Introduction
Hepatocellular carcinoma (HCC), one of the leading
causes for cancer-related deaths, is the most aggressive and lethal
of liver tumors (1). During the
last several decades, remarkable advances have been made regarding
the diagnosis and treatment of HCC (2). However, the long-term survival of HCC
patients is still poor (3). The
exact mechanisms responsible for the malignant growth and
metastatic behaviors of HCC cells remain largely uncovered. Thus,
it is worth investigating the novel oncogene, which plays an
essential role in the initiation and progression of HCC.
Immature colon carcinoma transcript-1 (ICT1),
previously named DS-1, is identified as a regulator during
colorectal cancer (CRC) cell differentiation in the study by van
Belzen et al (4). Further
studies report that ICT1 plays an important role in the progression
of CRC, lung, prostate and breast cancer, and that it has been
suggested as a potential biomarker (5–10). In
prostate cancer, ICT1 knockdown suppresses cancer cell
proliferation and viability, leading to cell cycle arrest at G2/M
phase and apoptosis (7). ICT1 is
overexpressed in CRC tissues and associates with poor prognosis of
patients (8). Furthermore, ICT1
promotes CRC progression by regulating cell cycle, migration and
apoptosis of cancer cells (8).
Similarly, the study by Wang et al (9) report that ICT1 acts as a potential
biomarker for diagnosis and treatment of lung cancer and
facilitates non-small cell lung cancer cell proliferation via
promoting cell cycle progression and inhibiting apoptosis.
Recently, Wang et al (10)
showed that ICT1 silencing restrains cell growth of breast cancer
by inducing cell cycle arrest and apoptosis. These above studies
suggest that ICT1 functions as an oncogene in different cancer
types. The present study aimed to investigate the clinical
significance, the biological function and the underlying mechanisms
of ICT1 in HCC.
This study demonstrates that ICT1 overexpression
correlates with malignant clinical features and poor prognosis of
HCC patients. Functionally, ICT1 promotes proliferation, cell cycle
progression and inhibits apoptosis of HCC cells. ICT1 regulates the
expressions of CDK1, cyclin B1, Bcl-2 and Bax, and is directly
targeted by microRNA-134 (miR-134) in HCC cells. In conclusion, the
present study supports the first evidence that ICT1 is a potential
prognostic biomarker and therapeutic target for HCC.
Materials and methods
Patients
Sixty-eight HCC tissues and matched tumor-adjacent
tissues were obtained from the The First Affiliated Hospital of
Xi'an Medical University. Tissue specimens were conserved in 10%
formalin for further investigation. No patients received
immunotherapy, radiotherapy or chemotherapy before surgery. Tumor
staging was based on the seventh Union for International Cancer
Control/American Joint Committee on Cancer (UICC/AJCC) staging
system (11). All samples were used
after obtaining informed consent. The Ethics Committee of The First
Affiliated Hospital of Xi'an Medical University approved the
protocols according to the Declaration of Helsinki.
Cell culture and transfection
The human immortalized normal hepatocyte cell line
(LO2) and human HCC cell lines including Hep3B, MHCC97L, MHCC97H
and HCCLM3 were obtained from Shanghai Institute of Biochemistry
and Cell Biology, Chinese Academy of Sciences (Shanghai, China).
All cell lines were cultivated in Dulbeccos minimum essential
medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) using 10%
fetal calf serum (FCS; Gibco, Grand Island, NY, USA) at 37°C in a
humidified atmosphere of 5% CO2.
Precursor miR-134, miR-205 and miR-340, and miR-134
inhibitor as well as corresponding negative control vectors were
purchased from GeneCopoeia (Guangzhou, China). ICT1 shRNA,
non-targeting (NT) shRNA, ICT1 overexpression vector and empty
vector (EV) were synthesized and obtained from Shanghai Genechem
Co., Ltd. (Shanghai, China). Vectors were transferred into cells
using Lipofectamine 2000 (Thermo Fisher Scientific) in accordance
with the manufacturers protocols.
