Introduction
Lung cancer is the primary cause of
cancer-associated mortality worldwide in men and the second leading
cause of cancer-related death in women (1). The main type of lung cancer is
non-small cell lung cancer (NSCLC) which accounts for ~85% of all
lung cancer cases (2).
Treatment options for lung cancer include surgery,
radiation therapy and chemotherapy. Although chemotherapy remains
an important form of treatment, novel drug development has focused
on molecular-targeted therapies. The effectiveness of such targeted
therapies rely on the presence or absence of alterations in gene(s)
that encode for the protein targeted by the agent or for proteins
involved in the molecular pathway targeted (3).
This targeted therapeutical approach necessitates
the molecular analysis of the tumor in order to select patients
with increased probability of response to the treatment given.
Until recently, the routine screening of tumors for mutations was
confined by limited tissue availability, cost, time and labor
demanding methods such as Sanger sequencing (4). More recently, the introduction of next
generation sequencing (NGS) methods has resolved this issue by
simultaneously sequencing thousands of short DNA sequences in a
massively parallel manner, thus offering a cost effective approach
for detecting multiple genetic alterations for many samples
simultaneously. Moreover, the sensitivity obtained by NGS is
superior to that of conventional sequencing technology, making
possible the detection of mutations that are present in very low
percentages in a background of normal DNA, which is extremely
important for the detection of somatic mutations (5,6).
Large scale application of this technology in tumor
molecular characterization led to the generation of databases that
catalog the NGS cancer genome. Such databases include the COSMIC
database, International Genome Consortium (ICGC) and the Cancer
Genome Atlas (TCGA) project (7–10).
Additionally, interpretation of somatic variants is feasible due to
several available knowledge resources and online interpretation
tools with My Cancer Genome being the first of such public somatic
variant interpretation resources (11). Alternatively, commercially available
software and bioinformatics tools such as Ion Torrent™ Oncomine™
Knowledgebase Reporter (https://www.thermofisher.com/) and QCI™ Interpret for
Somatic Cancer (www.qiagenbioinformatics.com) are now available for
clinical testing laboratories. These tools facilitate the
interpretation and reporting of genomic variants identified by NGS,
enabling the easy identification of actionable variants and the
association with treatment options and open clinical trials.
Currently, the most widely used platforms are those
offered by Illumina, Inc. (San Diego, CA, USA) and by Thermo Fisher
Scientific (Waltham, MA, USA). The Illumina platform involves
bridge amplification, to clonally amplify the fragments that are
then sequenced using sequencing-by-synthesis (SBS) chemistry. The
Thermo Fisher Scientific platform uses the Ion semiconductor
sequencing, detecting the protons released as nucleotides are
incorporated during synthesis. Both ΝGS platforms have all the
features required to carry out simultaneous analysis of a large
number of genes with actionable alterations in tumor tissue and
thus a more precise molecular characterization of the tumor. Such
targeted NGS panels for somatic mutation detection, include
actionable cancer genes aiming to increase the percentage of
patients with detectable actionable somatic gene alterations that
can be used to guide treatment decision (12–14).
The integration of these NGS multi-gene panels in
the health care setting is of great clinical importance and
increases the necessity of appropriate guidelines in order to
assure their optimal performance and accuracy (15,16).
In the present study, a custom 23 gene Ion AmpliSeq
Panel for DNA analysis and the Ion AmpliSeq™ RNA Fusion Lung Cancer
Research Panel for fusion RNA transcript analysis were applied in a
cohort of patients with NSCLC. The Ion AmpliSeq Panel for DNA
analysis was selected since it includes all the major mutations
occurring in lung cancer. It includes the same hot spot regions of
the 22 gene Colon and Lung V2 panel with the addition on certain
amplicons to detect exon 14 skipping mutations in the MET gene and
amplicons for exons 2 and 3 of the HRAS gene (13,14).
Since, these targeted NGS panels contain all important genes
related to targeted therapies in NSCLC, they were used in the the
mutation spectrum identification of tumors in newly diagnosed NSCLC
patients.
Materials and methods
Tumor samples
A total of 512 tumors collected at various
institutions between January 2014 and December 2015, from patients
with newly diagnosed non-small cell lung cancer (NSCLC), were
included in this study. All available clinical factors were
evaluated. Tumor classification was performed using the 2015 World
Health Organization (WHO) Classification of Lung Tumors (17). Informed consent was obtained from
all patients before testing. The study was approved by the Ethics
Committee of the Pulmonary Department, Oncology Unit, ‘G.
Papanikolaou’ General Hospital, Aristotle University of
Thessaloniki, Thessaloniki, Greece.
Tissue selection and DNA
extraction
Hematoxylin and eosin-stained sections of
formalin-fixed and paraffin-embedded (FFPE) tumor biopsies from all
samples were reviewed to ensure tumor cell content of >75% when
possible, and the tumor area was marked by a pathologist. Genomic
DNA was extracted from unstained 10-µm thick FFPE sections using
the QIAmp FFPE tissue kit (Qiagen, Antwerp, Belgium). After
extraction, the concentration of all samples was measured with the
use of a spectrophotometer (NanoDrop2000, Thermo Fisher
Scientific).
Ion AmpliSeq next generation
sequencing
The NGS for DNA analysis was conducted using a
custom 23 gene Ion AmpliSeq panel which was based on the Ion
AmpliSeq Colon and Lung Cancer Research Panel v2 with an additional
amplicon in the MET gene (to include the exon 14 skipping
mutations) and two amplicons of exons 2 and 3 of the HRAS gene.
Fusion RNA transcript analysis was performed using the Ion AmpliSeq
RNA Fusion Lung Cancer Research Panel (Thermo Fisher Scientific).
Details of the target regions of the 23-gene panel are available
upon request.
The genes analyzed include AKT1 (NM_05163),
ALK (NM_004304), BRAF (NM_004333), CTNNB1
(NM_001904), DDR2 (NM_001014796), EGFR (NM_005228),
ERBB2 (NM_004448), ERBB4 (NM_005235), FBXW7
(NM_033632), FGFR1 (NM_023110), FGFR2 (NM_022970),
FGFR3 (NM_000142), KRAS (NM_033360), MAP2K1
(NM_002755), MET (NM_001127500), NOTCH1 (NM_017617),
NRAS (NM_002524), PIK3CA (NM_006218), PTEN
(NM_000314), SMAD4 (NM_005359), STK11 (NM_000455),
TP53 (NM_000546) and HRAS (001130442).
The Ion AmpliSeq RNA Fusion Lung Cancer Research
Panel targets over 70 fusion transcripts associated with lung
cancer research. It enables the analysis of the major ALK, RET,
ROS1, and NTRK1 fusion transcripts, in addition to
targets designed to detect 5′ and 3′ ALK gene expression.
The panel also includes 5 positive control genes.
Library preparation
DNA or RNA concentrations were measured using the
Qubit™ 2.0 Fluorometer in combination with the Qubit dsDNA HS assay
kit (Thermo Fischer Scientific). cDNA was generated with
SuperScript® VILO™ cDNA Synthesis kit (Thermo Fischer
Scientific) from 10 ng of total RNA. For DNA library construction,
10 ng of DNA from each of the 502 FFPE samples was utilized.
An amplicon library was thus generated from total
DNA/cDNA using the Ion AmpliSeq Library kit 2.0 (Thermo Fischer
Scientific) according to the manufacturer's instructions. Briefly,
amplicon amplification was performed using Ion AmpliSeq HiFi Master
Mix (Thermo Fischer Scientific). The amplicons were then digested
with FUPA reagent and barcoded with the IonCode™ Barcode Adapters
1–384 kit (Thermo Fischer Scientific). Subsequently, the amplified
products were purified from the other reaction components using
Agencourt AMPure XP PCR purification system (Beckman Coulter, Inc.,
Brea, CA, USA).
RNA libraries were quantified using the Qubit 2.0
Fluorometer and the Qubit dsDNA HS assay kit. Each library (20 pM)
was multiplexed. For libraries that originated from genomic DNA,
the Ion Library Equalizer™ kit method was used for normalizing
library concentration at ~100 pM without the need for special
instrumentation for quantification. Finally, equal volumes of each
normalized DNA library and 20 pM of each RNA library were combined
and clonally amplified on Ion Sphere™ particles (ISP) by emulsion
PCR performed on the Ion One Touch™ 2 instrument with the Ion PI™
Hi-Q™ OT2 200 kit (Thermo Fisher Scientific) according to the
manufacturer's instructions.
Quality control was performed using the Ion Sphere
Quality Control kit (Thermo Fisher Scientific) to ensure that
10–30% of template positive ISP were generated in the emulsion PCR.
Finally, the template-positive Ion PI Ion Sphere™ Particles were
enriched using the Ion OneTouch™ ES instrument, loaded on an Ion PI
Chip v3 and sequenced on an Ion Proton™ Sequencer with the Ion PI
Hi-Q Sequencing 200 kit according to the manufacturer's
instructions.
NGS data analysis was performed with Ion Reporter™
5.0 software directly from within Torrent Suite™ 5.0.4 software
(Thermo Fisher Scientific) along with the commercial analysis
software Sequence Pilot version 4.3.0 (JSI Medical Systems GmbH,
Ettenheim, Germany). The coverage analysis was performed using the
coverage analysis plug-in v5.0.4.0. The statistics generated from
this plug in were used to evaluate the quality of each library in
the sequencing run. NGS amplification for each library was
considered successful when a minimum average of 500 reads or
greater was achieved across all target regions and the number of
mapped reads was >150,000. Copy number variation (CNV) analysis
was performed using the Ion Reporter Software directly from within
the Torrent Suite Software (Thermo Fisher Scientific). CNVs are
reported based on their copy number relative to the control sample
used. The software reports all possible CNVs assigning a score,
with scores >10 indicating high-confidence CNVs. This value is
used as threshold for identifying a copy number amplification.
