGGCTTGAGGAGTCT-3′; and iii) Bcl-2 forward,
5′-TCCCATCAAAACTCCTGTCTTT-3′ and reverse,
5′-GCAAGTGAATGAACACCTTCTC-3′; iv) glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) forward, 5′-CAAGCTCATTTCCTGGTATGAC-3′ and
reverse, 5′-CAGTGAGGGTCTCTCTCTTCCT-3′.
Protein extraction and western
blotting
Total cell protein was extracted according to a
previous study (15). After
denaturation, equal amounts of total protein were separated
electrophoretically on 10% SDS/polyacrylamide gels (SDS-PAGE) and
transferred onto polyvinylidene difluoride (PVDF) membranes
(Millipore, Bedford, MA, USA) using a tank transfer apparatus
(Bio-Rad Laboratories). The membranes were blocked with 5% skim
milk in Tris-buffered saline with Tween (TBS-T) for 2 h, then
incubated with primary antibodies overnight at 4°C. The following
day, after the incubation with horseradish peroxidase-conjugated
secondary antibodies, the blots were visualized with enhanced
chemiluminescence horseradish peroxidase (HRP) substrate
(Millipore) according to the manufacturer's instructions. The
intensity of each band was assessed using Image Lab 4.0 (Bio-Rad).
The following primary antibodies were used: anti-CENP-H (1:200;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti-Bax
(1:1,000), anti-Bcl-2 (both from Epitomics, Abcam, CA, USA,
1:2,000) and anti-cleaved caspase-3 (1:1,000; Cell Signaling
Technology, Beverly, MA, USA). To confirm consistent loading, mouse
anti-β-actin antibody (Santa Cruz Biotechnology, Inc.) diluted
1:1,000 was used as a primary antibody. The secondary antibodies
were purchased from Santa Cruz Biotechnology, Inc. and were diluted
1:5,000.
Lentiviral packaging and
infection
To knock down CENP-H expression, 2 lentiviruses with
different siRNA sequences were packaged using the LV-3 (pGLVH1/GFP
+ Puro) vector (GenePharma, Shanghai, China). The siRNA sequences
were as follows: CENP-H1, 5′-TGGTTGATGCAAGTGAAGA-3′; and CENP-H2,
5′-GCTTGAGAAGAATGTTGACAT-3′. Concomitantly, the negative controls
were also packaged. Puromycin (2.5 µg/ml; Sigma, St. Louis, MO,
USA) was used to kill the uninfected Hep3B cells. Hep3B cells that
were stably infected with CENP-H1 and CENP-H2 were named
LV3-CENP-H1 and LV3-CENP-H2, respectively, and the negative control
was named LV3-NC. Real-time PCR and western blotting were used to
detect knockdown efficiency, and the virus with the highest
knockdown efficiency was used in the subsequent experiments.
Proliferation assay
The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was performed to detect cell proliferation. Hep3B,
LV3-CENP-H1 and LV3-NC cells were separately seeded into 96-well
plates at a density of 2×103 cells/well (5 repeats/cell
line), after culture for 0–7 days; MTT reagent (Sigma), which was
freshly prepared, was added to each well. Following 4 h of
incubation, the supernatant was discarded, and dimethyl sulfoxide
(DMSO) (Sigma) was added to dissolve the crystals. All analyses
were performed in triplicate. The absorbance was assessed at a test
wavelength of 490 nm using an EnSpire™ Multilabel Reader
(PerkinElmer, Waltham, MA, USA).
Colony formation assay
Hep3B, LV3-CENP-H1 and LV3-NC cells were seeded in
fresh 6-well plates at a density of 500 cells/well in DMEM with 10%
FBS. After 10 days, the cells were washed with PBS, fixed in 4%
paraformaldehyde for 30 min, and stained with 0.5% crystal violet
for 20 min. Colonies were scored, and each experiment was repeated
3 times.
Cell apoptosis analysis using flow
cytometry
After 72 h of incubation, Hep3B, LV3-CENP-H1 and
LV3-NC cells were harvested, washed twice with cold PBS, diluted in
1X binding buffer, stained with recombinant human Annexin V-Alexa
Fluor 647 and PI using Annexin V-Alexa Fluor 647 apoptosis
detection kit (Beijing 4A Biotech Co., Ltd, Beijing, China)
according to the manufacturer's instructions, and then analyzed
using a FACSCanto™ II Flow Cytometer (BD Biosciences, San Jose, CA,
USA).
Transmission electron microscopy
(TEM)
After 72 h of incubation, Hep3B, LV3-CENP-H1 and
LV3-NC cells were harvested. The collected cells were first fixed
in 2.5% glutaraldehyde for 2 h at 4°C, then rinsed in 0.1 M
phosphate buffer (PB) (pH 7.4), and post-fixed in 1% osmium
tetraoxide for 2 h. After being washed in PB, the cells were
progressively dehydrated in a graded series of ethanol, and then
cleared in QY-1 (Nissin EM, Tokyo, Japan). Then, embedding in epoxy
resin 812 and sectioning were performed, and the ultrathin sections
(60-nm thickness) were stained with both uranyl acetate and lead
citrate. Finally, cellular ultramicroscopic structures were
examined by TEM (Hitachi, Tokyo, Japan).
Tumorigenicity in nude mice
Male 4–6 week-old BALB/c nude mice were purchased
from the Experimental Animal Center of Shanghai, and fed in
specific pathogen-free (SPF) animal rooms with constant temperature
and humidity at the Centre of Laboratory Animals of The Medical
College of Xi'an Jiaotong University. After 4 days of adaptive
feeding with an autoclaved chow diet with water, the mice were
randomly divided into 2 groups (4 mice/group), and 2×106
LV3-CENP-H/LV3-NC cells were subcutaneously injected into the right
thigh of each mouse. The body weights of the mice and tumor volumes
were assessed every 4–5 days, and the tumor volume was calculated
using the following formula: Tumor volume = (tumor length × tumor
width × tumor width)/2. After 4 weeks of observation of xenograft
tumor formation, the mice were sacrificed, and the tumor tissues
were obtained as soon as possible. After being washed with PBS,
some of the tumor samples were fixed with 4% formalin and
paraffin-embedded for histological studies, and others were
immediately snap-frozen in liquid nitrogen and stored at −80°C for
long-term preservation.