Immunohistochemistry (IHC)
The HCC tissues that were previously formalin-fixed
and paraffin-embedded were sliced into 4 µm sections, and underwent
deparaffination and then rehydration. Antigen retrieval,
suppression of endogenous peroxidase activity and 10% skim milk
blocking were performed before primary antibody incubation. ICT1
(AP20382b; Abgent, Inc., San Diego, CA, USA) and Ki-67 primary
antibody (ab15580; Abcam, Cambridge, MA, USA) was used for
incubation overnight at 4°C. The slides were subsequently incubated
with peroxidase conjugated secondary antibody (ZSGB Bio, Beijing,
China) for 90 min, and a peroxidase-labeled polymer, DAB solution
was used for signal development for 5 min. The sections were
counterstained with hematoxylin followed by dehydrating and
mounting. Staining intensity was scored as no staining, 0, weak
staining, 1, moderate staining, 2, and strong staining, 3. Staining
quantity was graded as <25%, 1, 25–75%, 2, and >75%, 3. IHC
score was manually confirmed by two independent experienced
pathologists using the formula: IHC score = staining intensity ×
staining quantity. IHC score ≥3 was considered as positive
expression of ICT1.
Quantitative real-time PCR
RNA was isolated using TRIzol (Invitrogen, Carlsbad,
CA, USA) following the manufacturers protocols. The first strand
cDNA was compounded using a Tianscript RT kit (Tiangen Biotech,
Co., Ltd., Beijing, China). PCR amplification for ICT1 mRNA was
performed with the SYBR Premix Ex Taq™ kit (Takara Bio, Shiga,
Japan) in ABI 7300 system (Applied Biosystems, Foster City, CA,
USA). GAPDH was employed as a reference gene to normalize the
expression of ICT1 mRNA. The primers used for ICT1 and GAPDH were
purchased from Sangon Biotech Co., Ltd. (Shanghai, China).
Colony formation assay
For cell proliferation, colony formation assay was
performed. HCC cells transfected with corresponding vectors were
seeded in 6-well plates and maintained in cell incubators for 14
days. The formed cell colonies were stained with crystal violet
solution. The number of cell colonies was counted to represent the
cell proliferation ability of HCC cells.
Cell cycle and apoptosis analysis
For cell cycle assay, HCC cells were collected 72 h
after transfection. These cells were fixed with 80% ethanol
overnight, and then were stained with propidium iodide (50 µg/ml;
BD Biosciences, Franklin Lakes, NJ, USA) at room temperature for 30
min. The percentage of cells in each cell cycle was measured with
FACSCalibur system (BD Biosciences). For apoptosis assay, HCC cells
after transfection were subjected to an apoptosis assay. The
percentage of apoptotic HCC cells were measured using Annexin
V/propidium iodide kit (BD Biosciences, San Diego, CA, USA) based
on the protocol provided by manufactures.
In vivo tumor growth assay
For tumor growth studies, nude mice were injected
subcutaneously with 1×106 HepG2 cells transfected with
ICT1 shRNA or NT shRNA. Tumor sizes were measured every 4 days
after subcutaneous injection. Three weeks later, the mice were
sacrificed by cervical dislocation under anesthesia, and tumors
were removed for the volume measurement. The protocols for animal
experiments were approved by the Ethics Committee of The First
Affiliated Hospital of Xi'an Medical University.
Luciferase reporter assay
Wild-type (wt) or mutant (mt) 3′-UTR of ICT1 was
amplified and cloned into pmiR-RB-REPORT™ Luciferase. Then, the
3′-UTR of ICT1 and corresponding miRNA vectors were co-transfected
into HepG2 cells, respectively. Forty-eight hours after the
co-transfection, the cells were lysed and detected using a
Dual-Luciferase® reporter assay kit (Promega, Madison,
WI, USA) based on the manufacturers protocols.
Western blotting
Total proteins were collected with RIPA lysis buffer
(Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and 40 µg
protein were subjected to 4–20% SDS gel electrophoresis
(Sigma-Aldrich) and were then transferred to PVDF membranes (Roche,
Indianapolis, IN, USA). Then, 5% milk blocked membranes were
incubated with ICT1 (Abgent), CDK1 (ab18; Abcam), cyclin B1
(#12231; Cell Signaling Technology, Beverly, MA, USA), Bax (#5023;
Cell Signaling Technology) or Bcl-2 (#15071; Cell Signaling
Technology) primary antibody and subsequently incubated with
matched secondary antibodies (Cell Signaling Technology). Then,
signals for each protein expression were detected with the Bio-Rad
Gel imaging system (Bio-Rad Laboratories, Hercules, CA, USA). GAPDH
(G8140; US Biological, Swampscott, MA, USA) was used as a loading
control.