High resolution melting curve and
Sanger sequencing analysis
EGFR exon 18, 19, 20, 21, KRAS and
NRAS exon 2,3,4 and BRAF exon 11 and 15 mutation
analysis was carried out by high resolution melting curve (HRM)
analysis followed by sequencing analysis as previously described
(18,19). For the Sanger sequencing reaction,
PCR amplification products were purified using the
NucleoFast® 96 PCR Clean-up kit (Macherey-Nagel, Düren,
Germany), according to the manufacturer's protocol. The purified
product (7 µl) was used for the sequencing reaction using the
BigDye® Terminator v1.1 Cycle Sequencing kit (Applied
Biosystems, Inc., Foster City, CA, USA). Sequencing reaction
products were purified prior to electrophoresis using the Montage™
SEQ96 Sequencing Reaction kit (EMD Millipore Corporation,
Billerica, MA, USA). Sequencing analysis was performed on an
Applied Biosystems 3130 Genetic Analyzer (Applied Biosystems,
Inc.).
TruSeq Custom Amplicon Targeted NGS
assay
TruSeq Custom Amplicon Library Preparation
(Illumina, Inc.) allows targeted sequencing of the genomic regions
spanning upwards of 600 kb with up to 1,536 amplicons in a single
multiplex reaction.
A pool of custom upstream and down primers was
designed. The oligos were specific for amplification of specific
regions involved in somatic mutations in different cancers. In
total, 17 targets were amplified, using 42 amplicons, according to
the manufacturer's protocol. The amplicons were located in BRAF,
NRAS, BRAF, HRAS, cKIT, PDGFRA and EGFR genes. The
library preparation was performed as previously described (18). Sequencing was carried out on the
MiSeq sequencer (Illumina, Inc.). NGS data analysis was performed
using Illumina's genomics computing environment BaseSpace.
Real-time PCR for ERBB2 and MET
amplification
Copy number variation analysis of MET and
ERBB2 gene amplification compared to a reference gene was
carried out using TaqMan® Copy Number Assays
(Hs01432482_cn and Hs00817646_cn) and TaqMan Copy Number Reference
Assay (using TERT as endogenous reference gene). The
real-time PCR quantification was performed on the Cobas 4800 (Roche
Molecular Biochemicals). DNAs obtained from FFPE tissues were used.
The qPCR reaction mixture contained 4 µl genomic DNA template
(diluted to a concentration of 5 ng/µl), 10 µl of 2X TaqMan
Genotyping Master Mix, 1 µl of the TaqMan copy number target assay
(MET or ERBB2), 1 µl of the TaqMan Copy Number
Reference Assay (TERT), and 4 µl of nuclease-free water.
Each sample was run in a minimum of three replicates. Calculation
of the relative amounts of the target gene compared to the
reference gene was performed by Cobas 4800 Relative Quantification
software (Roche Molecular Biochemicals). The final results are
expressed as a ratio of the target gene to the reference gene
copies in the sample, normalized with the ratio of target
gene:reference gene copies in the Calibrator DNA (which was set to
one). A ratio of <2.0 was assumed to be negative for gene
over-amplification, a ratio of ≥2.0 was assumed to be positive for
gene over-amplification.
Method sensitivity
The mutation detection limit was determined using
genomic DNA reference standards with defined allelic frequencies
(Horizon Diagnostics, Cambridge, UK).
Calculation of the NGS mutation detection limit was
carried out using two EGFR multiplex reference standards
that cover mutations at codons 719 (p.G719S), 746–750
(A746-E750del), 790 (p.T790M), 858 (p.L858R) and 861 (p.L861Q)
spanning exons 19, 20 and 21. These standards were manufactured
using five engineered EGFR mutant cell lines and mixed to
generate a 5% and 1% mutant EGFR allelic frequency,
respectively. A 3% reference standard was created by adding
equimolecular quantities of the 5% and 1% mutant EGFR
allelic frequency standards. Additionally, a third Quantitative
Multiplex FFPE Reference Standard (Horizon Diagnostics) was used.
This standard covers mutations at codons 719 (p.G719S), 746–750
(A746-E750del), 790 (p.T790M), 858 (p.L858R), with a mutant
EGFR allelic frequency of 24.5, 2, 1 and 3%, respectively. A
custom DNA Reference Standard (Horizon Diagnostics), containing
KRAS (p.G12D, p.G13D, p.Q61H), NRAS (p.Q61R) and
BRAF (p.V600E) mutations at a 5% mutant allelic frequency
was also used. All reference standards were run in triplicate in
three different runs.
Furthermore, a reference standard positive for
EML4-ALK (Variant 1), CCDC6-RET and
SLC34A2-ROS1 (Horizon) was used for the evaluation of the
Ion AmpliSeq RNA Fusion Lung Cancer Research Panel.
Results
Test performance and comparison of
methods
The performance of our customized AmpliSeq 23 gene
panel was first evaluated using high resolution melting curve (HRM)
analysis and sequencing in 100 consecutive samples in exons 18, 19,
20, 21 of the EGFR gene, exons 2, 3, 4 of the KRAS
and NRAS genes and exons 11 and 15 of the BRAF gene.
The concordance between HRM and NGS was 100% for all mutations
detected in a percentage >5%. These 100 samples were also
genotyped using a TruSeq Custom Amplicon assay (Illumina, Inc.)
that targets hotspot regions in 7 genes (KRAS, NRAS, BRAF, cKIT,
PDGFRA, EGFR, HRAS). A total of 21/100 (21%) samples tested
were not efficiently amplified using the TruSeq Custom Amplicon
assay, compared to only 1/100 (1%) of the samples tested using the
AmpliSeq panel. This was attributed to the better DNA quality and
quantity required by the TruSeq Custom Amplicon assay. In the 79
samples that were efficiently amplified by both methods, a full
concordance was observed in the genes that were common to both
panels (KRAS, NRAS, HRAS, BRAF, and EGFR). Two
samples, presented detectable mutations by both NGS platforms
(KRAS p.Q61H and EGFR p.L858R, respectively), but
were classified as normal using the HRM method. Since the mutation
rate in both samples was <4%, it was assumed that the
discordance was due to the lower sensitivity of the HRM method.
All three methods applied can reliably be used for
somatic mutation detection. However, the NGS-based approaches had
increased sensitivity compared to the HRM method. Between the two
NGS-based techniques used, the AmpliSeq technology had the
advantage of being compatible with lower DNA concentrations, thus
it was the method of choice for tumor molecular profiling in our
cohort.
The sensitivity of the AmpliSeq method was also
evaluated using 5 Horizon reference standards. The samples were
analyzed in triplicate in 3 different experiments. In every
experiment the same DNA was used for the construction of 3
different libraries. All EGFR, KRAS, NRAS and BRAF
mutations present at mutant allelic frequency >3% in the
reference standards were always detected by the AmpliSeq panel used
in all three experiments performed in triplicate. Concerning the 1%
EGFR reference standard, the performance was variable
between the 3 replicates as well as between the three experiments.
In all three experiments, p.G719S was not detected. The
A746-E750del mutation was detectable in 2/3 replicates in the first
experiment, in 2/3 replicates in the second and in 1/3 replicates
in the third experiment. Similarly, p.L861Q was detected in 1/3
replicates in the first, in 1/3 replicates in the second and in 2/3
replicates in the third experiment. p.T790M was detected in 2/3
replicates in the first experiment, in 1/3 replicates in the second
experiment and in 2/3 replicates in the third experiment. All
EGFR mutations present in the a third Quantitative Multiplex
FFPE Reference Standard (Horizon Diagnostics) were detected in all
three NGS runs with the exception of the 790 mutation (1% mutant
allelic frequency) which was inconsistently detected (in 50% of
cases). Based on these results, we defined the mutant allelic
frequency detection limit at 3%, since at this allelic frequency
all variants were consistently detected.
The performance evaluation of the AmpliSeq RNA
Fusion Lung Cancer Research Panel was carried out using 8 samples
previously identified as positive for ALK translocation (by
FISH) and one sample positive for ROS translocation. Additionally a
Reference Standard positive for EML4-ALK (Variant 1),
CCDC6-RET and SLC34A2-ROS1 was analyzed in triplicate
in three different experiments. In all cases the correct gene
translocation was detected.
Mutation distribution and patient
characteristics
A total of 512 NSCLC patients were included in this
study. In 10 patients, no amplification was obtained due to
insufficient DNA quantity/quality, thus they were excluded from the
study. In total, 502 NSCLC patients were eligible for NGS analysis
(including the 99 patients of the evaluation cohort). Among these,
374 (74.5%) were male and 128 (25.5%) were female. The mean age of
diagnosis was 66 years. The majority of tumors with known histology
were classified as adenocarcinomas (85%).