Immunohistochemistry analysis
Xenograft samples were immediately fixed in 4%
formalin once obtained from the mice. After paraffin-embedding,
4-µm thick paraffin sections were adhered to glass slides and
prepared for immunohistochemical assay using a standardized
streptavidin-peroxidase (SP) method as previously reported
(15). Following deparaffinization,
rehydration, antigen retrieval and blockage, the slides were
incubated with primary antibodies overnight at 4°C. The following
day, after secondary antibody incubation and dehydration, the
slides were examined under a microscope (Olympus, Tokyo, Japan).
The primary antibodies included anti-CENP-H (1:40; Santa Cruz
Biotechnology, Inc.), anti-Bax, anti-Bcl-2 (1:100) (both from
Epitomics), anti-cleaved caspase-3 (1:300) and anti-Ki-67 (1:200)
(both from Cell Signaling Technology). The secondary antibodies
were from Zhongshan Golden Bridge Biotechnology (Beijing,
China).
Statistical analysis
The experimental data are presented as the mean ±
SEM and were analyzed using the two-tailed Students t-test.
P<0.05 was considered statistically significant. Statistical
analysis was performed using SPSS 19.0 statistical software (SPSS,
Inc., Chicago, IL, USA).
Results
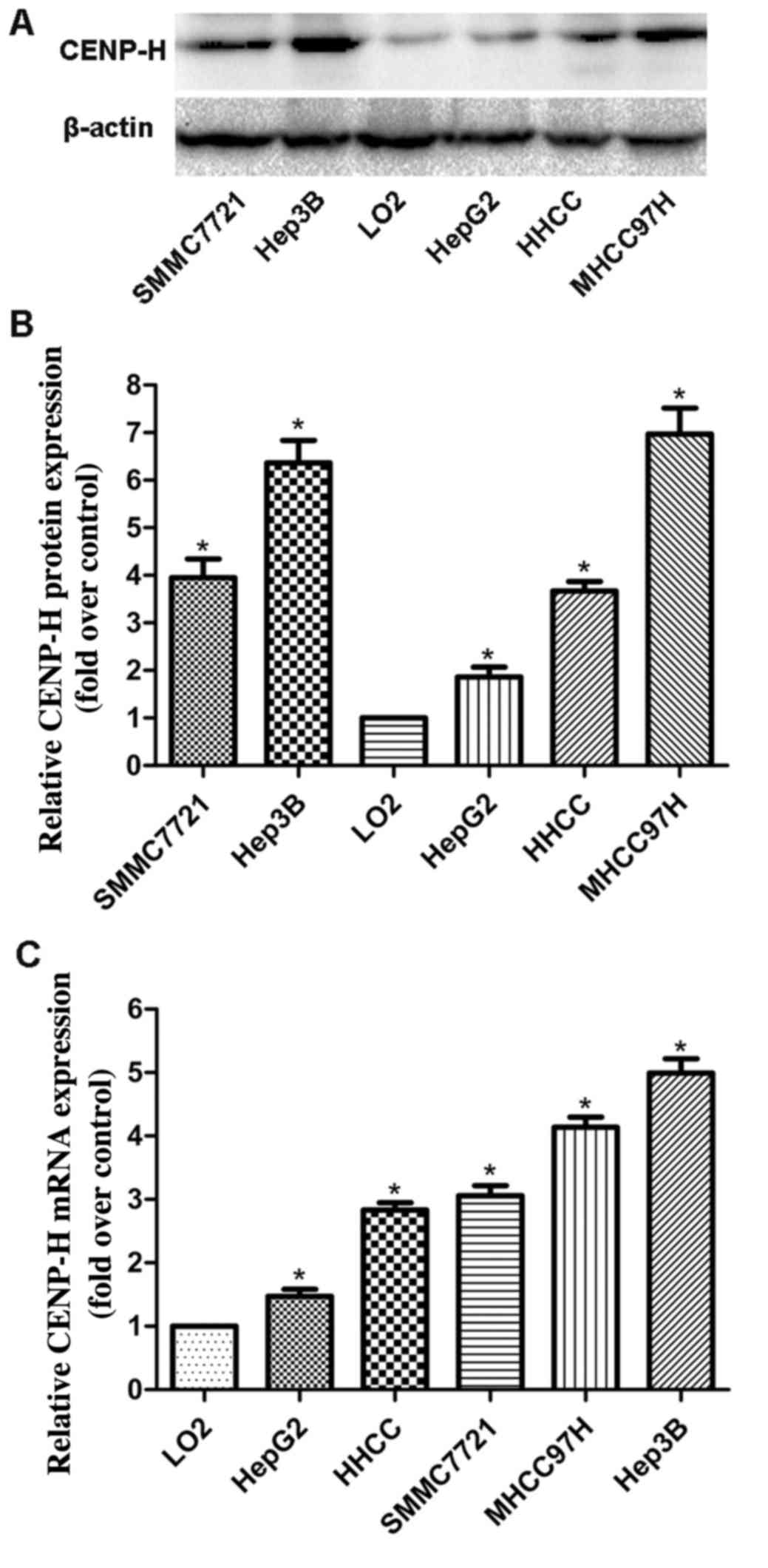
The expression of CENP-H in HCC cell
lines and immortalized human liver cells
To investigate the protein and gene expression
levels of CENP-H in different HCC cell lines and immortalized human
liver cells, total protein and mRNA were extracted from Hep3B,
HepG2, HHCC, SMMC7721, MHHC97H and L02 cells, separately. Western
blotting revealed that the CENP-H protein levels in all HCC cell
lines were higher than that in the L02 cells (Fig. 1A and B). Real-time PCR analysis
produced similar results as the western blotting assay (Fig. 1C). These results demonstrated that
CENP-H was upregulated at both the protein and mRNA levels in HCC
cell lines and suggest that it may play an important role in HCC
progression. Additionally, considering the high endogenous
expression of CENP-H in the Hep3B cell line, the Hep3B cell line
was chosen for the following knockdown experiments.