Statistical analysis
Data are presented as mean ± SD and analyzed by
GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA,
USA). Chi-squared test was employed to explore the association
between two variables. The Student's t-test and analysis of
variance (ANOVA) were carried out to analyze continuous variables.
Survival curves were constructed and differences among groups were
calculated using the Kaplan-Meier method and log-rank test. The
value of P<0.05 was considered to have statistical
significance.
Results
ICT1 is overexpressed in HCC
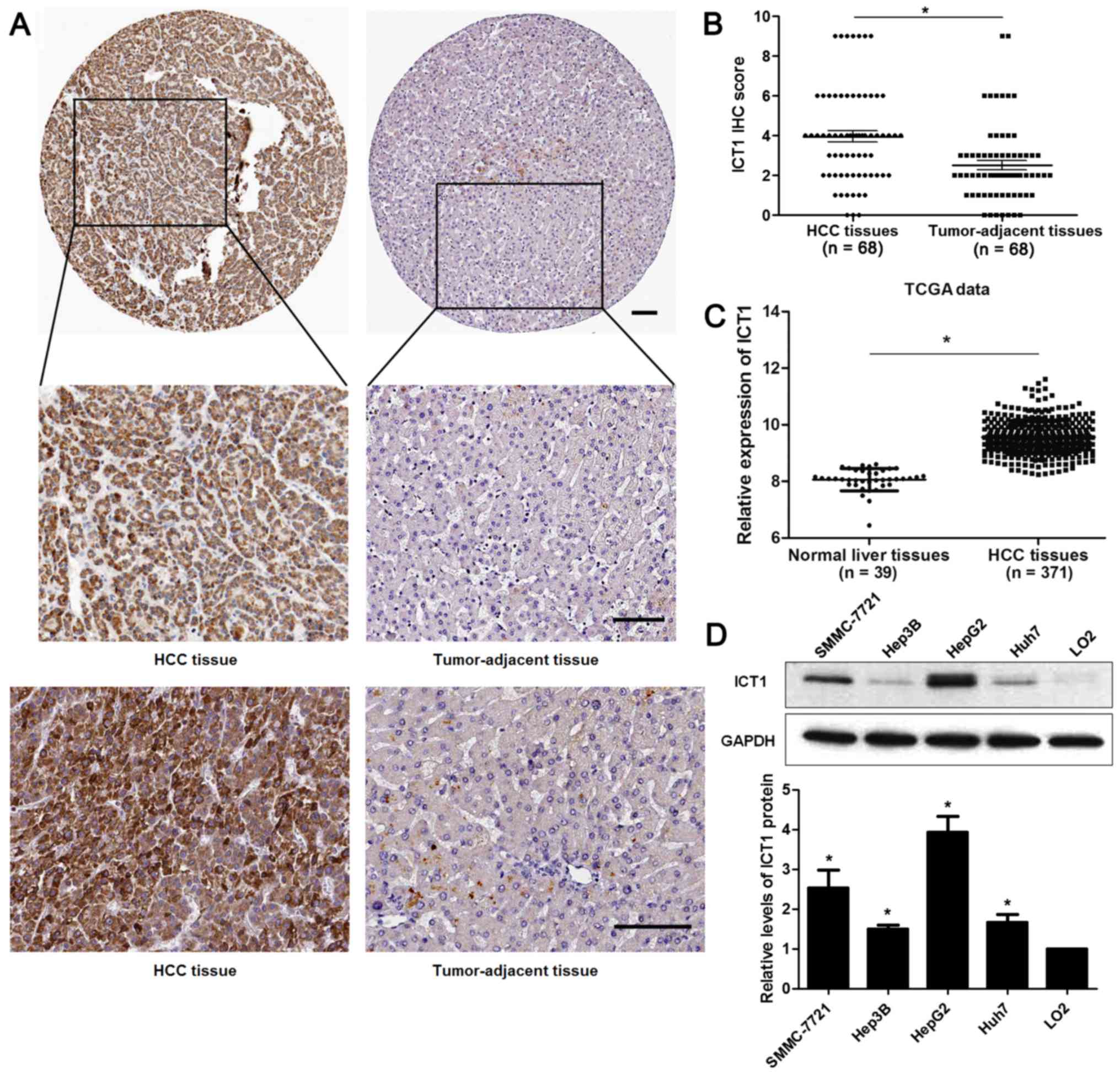
IHC staining was performed to detect the expression
of ICT1 in 68 pairs of HCC and matched tumor-adjacent tissues.