Mutation analysis of the 23 gene NGS panel revealed
the presence of at least one mutation in 74.5% of the tumors tested
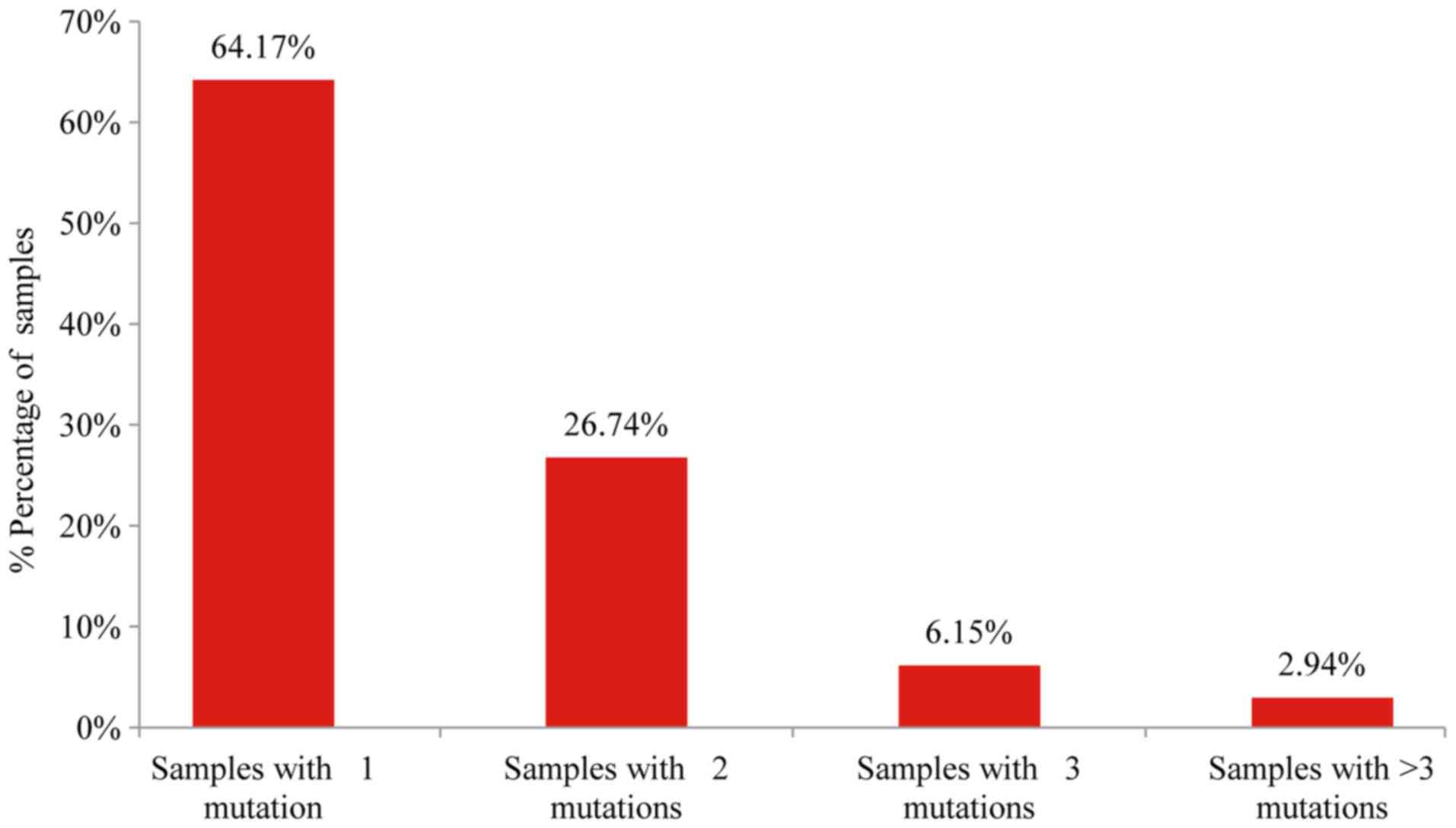
(374/502). Among these 64.2% (240/374) presented only one mutation,
26.74% (100/374) two, 6.1% (23/374) three and 2.9% (11/374) more
than three mutations (Fig. 1). In
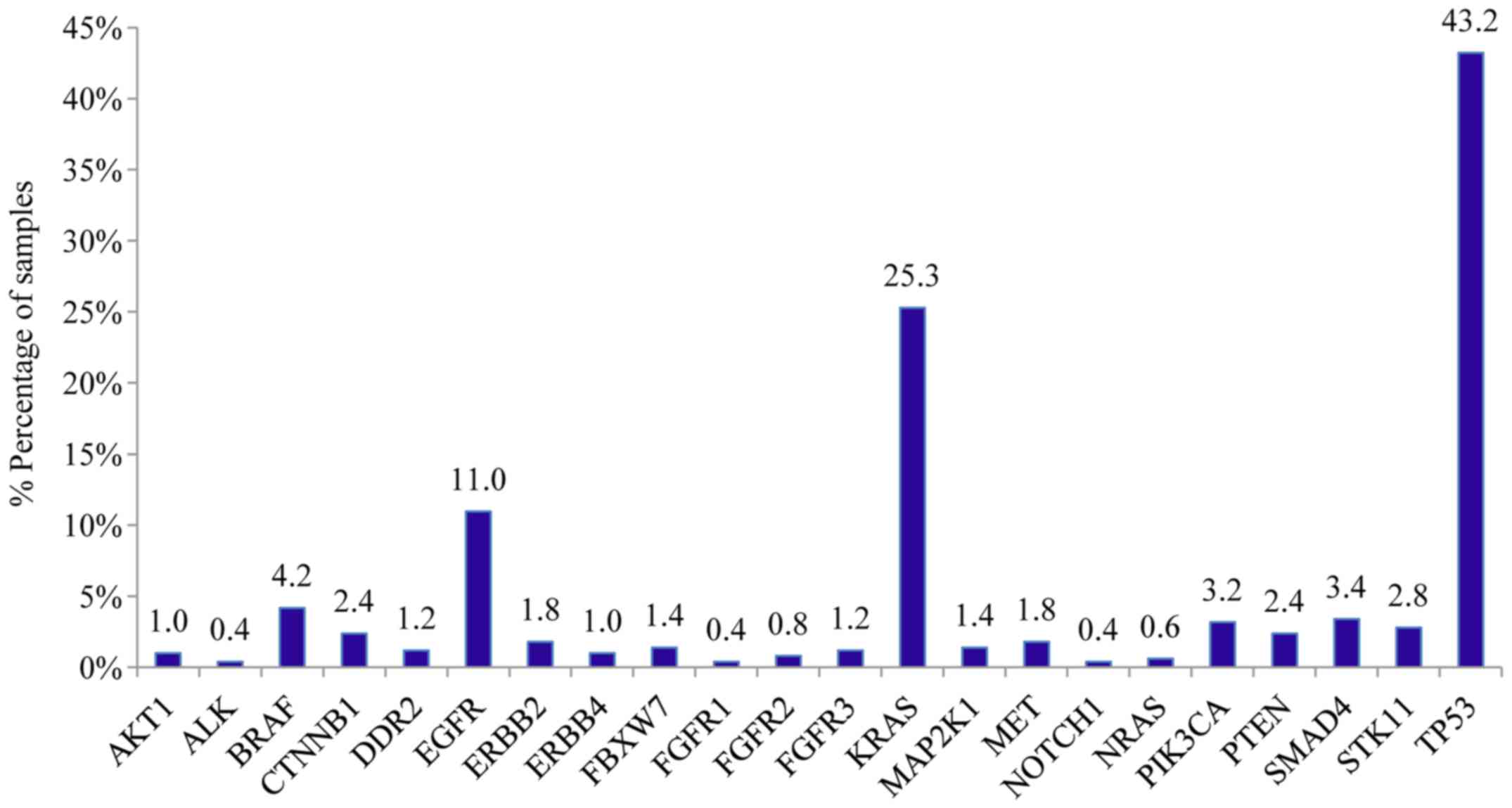
accordance with previous studies, TP53 mutations were the most
common alterations (detected in 43% of the patients), followed by
KRAS and EGFR gene mutations detected in 25% and 11%
of the cases, respectively (Fig. 2)
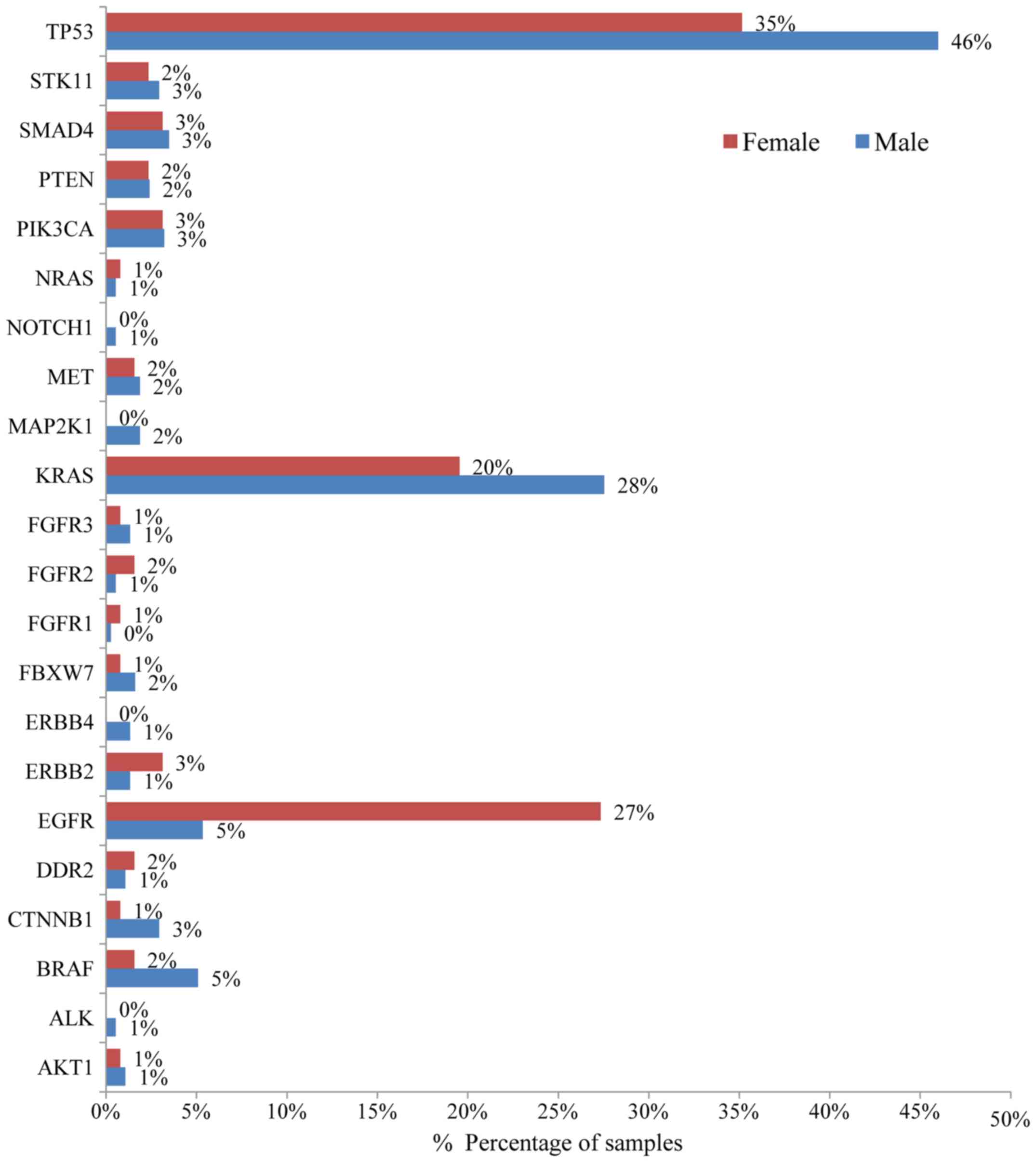
(20). The most notable difference
in mutation frequency between male and female patients was observed
as expected for the EGFR gene. In male patients the mutation
frequency was much lower (5%) than in female patients (27%),
indicating a gender-related EGFR mutation frequency
(P<0.001) (Fig. 3). EGFR
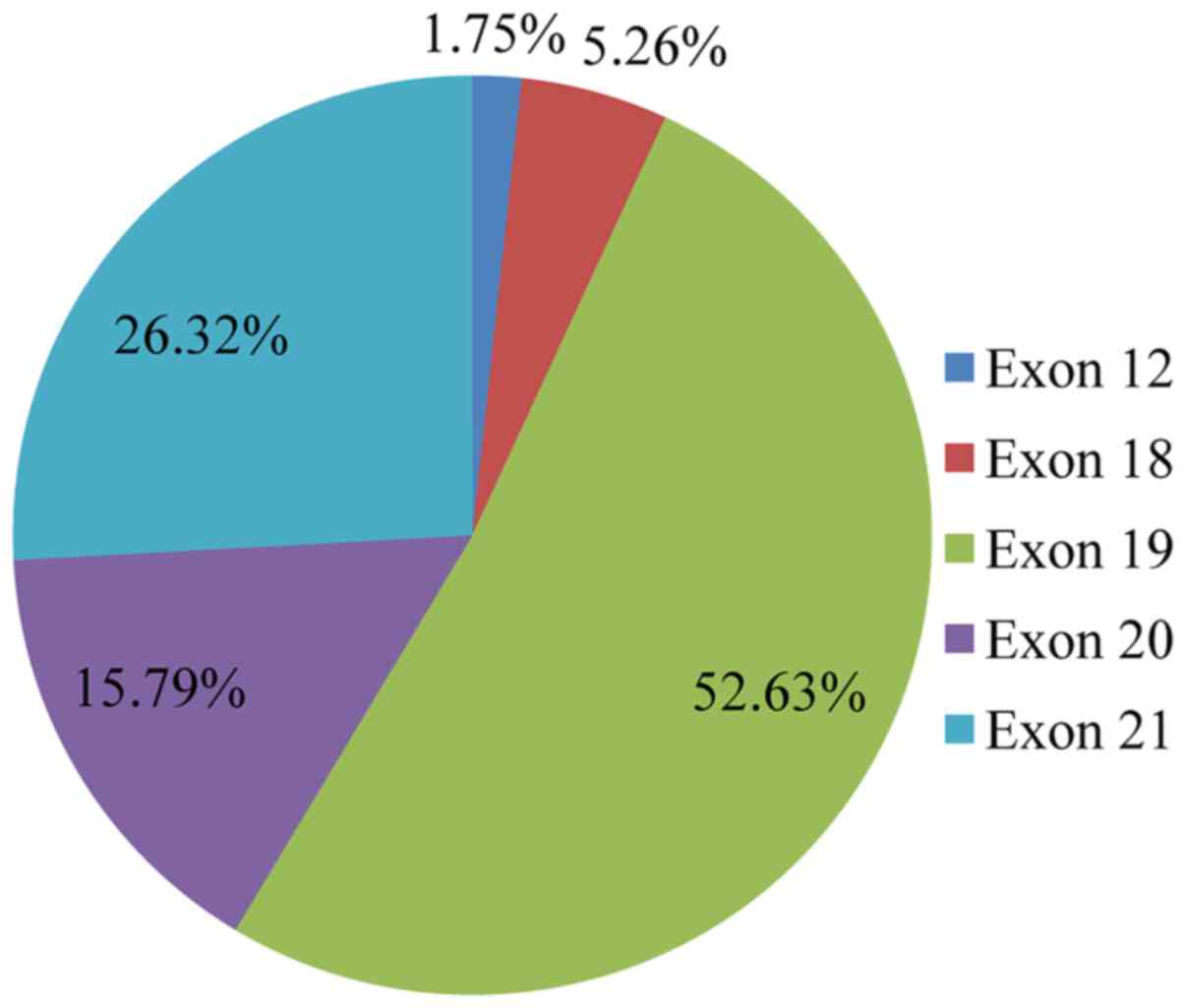