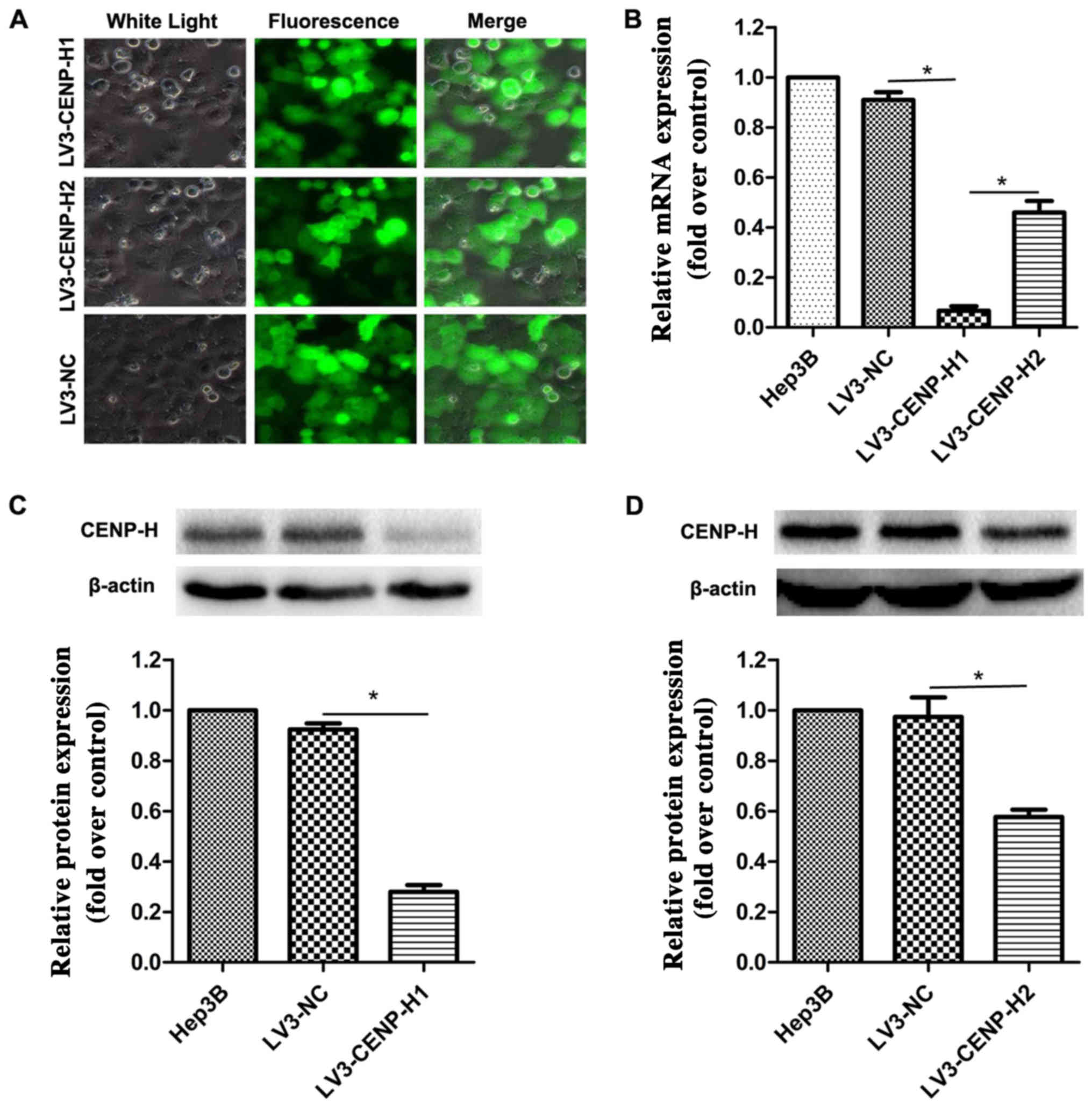
CENP-H knockdown inhibits cell
proliferation in Hep3B cells
To elucidate the biological function of CENP-H in
HCC cells, we packaged lentiviruses to infect Hep3B cells in order
to construct stable shRNA-mediated CENP-H knockdown. After 72 h of
incubation, the viral infection efficiency was >90% using
fluorescence microscopy (Fig. 2A).
To further validate the knockdown efficiency, real-time PCR and
western blotting were performed. The results confirmed that
compared with the negative control, both the siRNA sequences
significantly eliminated the endogenous CENP-H expression in the
Hep3B cells (Fig. 2B-D). Taking
knockdown efficiency into account, we chose LV3-CENP-H1 for the
subsequent biological experiments.
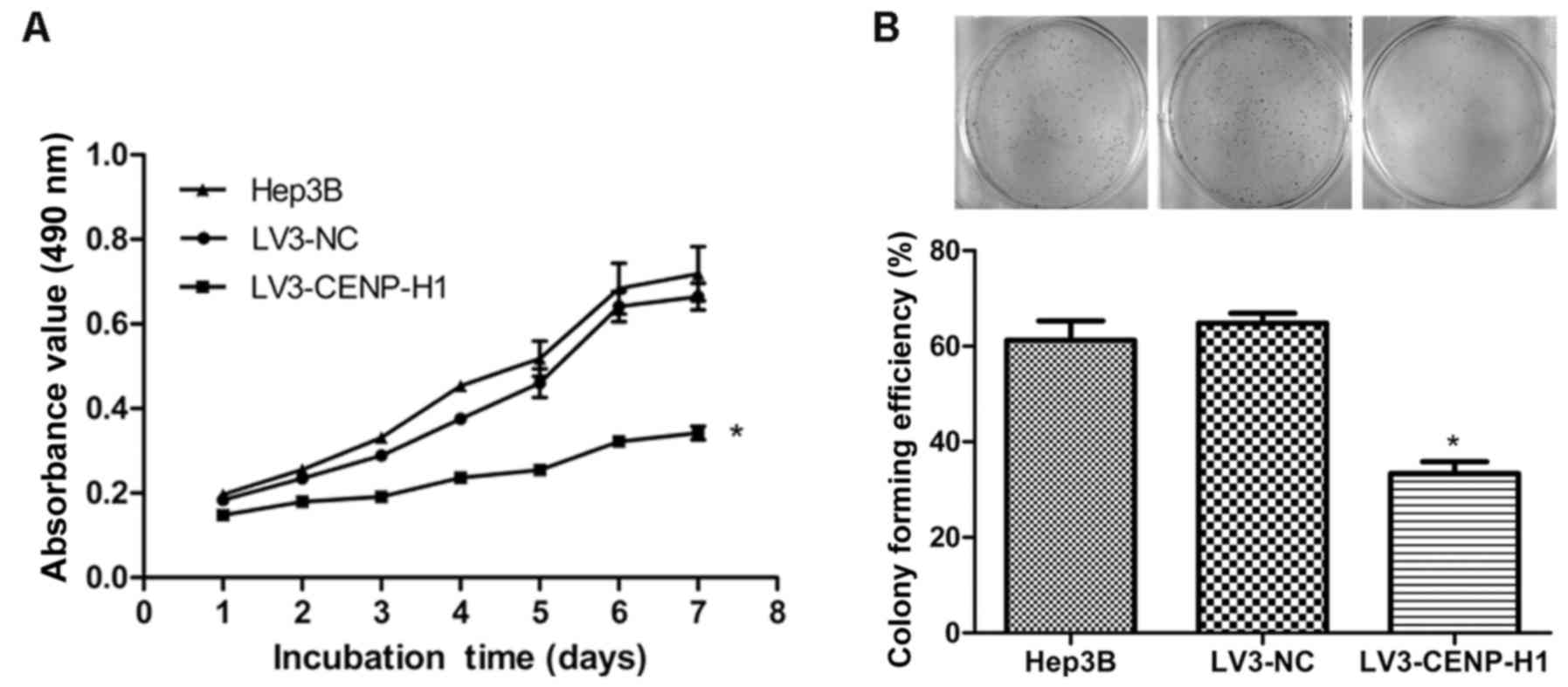
The MTT and colony formation assays were used to
detect the changes in the proliferative capacity of Hep3B after
CENP-H knockdown. The MTT results revealed that LV3-CENP-H1 cells
had a lower proliferative ability than the Hep3B and LV3-NC cells.