Forty-seven of 68 (69.12%) HCC tissues showed positive expression
of ICT1, while ICT1 signal was observed in only 27 of 68 (39.71%)
tumor-adjacent tissues (P<0.05; Fig.
1A). Our data indicated that the levels of ICT1 in HCC tissues
were significantly higher than those in matched non-cancerous
tissues (P<0.05; Fig. 1B). TCGA
data from R2: Genomics Analysis and Visualization Platform
(http://r2.amc.nl) showed that the expression of ICT1
is significantly upregulated in HCC tissues compared to normal
liver tissues (P<0.05; Fig. 1C),
which is consistent with our results. In addition, the expression
of ICT1 in HCC cell lines (SMMC-7721, Hep3B, HepG2 and Huh7) were
notably upregulated compared to LO2 cells (P<0.05, respectively;
Fig. 1D). Thus, the expression of
ICT1 is restrained in HCC.
Clinical significance of ICT1 in HCC
patients
Next, we investigated the clinical significance of
ICT1 in HCC patients. Sixty-eight HCC patients were divided into
ICT1 positive expression group (IHC score ≥3) and ICT1 negative
expression group (IHC score <3). Clinical association analysis
found that the ICT1 positive expression was correlated with larger
tumor size and advanced TNM tumor stage (P<0.05, respectively;
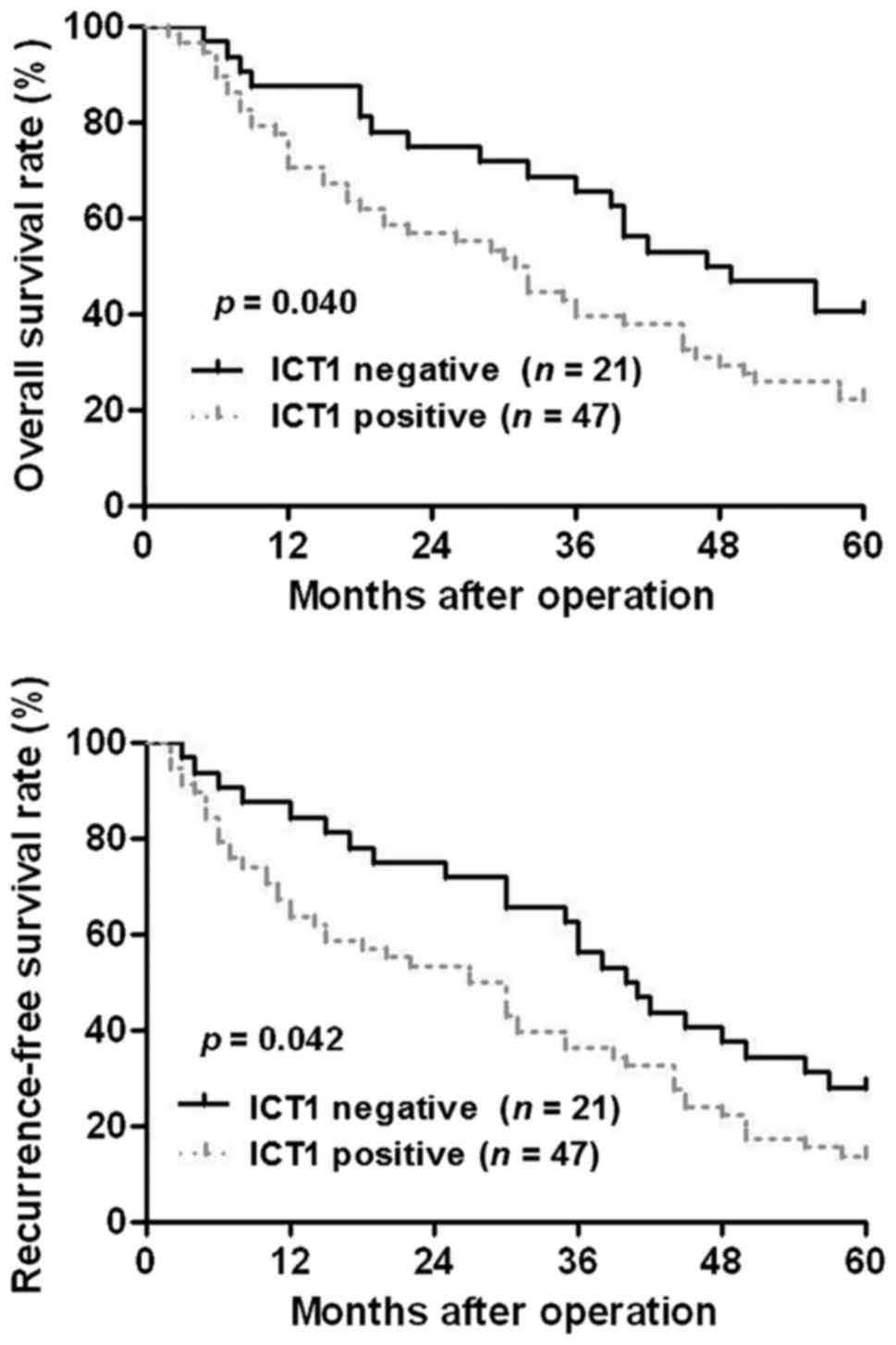
Table I). Notably, Kaplan-Meier
plots indicated that ICT1-positive expression in HCC patients
showed a significant reduced overall survival and recurrence-free
survival compared to ICT1 negative expression cases (P<0.05,
respectively; Fig. 2). These
results suggest that ICT1 may act as a promising prognostic marker
for HCC patients.
 | Table I.Correlation between the
clinicopathological features and ICT1 expression in hepatocellular
carcinoma (n=68). |
Table I.
Correlation between the
clinicopathological features and ICT1 expression in hepatocellular
carcinoma (n=68).
|
|
| ICT1 expression |
|
|---|
|
|
|
|
|
|---|
| Characteristics | Total no. of
patients | Positive (n=47) | Negative (n=21) | P-value |