mutation distribution was 1 in exon 12 (1.75%), 3 in exon 18
(5.26%), 30 in exon 19 (52.63%), 9 in exon 20 (15.79%) and 15 in
exon 21 (26.32%) (Fig. 4). In 1
sample, 2 concomitant exon 18 mutations were identified: G719S
(drug-sensitive)+E709A compound mutation. In vitro the
double-mutant receptor has been shown less sensitive to EGFR
TKIs (21). All except one
mutations detected in exon 19 were deletions. In one sample an
insensitizing primary mutation was identified in exon 19 (p.D761Y)
(22). p.L858R was the predominant
mutation in exon 21 accounting for 90% of the mutations detected in
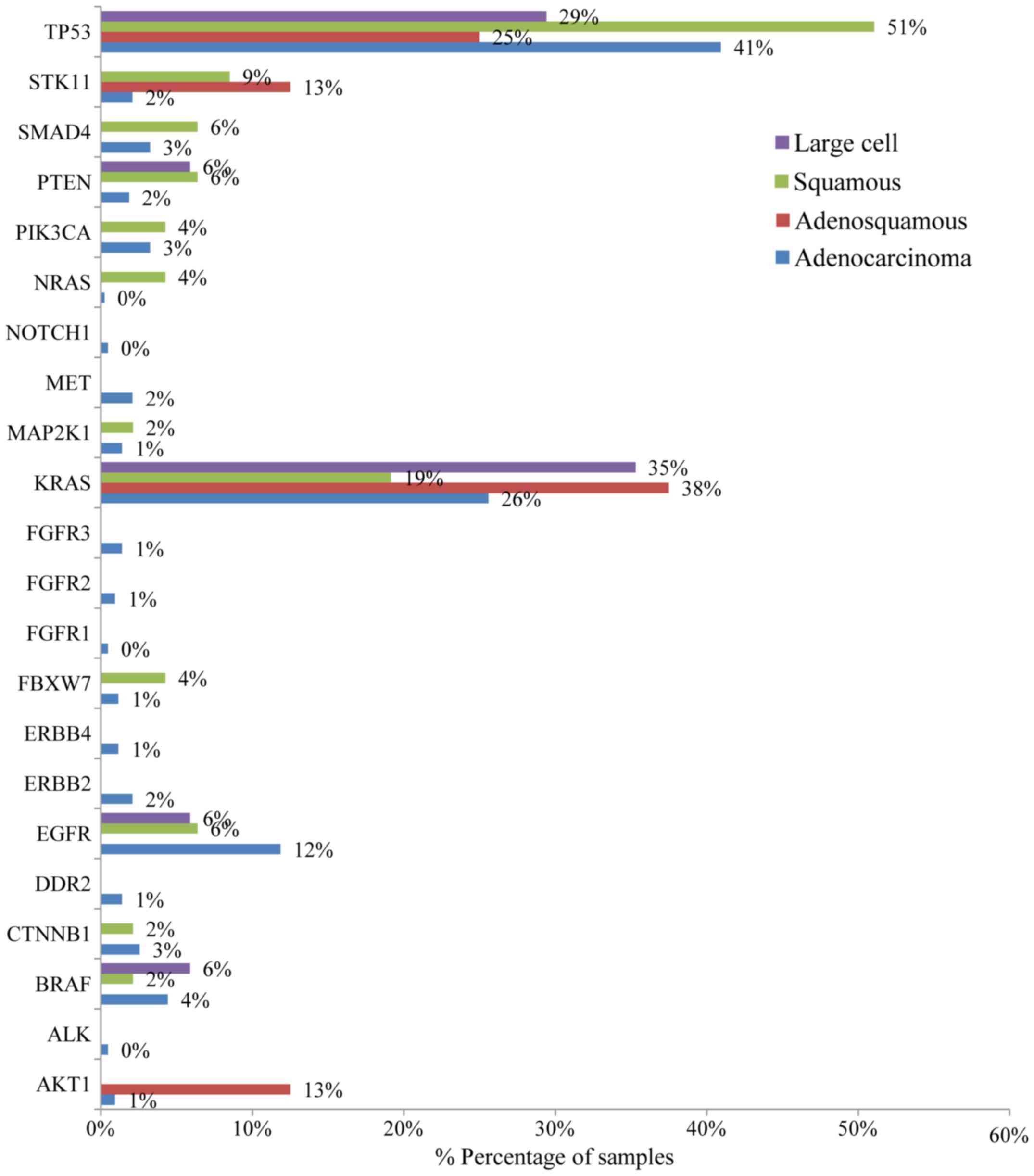
that exon. Histopathology of the tumor appeared to be related to
the EGFR mutation rates as well (Fig. 5). The greater mutation percentage
was observed in adenocarcinomas tumors. On the contrary,
adenosquamous, squamous and large cell NSCLC were associated with
reduced mutation rates. This was the case for both males and
female.