In addition, the proliferative abilities of the Hep3B and LV3-NC
cells did not significantly differ (Fig. 3A). CENP-H knockdown suppressed the
colony formation ability of the Hep3B cells (Fig. 3B). Both the MTT and colony formation
assay results demonstrated that CENP-H knockdown inhibits Hep3B
proliferation.
CENP-H knockdown induces apoptosis of
Hep3B cells
Flow cytometric and TEM assays were used to detect
cell apoptosis. The apoptotic index was increased from 3.13±0.42%
in the LV3-NC cells to 19.96±0.5% in the LV3-CENP-H1 cells after
CENP-H knockdown (Fig. 4A). Through
TEM, we observed chromatin condensation, mitochondrial swelling and
apoptotic bodies in the LV3-CENP-H1 cells (Fig. 4B). Both the flow cytometric and TEM
results indicated that CENP-H knockdown induced apoptosis of the
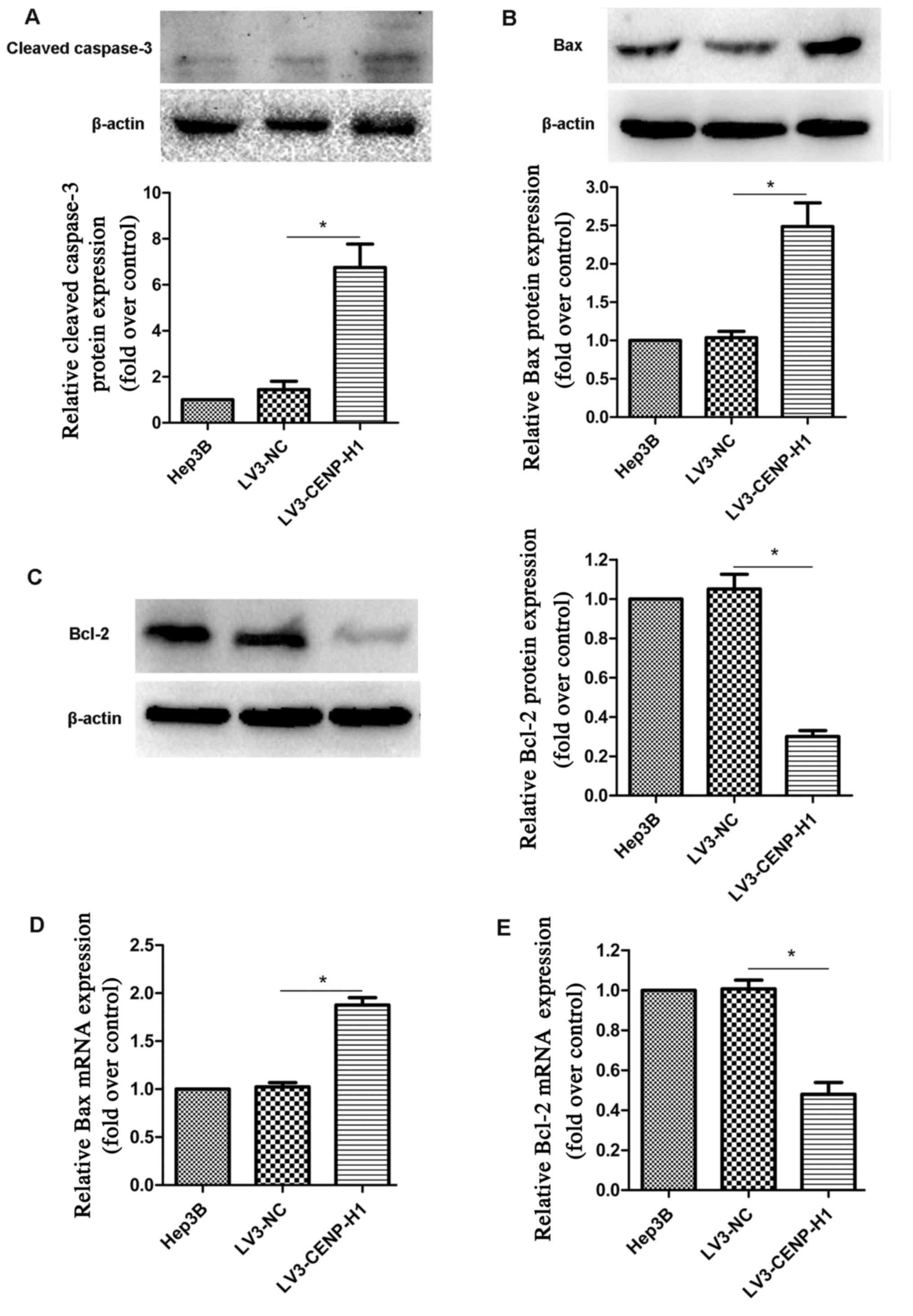
Hep3B cells. Based on western blotting, we found that cleaved
caspase-3 expression was increased in the LV3-CENP-H1 cells
compared with the level noted in the Hep3B and LV3-NC cells
(Fig. 5A).