|---|
| Age (years) |
|
|
| 0.723 |
|
<50 | 27 | 18 | 9 |
|
| ≥50 | 41 | 29 | 12 |
|
| Sex |
|
|
| 0.612 |
| Male | 49 | 33 | 16 |
|
|
Female | 19 | 14 | 5 |
|
| HBV |
|
|
| 0.635 |
|
Absent | 20 | 13 | 7 |
|
|
Present | 48 | 34 | 14 |
|
| Serum AFP level
(ng/ml) |
|
|
| 0.155 |
|
<400 | 24 | 14 | 10 |
|
| ≥400 | 44 | 33 | 11 |
|
| Tumor size (cm) |
|
|
| 0.020a |
|
<5 | 25 | 13 | 12 |
|
| ≥5 | 43 | 34 | 9 |
|
| No. of tumor
nodules |
|
|
| 0.445 |
| 1 | 54 | 39 | 15 |
|
| ≥2 | 14 | 8 | 6 |
|
| Cirrhosis |
|
|
| 0.373 |
|
Absent | 27 | 17 | 10 |
|
|
Present | 41 | 30 | 11 |
|
| Venous
infiltration |
|
|
| 0.880 |
|
Absent | 51 | 35 | 16 |
|
|
Present | 17 | 12 | 5 |
|
| Edmondson-Steiner
grading |
|
|
| 0.067 |
|
I+II | 48 | 30 | 18 |
|
|
III+IV | 20 | 17 | 3 |
|
| TNM tumor
stage |
|
|
| 0.004a |
|
I+II | 49 | 29 | 20 |
|
|
III+IV | 19 | 18 | 1 |
|
ICT1 regulates proliferation, cell
cycle progression and apoptosis of HCC cells