KRAS gene somatic mutations were also present
in a high percentage of NSCLC patients and are associated with
poorer prognosis and resistance to EGFR-TKIs (19,23).
In our cohort, a KRAS mutation was observed in 25.3%
(127/502) of all tumors analyzed. Among the KRAS mutant
patients, the majority of mutations was observed in exon 2 of the
KRAS gene (111/127, 87.4%), while a KRAS exon 3
mutation was observed in 7.81% (10/127) and an exon 4 mutation was
observed in 4.69% (6/127) of the cases.
Of note, 79.4% of the KRAS exon 2 mutations
detected were transversion mutations (substitution of a purine for
a pyrimidine or conversely, G→T or G→C), which are known to be
smoking-associated (24), while the
percentage of transition mutations (substitution of a purine for a
purine, e.g., G→A or a pyrimidine for a pyrimidine, C→T) was
limited to 20.6% of the patients.
BRAF is an important new therapeutic target
in NSCLC (25–27). In our study, a mutation was observed
in 4.18% (21/502) of the cases. Among the BRAF mutant cases,
the majority was observed in exon 11 of BRAF (12/21,
57.14%), while in 42.86% (9/21) of the BRAF mutant patients
an exon 15 mutation was observed. However, only in 4 cases the
V600E mutation which is associated with targeted therapy was
detected. Thus, the V600E mutation percentage in NSCLC patients was
only 0.8% (4/502).
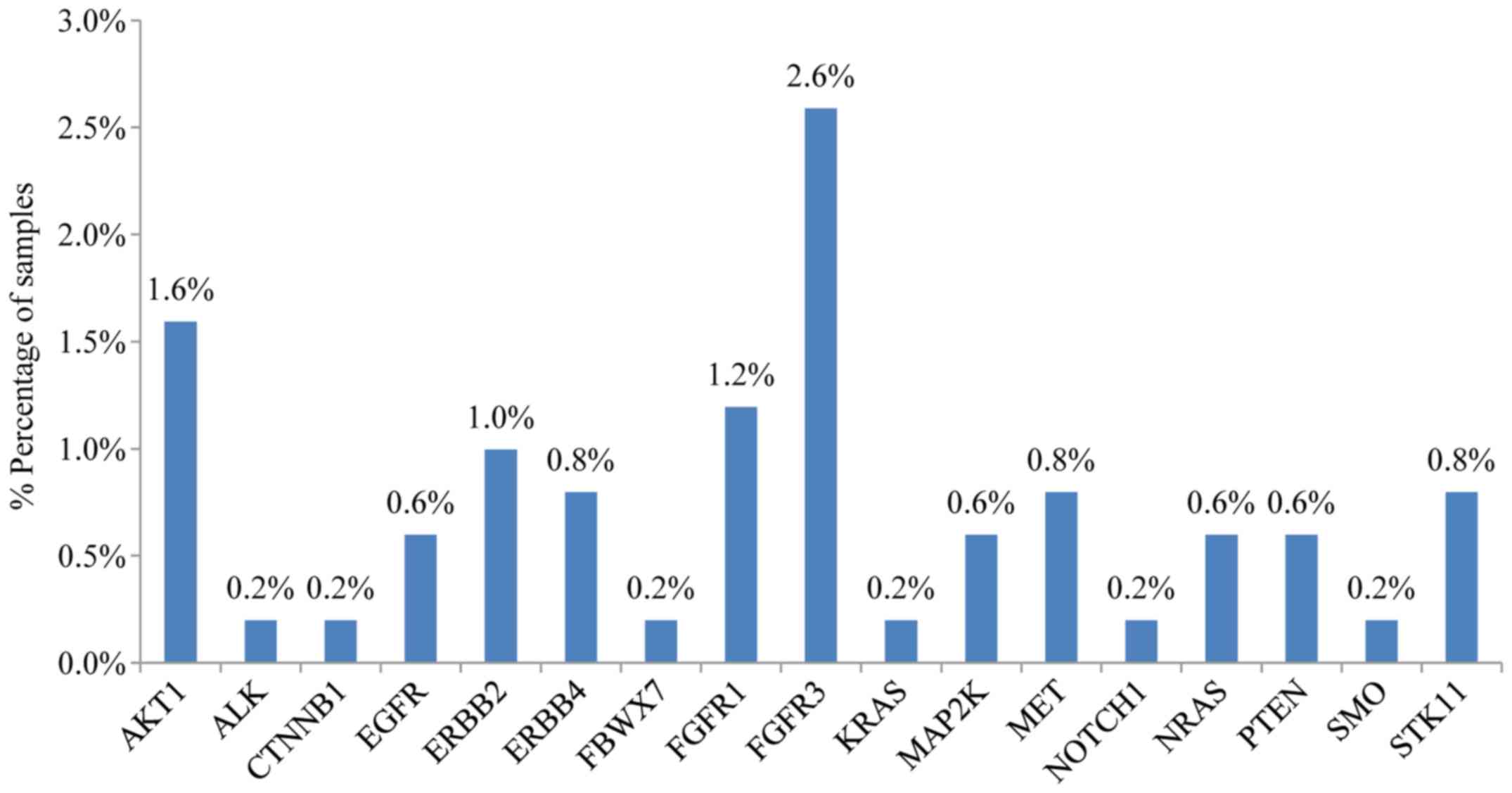
Other genes with a significant mutation frequency in
the NSCLC cohort tested included ERBB2, MET, CTNBB1 and
PTEN (with mutation rates for each one of ~2%), PIK3CA,
SMAD4, STK11 (each one with mutation frequency ~2%). At a
frequency ~1%, mutations were also observed for AKT1, DDR2,
ERBB4, FBWX7, FGFR1, FGFR3, MAP2K1 and NRAS.
In addition to the presence of somatic mutations,
this 23 gene panel was also accessed for the detection of copy
number variations (CNV) in cancer-driver genes. Regarding NSCLC,
copy number amplifications (CNA) have been observed for MET,
ERBB2, FGFR1 and FGFR3 that represent viable treatment
targets for approved or investigational therapies (28–31)
(Fig. 6). The presence of
MET and ERBB2 amplification was also confirmed by
real-time PCR in all samples.
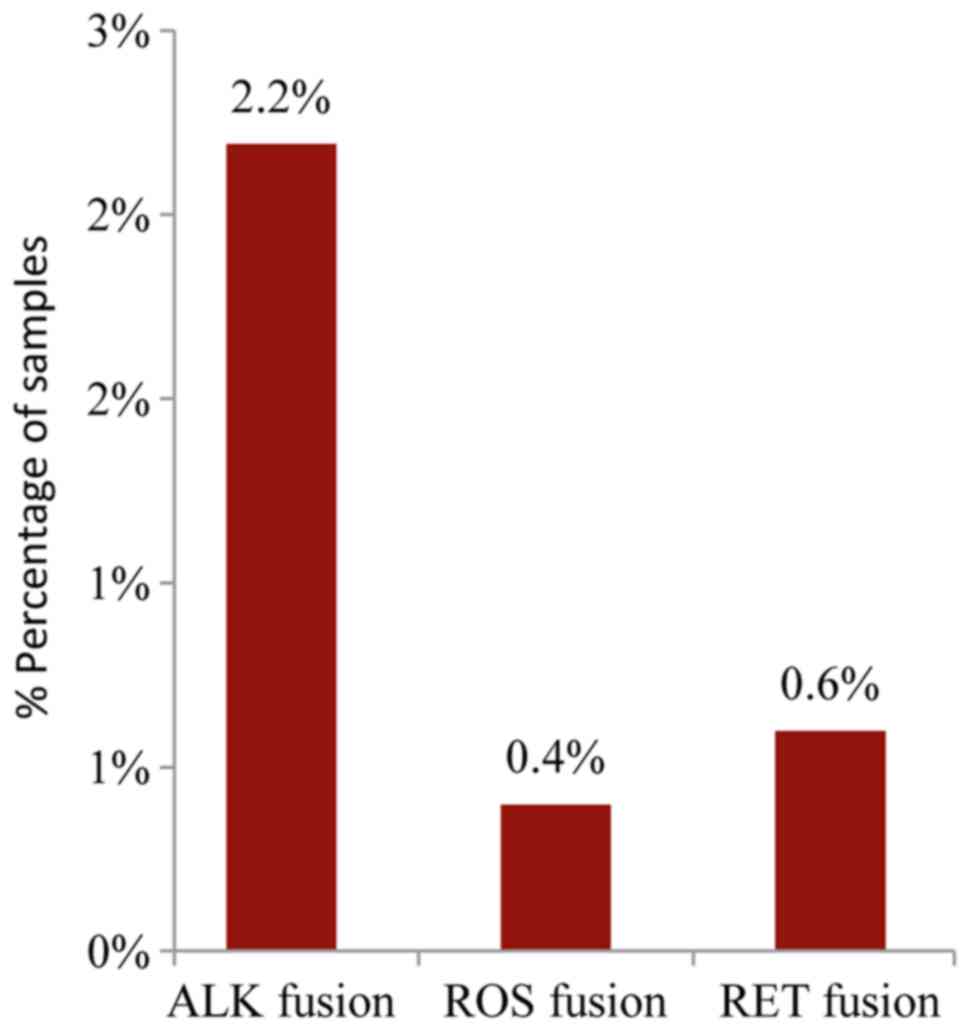
The analysis of the major ALK, RET, ROS1, and
NTRK1 fusion transcripts using the Ion AmpliSeq RNA Fusion
Lung Cancer Research Panel, revealed the presence of an ALK fusion
in 2.2% (11/502) of the samples while a ROS or RET
fusion was present in 0.4% and 0.6% of the cases, respectively
(2/502 and 3/502) (Fig. 7). A
complete list of gene mutation fusions and copy number
amplifications are available upon request.