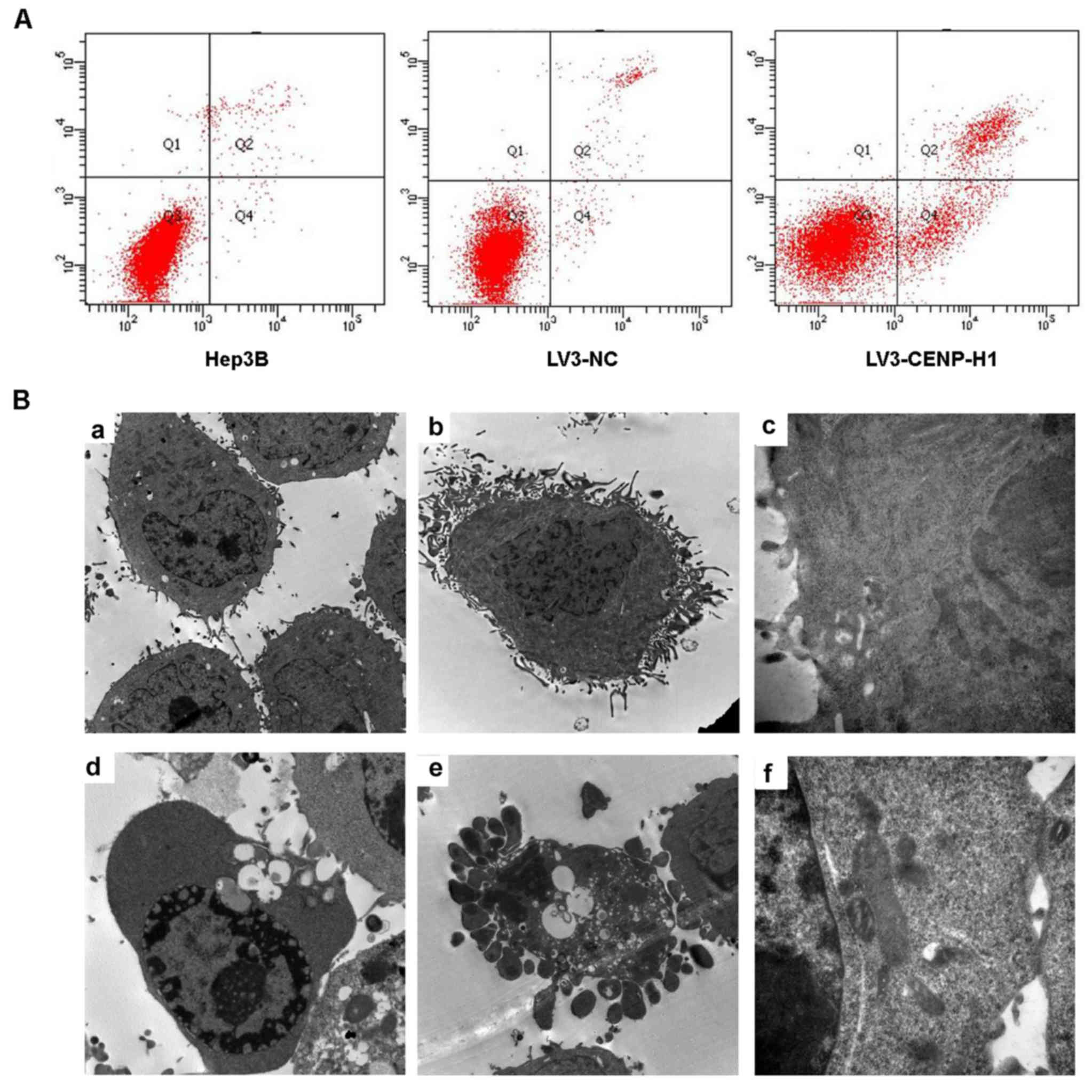 | Figure 4.CENP-H knockdown induces apoptosis of
Hep3B cells. (A) CENP-H knockdown induced apoptosis of Hep3B cells.
(B) Representative transmission electron micrographs demonstrating
the morphology and ultrastructure of CENP-H knockdown Hep3B cells.
a, Magnification (×8,000), cell morphology of Hep3B cells; b,
magnification (×8,000), cell morphology of LV3-NC cells; c,
magnification (×40,000), cell ultrastructure in LV3-NC cells; d,
magnification (×8,000), chromatin margination and vacuolization in
LV3-CENP-H1 cells; e, magnification (×8,000), apoptotic bodies in
LV3-CENP-H1 cells; f, magnification (×40,000), obvious
mitochondrial swelling in LV3-CENP-H1 cells. Values are expressed
as the mean ± standard error; *P<0.05 vs. the LV3-NC and Hep3B
cells. CENP-H, centromere protein-H. |
Based on swollen mitochondria observed using TEM,
the apoptosis-related factors involved in the mitochondrial
apoptotic pathway were detected. Both protein and mRNA expression
levels of Bax were increased in the LV3-CENP-H1 cells compared to
these levels in the Hep3B and LV3-NC cells (Fig. 5B and D). Conversely, the protein and
mRNA expression levels of Bcl-2 were downregulated after CENP-H
knockdown (Fig. 5C and E).