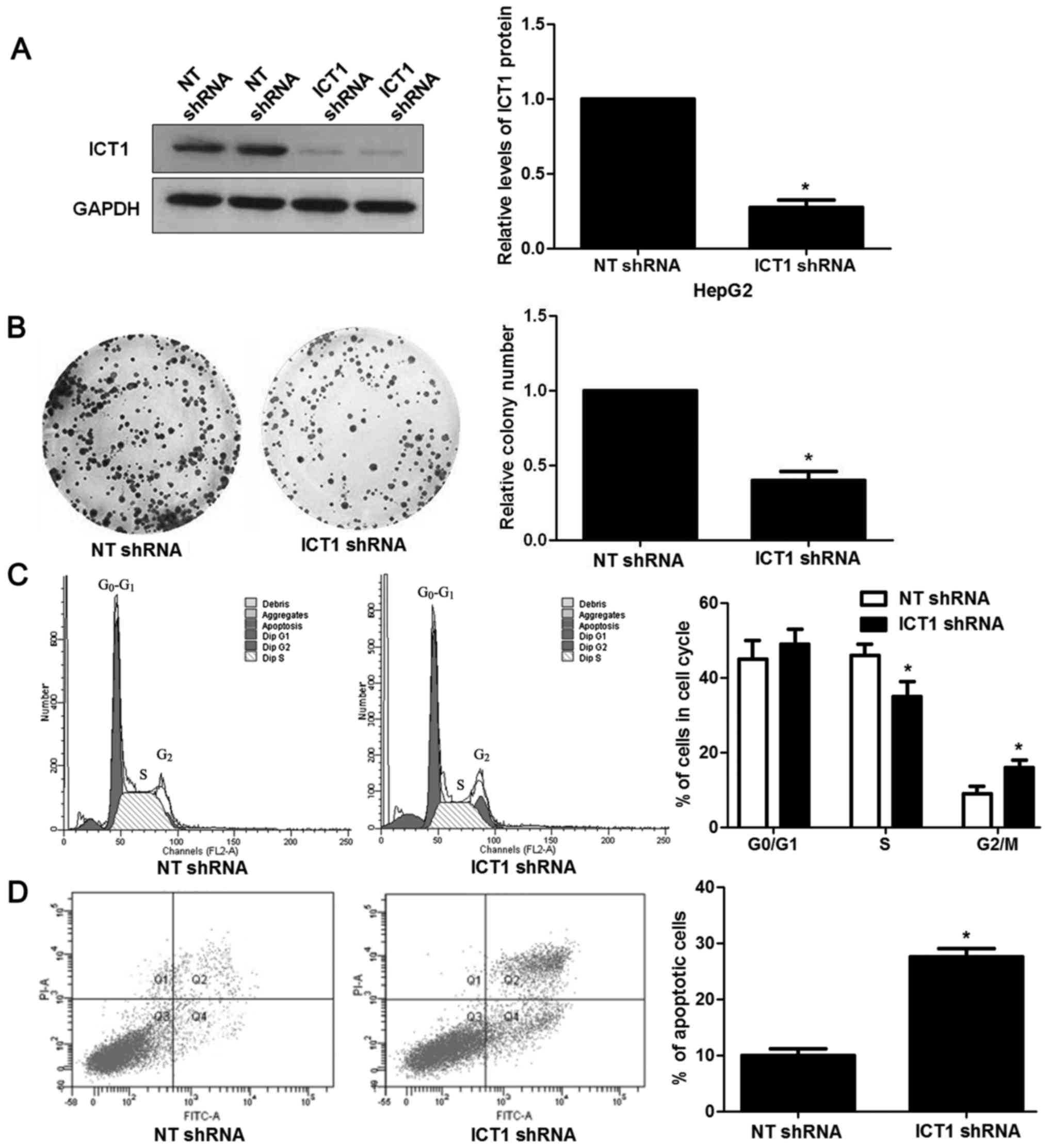
Next, we investigated the biological function of
ICT1 in HCC cells. HepG2 cells showed the highest level of ICT1,
while Hep3B cells showed the lowest level of ICT1. Thus, HepG2 and
Hep3B were used for loss- and gain-of-function experiments,
respectively. Loss-of-function experiments were performed in HepG2
cells after ICT1 knockdown (P<0.05; Fig. 3A). Colony formation assays revealed
that ICT1 knockdown prevented proliferation of HepG2 cells
(P<0.05; Fig. 3B). In addition,
ICT1 silencing resulted in cell cycle arrest at G2/M phase and
increased apoptosis in HepG2 cells (P<0.05, respectively;
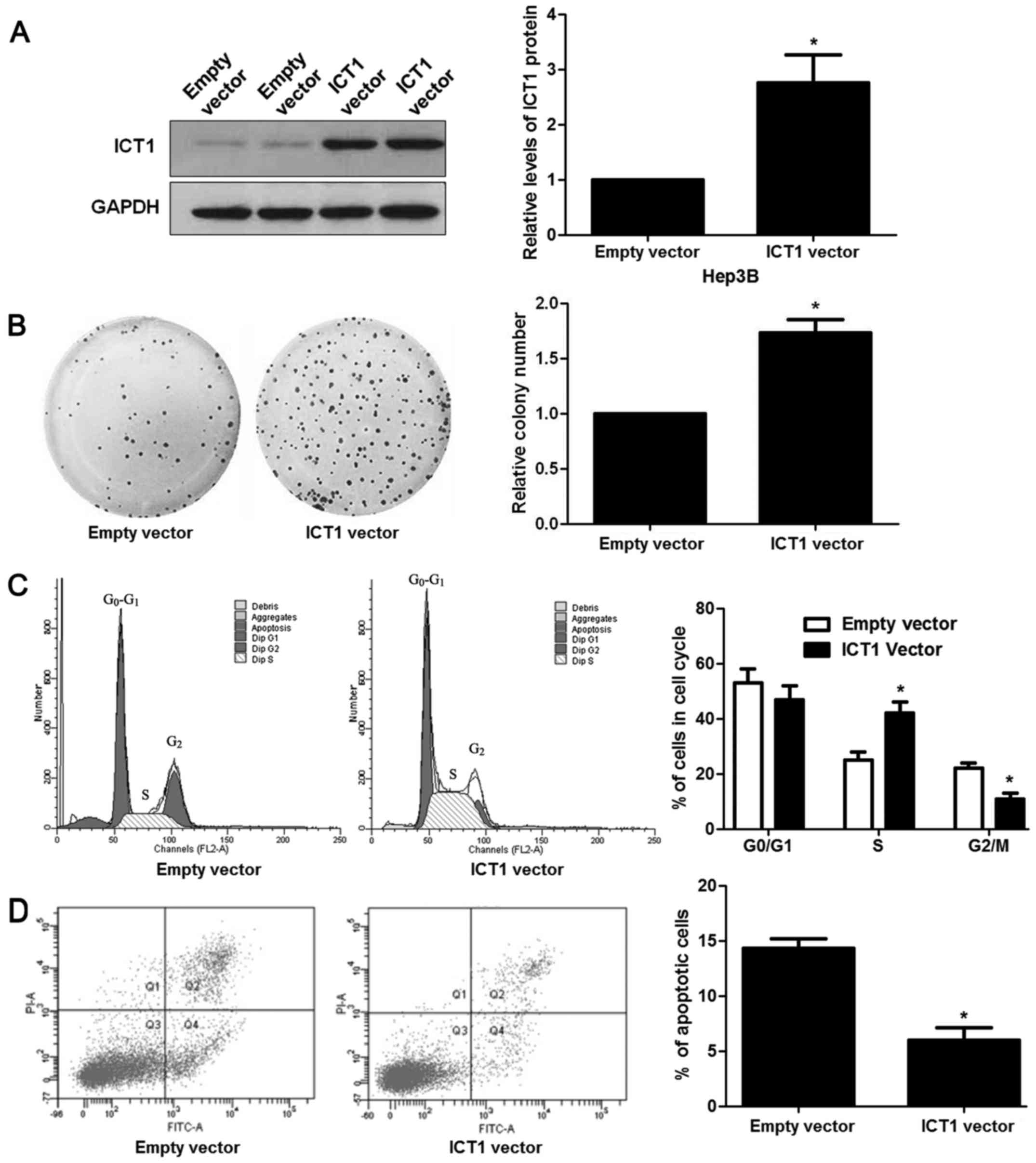
Fig. 3C and D). Then, ICT1
overexpression was confirmed by immunoblotting in Hep3B cells
(P<0.05; Fig. 4A). Further
experiments disclosed that ICT1 restoration contributed to
proliferation and cell cycle progression, and reduced apoptosis of
Hep3B cells (P<0.05, respectively; Fig. 4B-D).