Discussion
During the past few years, cancer treatment has been
redirected toward the use of targeted therapeutics, with their
effectiveness depending on tumor-specific alterations.
Consequently, the management of neoplastic diseases has changed
substantially, since the treatment decision is no longer unspecific
but depends on the molecular profile of the tumor for patient
selection (32).
A main prerequisite for the accurate molecular
characterization of the tumor is the selection of the optimal
analysis method. Single gene analysis methods are time consuming
and require a large amount of tumor DNA, which in the majority of
the cases is not available. Furthermore, the number of genetic
alterations associated with targeted therapies is constantly
increasing. Thus, global analysis of the genetic profile of tumors
is crucial and cannot be achieved by sequential analysis of a few
genes (33).
Targeted NGS, involving gene panels, is a quick and
cost effective multiple gene analysis method that allows a more
comprehensive tumor mutation profiling. The selection of the
optimal NGS methodology should be based on its performance using a
limited and bad quality DNA extracted from FFPE tissues.
Additionally, a careful selection of the genes included in the
panels should be conducted in order to include all important
treatment-related targets. Thus, targeted NGS panels for somatic
mutation detection include actionable cancer genes and allow the
determination of the tumor molecular profile of the patient
(34,35).
In the present study, a targeted amplicon-based NGS
assay was used for simultaneous analysis of tumor-specific
alterations in 23 genes commonly mutated in lung and colorectal
cancers. This panel used the Ion AmpliSeq technology offered by
Thermo Fisher Scientific and was selected since it has several
advantages that make it an attractive option of diagnostic utility.
AmpliSeq-based panels are currently available or can be custom
made. They are cost effective, have good performance, are
compatible with low DNA concentration and have very low failure
rates even with DNAs of relative low quality such as those
extracted from FFPE samples. Consequently, they can be an
attractive option and of clinical value for somatic mutation
analysis (13,14,35).
The gene panel used in this study contained all
important genes with actionable mutations related to NSCLC.
Therefore, it was deemed optimal for clinical use in this tumor
type. Additionally, it exhibited high rates (100%) of sensitivity
and specificity at a low mutation frequency of 3%. This is
extremely important for somatic mutation detection since, due to
tumor heterogeneity, the mutation rate in tumor tissue could be
extremely low. Furthermore, it allows the detection of different
types of alteration in one workflow (mutations, copy number
changes, fusions, expression).
The aim of such an approach is the determination of
the molecular profile (gene mutations) of the tumor, the
determination of the drug (if it exists) that targets the mutated
gene(s) or the pathway that the gene(s) is involved in and the
determination of gene interaction (in case of multiple mutations).
Moreover, patients can be assigned to clinical trials associated
with their molecular profile and consequently have more potential
treatment options in addition to FDA-approved targeted therapies.
This could be particularly important for patients who have failed
conventional therapy. In fact, there is an increasing number of
clinical trials in which patient access depends on the molecular
profile of the tumor determined by NGS technology (36–38).
There are two main approaches. The so-called ‘basket’ trials enroll
patients with multiple tumor types and assign different therapies
based on their tumor genetic alterations. The National Cancer
Institute (NCI)-Molecular Analysis for Therapy Choice (NCI MATCH)
Trial (39), and the Targeted Agent
and Profiling Utilization Registry (TAPUR) (40), which is sponsored by the American
Society of Clinical Oncology (ASCO) are two main examples of such
trials in previously treated advanced solid tumors. Alternatively,
the ‘umbrella’ trials enroll patients with a single tumor type, and
assign various treatments according to their tumor molecular
profile. Examples of such an approach constitute the Lung Master
Protocol (Lung MAP) trial in recurrent stage IV squamous cell lung
cancer (41), the National Lung
Matrix Trial (42) and the
Biomarker-Integrated Targeted Therapy Study in Previously Treated
Patients with Advanced NSCLC (BATTLE-2) (43).
NSCLC is a tumor type with the most available
targeted therapies. Genes with approved targeted agents include
EGFR, ALK, and ROS1 (3,44).
Additionally, specific genetic alterations in NSCLC are now
recognized as optimal targets for agents approved in other tumor
types. These agents target genes that are frequently altered in
NSCLC such as KRAS, ERBB2, MET and RET, that are
recommended as emerging new biomarkers by the National
Comprehensive Cancer Network (NCCN) guidelines. Furthermore,
potential targets, such as BRAF, PIK3CA, FGFR1, DDR2, PTEN
are currently being tested in clinical trials (45,46).
Therefore, the number of predictive biomarkers for novel targeted
drugs entering into clinical practice is rapidly increasing. The
selected gene panel includes the most commonly mutated genes in
lung and colorectal cancer. Mutational status of some of the genes
tested may be associated with the activity of certain approved
drugs. This is obvious when accessing data from MyCancer Genome
knowledge database (www.mycancergenome.org/), that provides reliable
information concerning important cancer-related genes and their
correlation with treatment options. As indicated in Table I, multiple approved therapies
targeting important cancer-related genes or their pathways are
available. However, each agent has received approval for a
particular tumor type(s) and not all agents targeting specific
genes have sufficient clinical evidence in a patient tumor type. In
principal, the molecular profile of a tumor could be indicative of
the treatment that should be followed if an approved targeted
therapy can be related to the patients genetic profile or if
clinical trials exist (37,38). Thus, particular caution should be
made when using the genetic information retrieved from these
panels.
 | Table I.Approved agents targeting important
cancer-related genes or their pathways. |
Table I.
Approved agents targeting important
cancer-related genes or their pathways.
| Agent | Target(s) | FDA-approved
indication(s) |
|---|
| Cetuximab
(Erbitux) | EGFR
(HER1/ERBB1) | Colorectal cancer
(KRAS wild-type), squamous cell cancer of the head and
neck |
| Panitumumab
(Vectibix) | EGFR
(HER1/ERBB1) | Colorectal cancer
(KRAS wild-type) |
| Regorafenib
(Stivarga) | KIT, PDGFRβ, RAF,
RET, VEGFR1/2/3 | Colorectal cancer,
gastrointestinal stromal tumors |
| Crizotinib
(XALKori) | ALK, MET,
ROS1 | Non-small cell lung
cancer (with ALK fusion or ROS1 gene alteration) |
| Alectinib
(Alecensa) | ALK | Non-small cell lung
cancer (with ALK fusion) |
| Ceritinib
(Zykadia) | ALK | Non-small cell lung
cancer (with ALK fusion) |
| Gefitinib
(Iressa) | EGFR
(HER1/ERBB1) | Non-small cell lung
cancer [with EGFR exon 19 deletions or exon 21 substitution
(L858R) mutations] |
| Afatinib
(Gilotrif) | EGFR (HER1/ERBB1),
HER2 (ERBB2/neu) | Non-small cell lung
cancer [with EGFR exon 19 deletions or exon 21 substitution
(L858R) mutations] |
| Erlotinib
(Tarceva) | EGFR
(HER1/ERBB1) | Non-small cell lung
cancer [with EGFR exon 19 deletions or exon 21 substitution
(L858R) mutations], pancreatic cancer |
| Osimertinib
(Tagrisso) | EGFR | Non-small cell lung
cancer (with EGFR T790M mutation) |
| Necitumumab
(Portrazza) | EGFR
(HER1/ERBB1) | Squamous non-small
cell lung cancer |
| Ponatinib
(Iclusig) | ABL, FGFR1-3, FLT3,
VEGFR2 | Chronic myelogenous
leukemia, acute lymphoblastic leukemia (Philadelphia
chromosome-positive) |
| DaBRAFenib
(Tafinlar) | BRAF | Melanoma (with
BRAF V600 mutation) |
| Vemurafenib
(Zelboraf) | BRAF | Melanoma (with
BRAF V600 mutation) |
| Vandetanib
(Caprelsa) | EGFR (HER1/ERBB1),
RET, VEGFR2 | Medullary thyroid
cancer |
| Cabozantinib
[Cabometyx (tablet), Cometriq (capsule)] | FLT3, KIT, MET,
RET, VEGFR2 | Medullary thyroid
cancer, renal cell carcinoma |
| Ado-trastuzumab
emtansine (Kadcyla) | HER2
(ERBB2/neu) | Breast cancer
(HER2+) |
| Pertuzumab
(Perjeta) | HER2
(ERBB2/neu) | Breast cancer
(HER2+) |
| Trastuzumab
(Herceptin) | HER2
(ERBB2/neu) | Breast cancer
(HER2+), gastric cancer (HER2+) |
| Lapatinib
(Tykerb) | HER2 (ERBB2/neu),
EGFR (HER1/ERBB1) | Breast cancer
(HER2+) |
| Cobimetinib
(Cotellic) | MEK | Melanoma (with
BRAF V600E or V600K mutation) |
| Trametinib
(Mekinist) | MEK | Melanoma (with
BRAF V600 mutation) |
| Everolimus
(Afinitor) | mTOR | Pancreatic,
gastrointestinal, or lung origin neuroendocrine tumor, renal cell
carcinoma, breast cancer (HR+, HER2−),
non-resectable subependymal giant cell astrocytoma associated with
tuberous sclerosis |
| Temsirolimus
(Torisel) | mTOR | Renal cell
carcinoma |
| Sorafenib
(Nexavar) | VEGFR, PDGFR, KIT,
RAF | Hepatocellular
carcinoma, thyroid carcinoma, renal cell carcinoma |
Molecular tumor profiling using our multigene panel
was feasible for the majority of the patients tested with a limited
amplification failure rate of approximately 2%. At least one DNA
mutation was detected in 374 patients (74.5%). In 16 patients,
(3.2%) an RNA fusion was identified. In accordance to previous
studies, the most commonly mutated gene in our cohort was the TP53
gene (43%), followed by KRAS and EGFR gene mutations
detected in 25 and 11% of the cases (20). As expected, great differences in
EGFR mutation frequency were observed between male (5%) and
female patients (27%) (18). The
mutual exclusivity of EGFR, KRAS, BRAF and HER2
genes, as well as ALK, ROS1 and RET fusions has been
already described and it was also confirmed in our cohort (47).