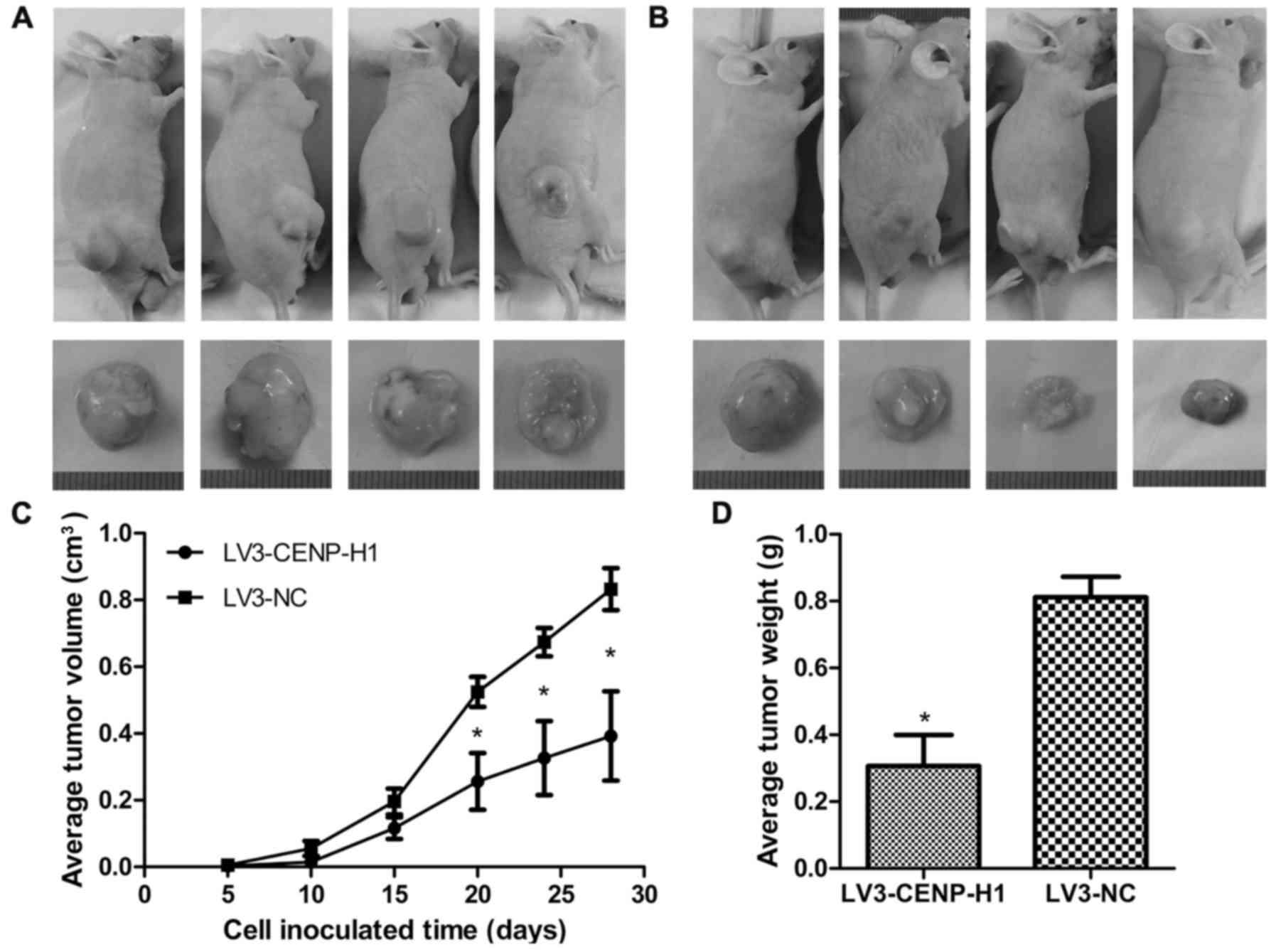
CENP-H knockdown suppresses tumor
growth in a Hep3B cell xenograft model
The Hep3B xenograft model was used to determine
whether CENP-H knockdown affects tumor growth in vivo. The
LV3-CENP-H1 group of animals formed much smaller tumors from 20
days after cell inoculation than the control group. After
sacrifice, we confirmed that the average tumor weight in the
LV3-CENP-H1 group was lighter than in the control. In addition, the
average tumor volume in the LV3-CENP-H1 group was also smaller than
in the control (Fig. 6).
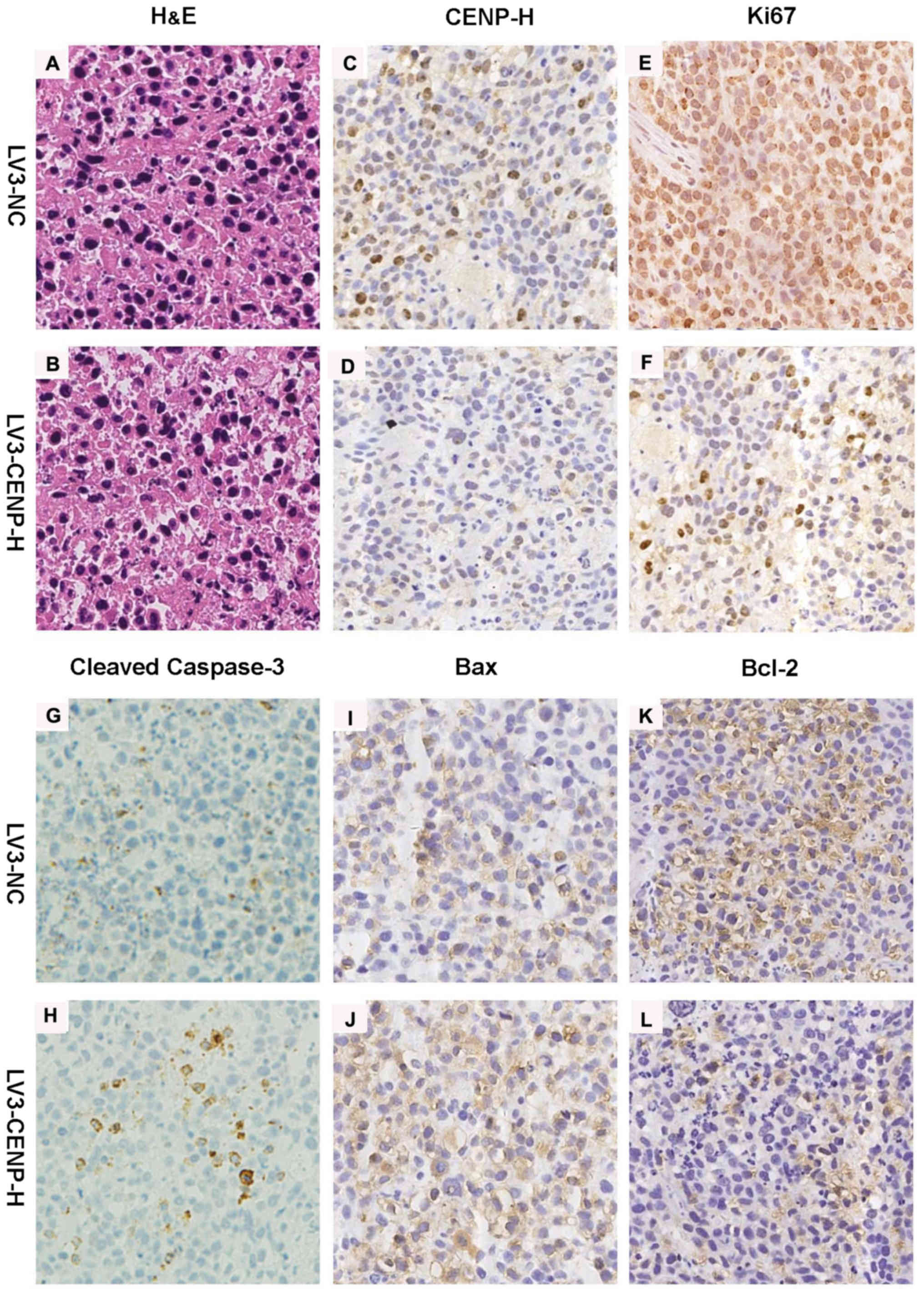
Immunohistochemistry for CENP-H confirmed the decreased staining in
the LV3-CENP-H1 group of tumors compared with the LV3-NC tumors.