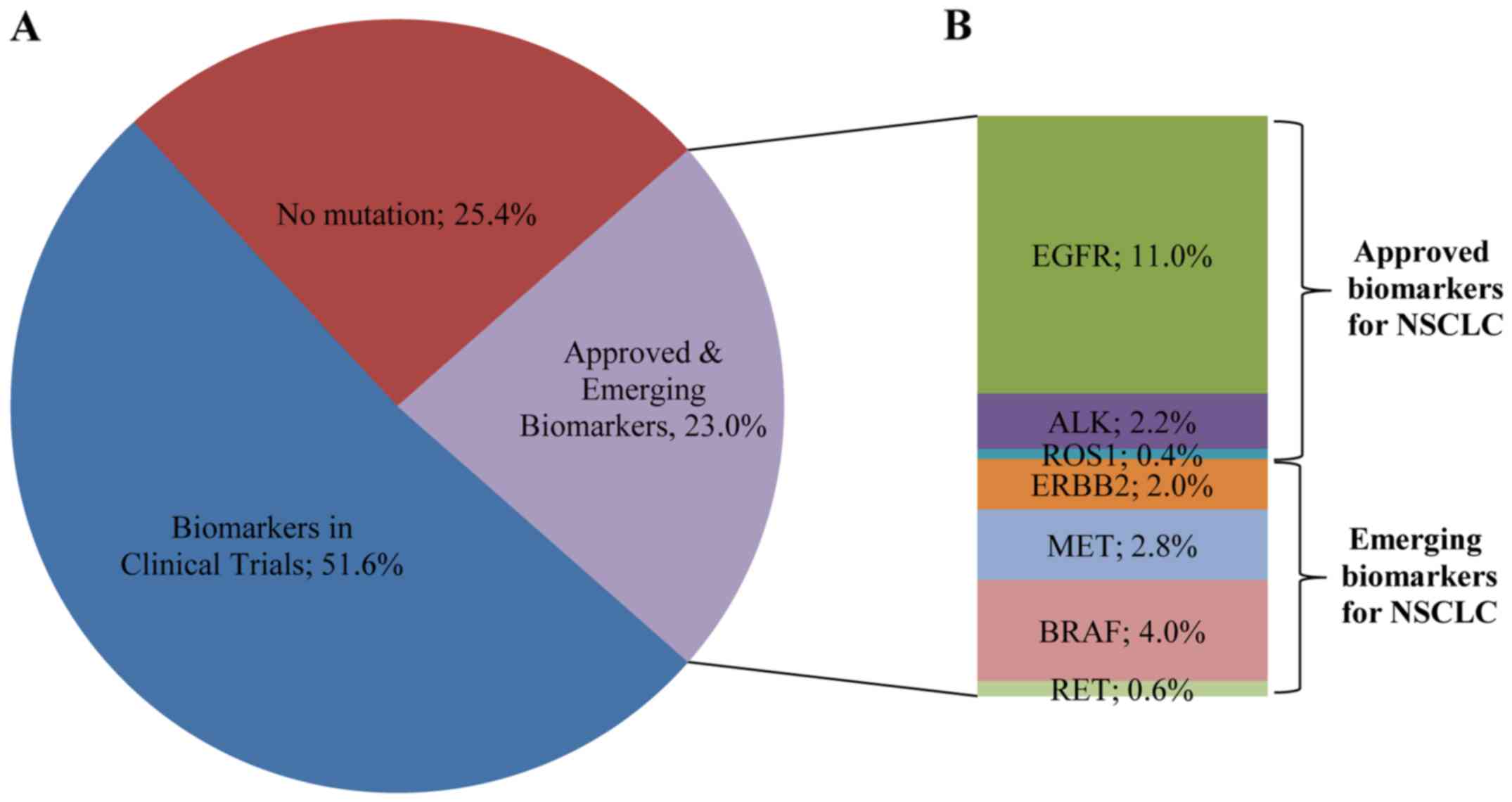
In the present study, 23% of the patients presented
a mutation in a gene associated with approved or emerging
treatments. More specifically, 13.5% (68/502) of the patients
presented a mutation in a gene with approved targeted therapy
(EGFR, ALK, ROS1) and 9.4% (47/502) of the patients had an
alteration in a gene related to emerging targeted therapies
according to the NCCN guidelines (Fig.
8). These alterations include ERBB2, BRAF and MET
mutations, MET amplification and RET rearrangements.
Thus, the information provided by this multigene analyses,
facilitates a physician's decision concerning the selection of the
appropriate treatment options. The remaining 51.6% (259/502) of the
patients had a mutation in a gene that could be related to an off
label therapy or indicate access to a clinical trial (Fig. 8). The identification of such
alterations could be valuable in cases with advanced tumors that
have progressed on standard therapies since it offers these
patients the opportunity to be enrolled in ongoing clinical
trials.
In conclusion, the multigene NGS panel applied was
able to identify alterations in a cancer-driver gene (including
point mutations, gene rearrangements and MET amplifications)
in 77.6% of the tumors tested. The method used has a vast
applicability in tumor molecular characterization of NSCLC,
allowing the simultaneous detection of actionable DNA alterations
in 23 genes in addition to 4 gene fusions in these patients.
Furthermore, due to its cost and time effectiveness, it can be
efficiently applied for molecular characterization of tumors in
order to increase the percentage of patients with gene alterations
related to approved, emerging or ongoing clinical trials for
targeted therapy.
References
|
1
|
Torre LA, Siegel RL and Jemal A: Lung
Cancer Statistics. Adv Exp Med Biol. 893:1–19. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Steliarova-Foucher E,
Lortet-Tieulent J, Rosso S, Coebergh JW, Comber H, Forman D and
Bray F: Cancer incidence and mortality patterns in Europe:
Estimates for 40 countries in 2012. Eur J Cancer. 49:1374–1403.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsao AS, Scagliotti GV, Bunn PA Jr,
Carbone DP, Warren GW, Bai C, de Koning HJ, Yousaf-Khan AU,
McWilliams A, Tsao MS, et al: Scientific advances in lung cancer
2015. J Thorac Oncol. 11:613–638. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gagan J and Van Allen EM: Next-generation
sequencing to guide cancer therapy. Genome Med. 7:802015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Diaz Z, Aguilar-Mahecha A, Paquet ER,
Basik M, Orain M, Camlioglu E, Constantin A, Benlimame N, Bachvarov
D, Jannot G, et al: Next-generation biobanking of metastases to
enable multidimensional molecular profiling in personalized
medicine. Mod Pathol. 26:1413–1424. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meldrum C, Doyle MA and Tothill RW:
Next-generation sequencing for cancer diagnostics: A practical
perspective. Clin Biochem Rev. 32:177–195. 2011.PubMed/NCBI
|
|
7
|
Pavlopoulou A, Spandidos DA and
Michalopoulos I: Human cancer databases (Review). Oncol Rep.
33:3–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Forbes SA, Beare D, Gunasekaran P, Leung
K, Bindal N, Boutselakis H, Ding M, Bamford S, Cole C, Ward S, et
al: COSMIC: Exploring the world's knowledge of somatic mutations in
human cancer. Nucleic Acids Res. 43:D805–D811. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chin L, Hahn WC, Getz G and Meyerson M:
Making sense of cancer genomic data. Genes Dev. 25:534–555. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wu TJ, Schriml LM, Chen QR, Colbert M,
Crichton DJ, Finney R, Hu Y, Kibbe WA, Kincaid H, Meerzaman D, et
al: Generating a focused view of disease ontology cancer terms for
pan-cancer data integration and analysis. Database (Oxford).
2015:bav0322015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dumur CI: Available resources and
challenges for the clinical annotation of somatic variations.
Cancer Cytopathol. 122:730–736. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin
D, Lu L and Law M: Comparison of next-generation sequencing
systems. J Biomed Biotechnol. 2012:2513642012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tops BB, Normanno N, Kurth H, Amato E,
Mafficini A, Rieber N, Le Corre D, Rachiglio AM, Reiman A, Sheils
O, et al: Development of a semi-conductor sequencing-based panel
for genotyping of colon and lung cancer by the Onconetwork
consortium. BMC Cancer. 15:262015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
D'Haene N, Le Mercier M, De Nève N,
Blanchard O, Delaunoy M, El Housni H, Dessars B, Heimann P,
Remmelink M, Demetter P, et al: Clinical validation of targeted
next generation sequencing for colon and lung cancers. PLoS One.
10:e01382452015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jennings LJ, Arcila ME, Corless C,
Kamel-Reid S, Lubin IM, Pfeifer J, Temple-Smolkin RL, Voelkerding
KV and Nikiforova MN: Guidelines for validation of next-generation
sequencing-based oncology panels: A joint consensus recommendation
of the Association for Molecular Pathology and College of American
Pathologists. J Mol Diagn. 19:341–365. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shahsiah R, DeKoning J, Samie S,
Latifzadeh SZ and Kashi ZM: Validation of a next generation
sequencing panel for detection of hotspot cancer mutations in a
clinical laboratory. Pathol Res Pract. 213:98–105. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Travis WD, Brambilla E, Nicholson AG,
Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E,
Flieder DB, et al: WHO Panel: The 2015 World Health Organization
Classification of Lung Tumors: Impact of Genetic, Clinical and
Radiologic Advances Since the 2004 Classification. J Thorac Oncol.
10:1243–1260. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Papadopoulou E, Tsoulos N, Tsirigoti A,
Apessos A, Agiannitopoulos K, Metaxa-Mariatou V, Zarogoulidis K,
Zarogoulidis P, Kasarakis D, Kakolyris S, et al: Determination of
EGFR and KRAS mutational status in Greek non-small-cell lung cancer
patients. Oncol Lett. 10:2176–2184. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Negru S, Papadopoulou E, Apessos A,
Stanculeanu DL, Ciuleanu E, Volovat C, Croitoru A, Kakolyris S,
Aravantinos G, Ziras N, et al: KRAS, NRAS and BRAF mutations in
Greek and Romanian patients with colorectal cancer: A cohort study.
BMJ Open. 4:e0046522014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scoccianti C, Vesin A, Martel G, Olivier
M, Brambilla E, Timsit JF, Tavecchio L, Brambilla C, Field JK and
Hainaut P: European Early Lung Cancer Consortium: Prognostic value
of TP53, KRAS and EGFR mutations in nonsmall cell lung cancer: The
EUELC cohort. Eur Respir J. 40:177–184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tam IY, Leung EL, Tin VP, Chua DT, Sihoe
AD, Cheng LC, Chung LP and Wong MP: Double EGFR mutants containing
rare EGFR mutant types show reduced in vitro response to gefitinib
compared with common activating missense mutations. Mol Cancer
Ther. 8:2142–2151. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Costa DB: Kinase inhibitor-responsive
genotypes in EGFR mutated lung adenocarcinomas: Moving past common
point mutations or indels into uncommon kinase domain duplications
and rearrangements. Transl Lung Cancer Res. 5:331–337. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kempf E, Rousseau B, Besse B and Paz-Ares
L: KRAS oncogene in lung cancer: Focus on molecularly driven
clinical trials. Eur Respir Rev. 25:71–76. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ahrendt SA, Decker PA, Alawi EA, Zhu Yr
YR, Sanchez-Cespedes M, Yang SC, Haasler GB, Kajdacsy-Balla A,
Demeure MJ and Sidransky D: Cigarette smoking is strongly
associated with mutation of the K-ras gene in patients with primary
adenocarcinoma of the lung. Cancer. 92:1525–1530. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cardarella S, Ogino A, Nishino M, Butaney
M, Shen J, Lydon C, Yeap BY, Sholl LM, Johnson BE and Jänne PA:
Clinical, pathologic, and biologic features associated with BRAF
mutations in non-small cell lung cancer. Clin Cancer Res.
19:4532–4540. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Villaruz LC, Socinski MA, Abberbock S,
Berry LD, Johnson BE, Kwiatkowski DJ, Iafrate AJ, Varella-Garcia M,
Franklin WA, Camidge DR, et al: Clinicopathologic features and
outcomes of patients with lung adenocarcinomas harboring BRAF
mutations in the Lung Cancer Mutation Consortium. Cancer.
121:448–456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nguyen-Ngoc T, Bouchaab H, Adjei AA and
Peters S: BRAF alterations as therapeutic targets in non-small-cell
lung cancer. J Thorac Oncol. 10:1396–1403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Dutt A, Ramos AH, Hammerman PS, Mermel C,
Cho J, Sharifnia T, Chande A, Tanaka KE, Stransky N, Greulich H, et
al: Inhibitor-sensitive FGFR1 amplification in human non-small cell
lung cancer. PLoS One. 6:e203512011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jorge SE, Schulman S, Freed JA, VanderLaan
PA, Rangachari D, Kobayashi SS, Huberman MS and Costa DB: Responses
to the multitargeted MET/ALK/ROS1 inhibitor crizotinib and
co-occurring mutations in lung adenocarcinomas with MET
amplification or MET exon 14 skipping mutation. Lung Cancer.
90:369–374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mar N, Vredenburgh JJ and Wasser JS:
Targeting HER2 in the treatment of non-small cell lung cancer. Lung
Cancer. 87:220–225. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Collisson EA, Campbell JD, Brooks AN,
Berger AH, Lee W, Chmielecki J, Beer DG, Cope L, Creighton CJ,
Danilova L, et al: Cancer Genome Atlas Research Network:
Comprehensive molecular profiling of lung adenocarcinoma. Nature.
511:543–550. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hirsch FR, Suda K, Wiens J and Bunn PA Jr:
New and emerging targeted treatments in advanced non-small-cell
lung cancer. Lancet. 388:1012–1024. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kamps R, Brandão RD, Bosch BJ, Paulussen
AD, Xanthoulea S, Blok MJ and Romano A: Next-generation sequencing
in oncology: Genetic diagnosis, risk prediction and cancer
classification. Int J Mol Sci. 18:E3082017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takeda M, Sakai K, Terashima M, Kaneda H,
Hayashi H, Tanaka K, Okamoto K, Takahama T, Yoshida T, Iwasa T, et
al: Clinical application of amplicon-based next-generation
sequencing to therapeutic decision making in lung cancer. Ann
Oncol. 26:2477–2482. 2015.PubMed/NCBI
|
|
35
|
de Leng WW, Gadellaa-van Hooijdonk CG,
Barendregt-Smouter FA, Koudijs MJ, Nijman I, Hinrichs JW, Cuppen E,
van Lieshout S, Loberg RD, de Jonge M, et al: Targeted next
generation sequencing as a reliable diagnostic assay for the
detection of somatic mutations in tumours using minimal DNA amounts
from formalin fixed paraffin embedded material. PLoS One.
11:e01494052016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Siu LL, Conley BA, Boerner S and LoRusso
PM: Next-generation sequencing to guide clinical trials. Clin
Cancer Res. 21:4536–4544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rashdan S and Gerber DE: Going into
BATTLE: Umbrella and basket clinical trials to accelerate the study
of biomarker-based therapies. Ann Transl Med. 4:5292016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sundar R, Chénard-Poirier M, Collins DC
and Yap TA: Imprecision in the era of precision medicine in
non-small cell lung cancer. Front Med (Lausanne).
4:392017.PubMed/NCBI
|
|
39
|
Colwell J: NCI-MATCH trial draws strong
interest. Cancer Discov. 6:3342016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Markman M: Maurie Markman on the
Groundbreaking TAPUR Trial. Oncology (Williston Park). 31:158–168.
2017.PubMed/NCBI
|
|
41
|
Herbst RS, Gandara DR, Hirsch FR, Redman
MW, LeBlanc M, Mack PC, Schwartz LH, Vokes E, Ramalingam SS,
Bradley JD, et al: Lung master protocol (Lung-MAP)-A
biomarker-driven protocol for accelerating development of therapies
for squamous cell lung cancer: SWOG S1400. Clin Cancer Res.
21:1514–1524. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Middleton G, Crack LR, Popat S, Swanton C,
Hollingsworth SJ, Buller R, Walker I, Carr TH, Wherton D and
Billingham LJ: The National Lung Matrix Trial: Translating the
biology of stratification in advanced non-small-cell lung cancer.
Ann Oncol. 26:2464–2469. 2015.PubMed/NCBI
|
|
43
|
Kim C and Giaccone G: Lessons learned from
BATTLE-2 in the war on cancer: The use of Bayesian method in
clinical trial design. Ann Transl Med. 4:4662016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chan BA and Hughes BG: Targeted therapy
for non-small cell lung cancer: Current standards and the promise
of the future. Transl Lung Cancer Res. 4:36–54. 2015.PubMed/NCBI
|
|
45
|
Rothschild SI: Targeted therapies in
non-small cell lung cancer-beyond EGFR and ALK. Cancers (Basel).
7:930–949. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zer A and Leighl N: Promising targets and
current clinical trials in metastatic non-squamous NSCLC. Front
Oncol. 4:3292014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kohno T, Nakaoku T, Tsuta K, Tsuchihara K,
Matsumoto S, Yoh K and Goto K: Beyond ALK-RET, ROS1 and other
oncogene fusions in lung cancer. Transl Lung Cancer Res. 4:156–164.
2015.PubMed/NCBI
|