Similarly to the in vitro experiment results, the
LV3-CENP-H1 tumors exhibited increased staining for cleaved
caspase-3 and Bax, compared with the LV3-NC tumors. In addition,
the expression of Bcl-2 decreased in the LV3-CENP-H1 group.
Immunohistochemistry for Ki-67 was used to detect the proliferative
activities of both the LV3-CENP-H1 and LV3-NC tumors. As expected,
the LV3-CENP-H1 tumors had a lower Ki-67 expression than the LV3-NC
tumors (Fig. 7).
Discussion
HCC is not only the sixth most common neoplasm, but
is also the third leading cause of cancer-related deaths worldwide
(16). Hepatocarcinogenesis is a
complex multistep process in which many genes and signaling
pathways are involved. In our previous research, we found that
CENP-H expression is not only upregulated in HCC tissues at both
the mRNA and protein levels but is also closely related to tumor
size, histological grade, and clinical stage. Additionally, CENP-H
expression based on immunohistochemistry was negatively associated
with patient prognosis (15). In
the present study we further confirmed that the expression levels
of CENP-H were higher in the HCC cell lines than that in the L02
immortalized human liver cells. CENP-H knockdown inhibited the
proliferation of Hep3B cells and induced their apoptosis in
vitro. Furthermore, tumor growth in the Hep3B xenograft model
was inhibited after CENP-H knockdown. The role of CENP-H in HCC
growth may be mediated through the mitochondrial apoptotic
signaling pathway. All of these results confirmed that CENP-H may
play a key role in the regulation of HCC growth.
Uncontrolled proliferation is an obvious
characteristic of cancers, and tumor growth can be inhibited by
controlling the proliferation of tumor cells. Orthaus et al
(17) reported that depletion of
CENP-H resulted in the decreased number of living cells and
increased cell death in human HEp-2 cells by RNAi knockdown of
CENP-H. Compared with the control cells, the population doubling
time was shorter and the colony formation ability was decreased in
Tca8113/CENP-H RNAi cells, which highlighted the compromised
viability caused by the downregulation of CENP-H (18). Consistent with these studies
(12,17,18),
our MTT and colony formation data also revealed that shRNA-mediated
CENP-H knockdown caused Hep3B proliferation inhibition. Similarly
to the in vitro experiments, LV3-CENP-H1 cells exhibited
decreased tumorigenicity in nude mice in vivo. Ki-67, a
proliferation marker of many malignancies including HCC (19), was found to be expressed at lower
levels in the LV3-CENP-H1 subcutaneous xenograft tumors. These
results collectively demonstrated that CENP-H is closely associated
with the proliferation of HCC Hep3B cells.
In the present study, we also analyzed the effects
of CENP-H knockdown on the apoptosis of Hep3B cells by flow
cytometry and TEM. Both results revealed that CENP-H knockdown
increased apoptosis in the Hep3B cells. Moreover, the cleaved
caspase-3 protein expression increased after CENP-H knockdown based
on western blotting. Survivin, a member of the inhibitor of
apoptosis protein family, has been implicated as a protector
against cell apoptosis; the aberrant high expression of this factor
in cancers is closely correlated with advanced disease and poor
overall survival (20,21). According to previous studies, CENP-H
siRNA decreases the protein expression of survivin, indicating
apoptosis induction (12,18). Wang et al (22) found that siRNA knockdown of CENP-H
mRNA promoted FaDu cell apoptosis. All of these results are
consistent with our findings.
Considering the changes in mitochondria observed
under TEM, the factors involved in the mitochondrial apoptotic
pathway were examined by western blotting and real-time PCR
(23). The results demonstrated
that CENP-H knockdown not only induced the activation of caspase-3
in Hep3B cells with increased protein expression of cleaved
caspase-3 but also resulted in a Bax/Bcl-2 imbalance with a
significant increase of Bax expression and an obvious decrease in
Bcl-2 expression at both the mRNA and protein levels. All of these
results were further ascertained in subcutaneous xenograft models.
CENP-A, another inner centromere protein, has been shown to be
upregulated in HCC. CENP-A siRNA induced apoptosis in HepG2 cells
with increased expression of Bax and decreased expression of Bcl-2
(24), which was consistent with
our results. Bax and Bcl-2 are two of the most important members of
the Bcl-2 family; anti-apoptotic Bcl-2 protein prevents apoptosis
by inhibiting the activation of pro-apoptotic Bax protein (25). Increased Bax/Bcl-2 ratio results in
the release of cytochrome c, which cooperates with cytosolic
factors to activate the caspases (25–27).
Once the executioner caspase-3 is activated, apoptosis is
inevitable (28,29). In the present study, we found that
apoptosis was accompanied by a Bax/Bcl-2 imbalance and activation
of caspase-3 at both the cellular and tissue levels after CENP-H
knockdown. These results collectively suggested that targeting
CENP-H induced HCC cell apoptosis through the mitochondrial
apoptotic pathway.
In conclusion, CENP-H was upregulated in HCC cells,
and CENP-H knockdown inhibited Hep3B cell proliferation both in
vitro and in vivo. The mitochondrial apoptotic pathway
mediated by a Bax/Bcl-2 imbalance may be involved in apoptosis
induction by CENP-H knockdown. All of these data suggested that
CENP-H may be a potential therapeutic target for HCC.
Acknowledgements
The present study was supported by the International
Cooperation Project of Shaanxi Province (2015KW-043). The present
study also received support from the National Natural Science
Foundation of China (no. 30771895). We thank the staff of the
Transform Medical Center, Xi'an Jiaotong University for their
technical assistance in these studies.
References
|
1
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zender L, Villanueva A, Tovar V, Sia D,
Chiang DY and Llovet JM: Cancer gene discovery in hepatocellular
carcinoma. J Hepatol. 52:921–929. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schvartzman JM, Sotillo R and Benezra R:
Mitotic chromosomal instability and cancer: Mouse modelling of the
human disease. Nat Rev Cancer. 10:102–115. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Westhorpe FG and Straight AF: Functions of
the centromere and kinetochore in chromosome segregation. Curr Opin
Cell Biol. 25:334–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pfau SJ and Amon A: Chromosomal
instability and aneuploidy in cancer: From yeast to man. EMBO Rep.
13:515–527. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fukagawa T, Mikami Y, Nishihashi A,
Regnier V, Haraguchi T, Hiraoka Y, Sugata N, Todokoro K, Brown W
and Ikemura T: CENP-H, a constitutive centromere component, is
required for centromere targeting of CENP-C in vertebrate cells.
EMBO J. 20:4603–4617. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomonaga T, Matsushita K, Ishibashi M,
Nezu M, Shimada H, Ochiai T, Yoda K and Nomura F: Centromere
protein H is up-regulated in primary human colorectal cancer and
its overexpression induces aneuploidy. Cancer Res. 65:4683–4689.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo XZ, Zhang G, Wang JY, Liu WL, Wang F,
Dong JQ, Xu LH, Cao JY, Song LB and Zeng MS: Prognostic relevance
of Centromere protein H expression in esophageal carcinoma. BMC
Cancer. 8:2332008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Quan T, He B, Liu T, Li W, Wu S, Jiang Q,
Liu W, Liu H and Xu X: Role of centromere protein H in human
gastric cancer cell proliferation. Nan Fang Yi Ke Da Xue Xue Bao.
32:265–269. 2012.(In Chinese). PubMed/NCBI
|
|
12
|
He WL, Li YH, Yang DJ, Song W, Chen XL,
Liu FK, Wang Z, Li W, Chen W, Chen CY, et al: Combined evaluation
of centromere protein H and Ki-67 as prognostic biomarker for
patients with gastric carcinoma. Eur J Surg Oncol. 39:141–149.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liao WT, Feng Y, Li ML, Liu GL, Li MZ,
Zeng MS and Song LB: Overexpression of centromere protein H is
significantly associated with breast cancer progression and overall
patient survival. Chin J Cancer. 30:627–637. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Weng MY, Li L, Hong SJ and Feng SY:
Clinical significance of CENP-H expression in uterine cervical
cancer. Cancer Biol Med. 9:192–196. 2012.PubMed/NCBI
|
|
15
|
Lu G, Shan T, He S, Ren M, Zhu M, Hu Y, Lu
X and Zhang D: Overexpression of CENP-H as a novel prognostic
biomarker for human hepatocellular carcinoma progression and
patient survival. Oncol Rep. 30:2238–2244. 2013.PubMed/NCBI
|
|
16
|
Forner A, Llovet JM and Bruix J:
Hepatocellular carcinoma. Lancet. 379:1245–1255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Orthaus S, Ohndorf S and Diekmann S: RNAi
knockdown of human kinetochore protein CENP-H. Biochem Biophys Res
Commun. 348:36–46. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liao WT, Yu CP, Wu DH, Zhang L, Xu LH,
Weng GX, Zeng MS, Song LB and Li JS: Upregulation of CENP-H in
tongue cancer correlates with poor prognosis and progression. J Exp
Clin Cancer Res. 28:742009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Luo Y, Ren F, Liu Y, Shi Z, Tan Z, Xiong
H, Dang Y and Chen G: Clinicopathological and prognostic
significance of high Ki-67 labeling index in hepatocellular
carcinoma patients: A meta-analysis. Int J Clin Exp Med.
8:10235–10247. 2015.PubMed/NCBI
|
|
20
|
Athanasoula KC, Gogas H, Polonifi K,
Vaiopoulos AG, Polyzos A and Mantzourani M: Survivin beyond
physiology: Orchestration of multistep carcinogenesis and
therapeutic potentials. Cancer Lett. 347:175–182. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Andersen MH, Svane IM, Becker JC and
Straten PT: The universal character of the tumor-associated antigen
survivin. Clin Cancer Res. 13:5991–5994. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang JX, Zhang YY, Yu XM, Jin T and Pan
XL: Role of centromere protein H and Ki67 in relapse-free survival
of patients after primary surgery for hypopharyngeal cancer. Asian
Pac J Cancer Prev. 13:821–825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lakhani SA, Masud A, Kuida K, Porter GA
Jr, Booth CJ, Mehal WZ, Inayat I and Flavell RA: Caspases 3 and 7:
Key mediators of mitochondrial events of apoptosis. Science.
311:847–851. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Zhu Z, Zhang S, Yu D, Yu H, Liu L,
Cao X, Wang L, Gao H and Zhu M: ShRNA-targeted centromere protein A
inhibits hepatocellular carcinoma growth. PLoS One. 6:e177942011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lindsay J, Esposti MD and Gilmore AP:
Bcl-2 proteins and mitochondria - specificity in membrane targeting
for death. Biochim Biophys Acta. 1813:532–539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Autret A and Martin SJ: Emerging role for
members of the Bcl-2 family in mitochondrial morphogenesis. Mol
Cell. 36:355–363. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Howells CC, Baumann WT, Samuels DC and
Finkielstein CV: The Bcl-2-associated death promoter (BAD) lowers
the threshold at which the Bcl-2-interacting domain death agonist
(BID) triggers mitochondria disintegration. J Theor Biol.
271:114–123. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Taylor RC, Cullen SP and Martin SJ:
Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol
Cell Biol. 9:231–241. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brancolini C, Lazarevic D, Rodriguez J and
Schneider C: Dismantling cell-cell contacts during apoptosis is
coupled to a caspase-dependent proteolytic cleavage of
beta-catenin. J Cell Biol. 139:759–771. 1997. View Article : Google Scholar : PubMed/NCBI
|