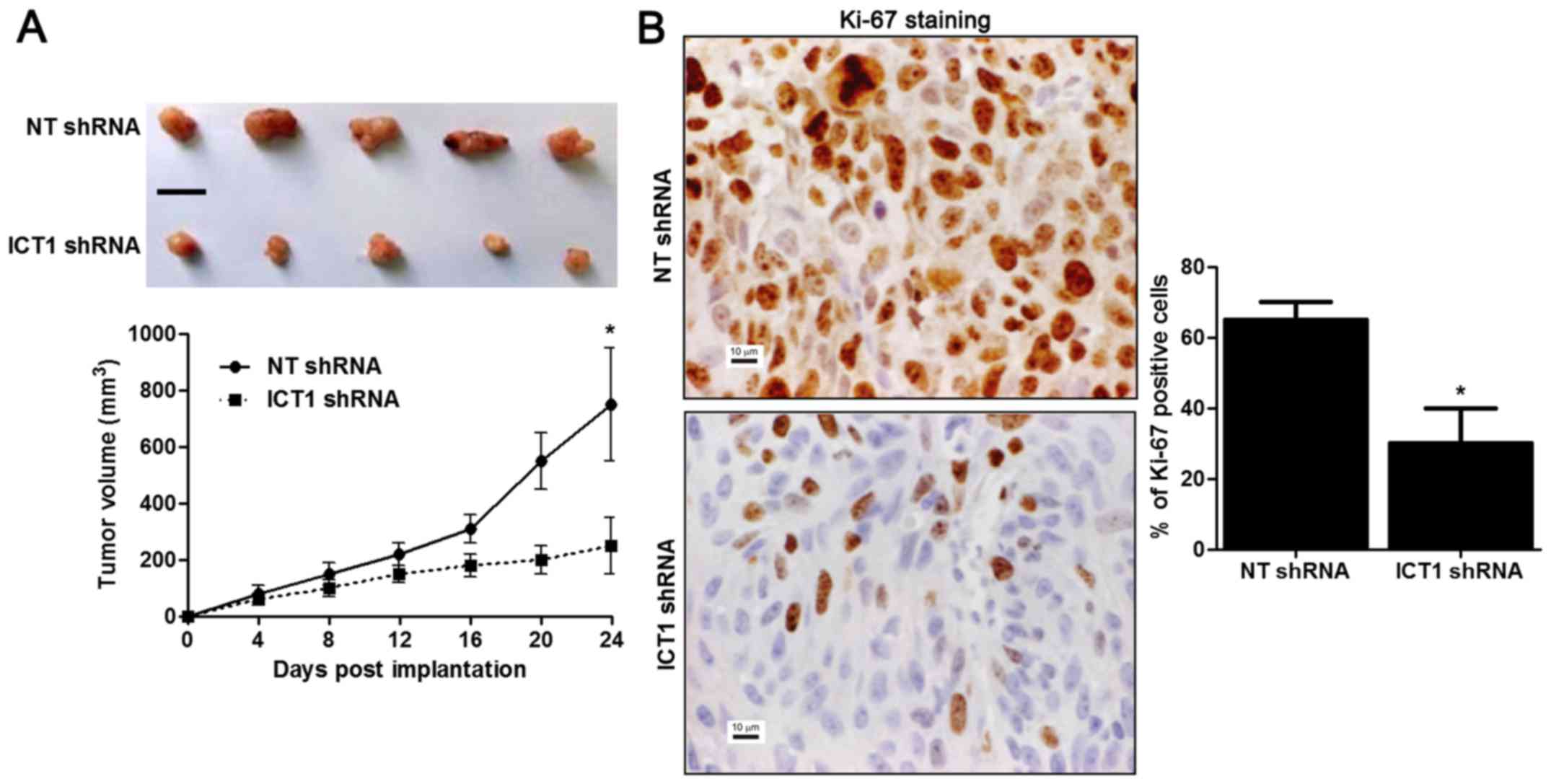
ICT1 knockdown restrains growth of HCC
in mice
HepG2 cells that were transfected with NT shRNA or
ICT1 shRNA were implanted into nude mice via subcutaneous
injection. Tumor growth curves showed that ICT1 knockdown reduced
the volume of subcutaneous tumor nodules in nude mice with HCC
(P<0.05; Fig. 5A). Furthermore,
Ki-67 staining was performed to detect the proliferative index of
HCC in vivo. The percentage of Ki-67 positive cancer cells
in ICT1 knockdown group was notably reduced compared to control
group (P<0.05; Fig. 5B). These
results indicate that ICT1 inhibits the progression of HCC via
regulating proliferation, cell cycle progression and apoptosis.
ICT1 modulates the levels of cell
cycle and apoptosis-associated proteins in HCC cells
Previous studies report that ICT1 regulate cell
growth of breast, CRC, prostate and lung cancer by modulating cell
cycle and apoptosis-associated proteins including CDK1, cyclin B1,
Bax and Bcl-2 (7–10). Thus, western blot analysis was
performed to detect the levels of CDK1, cyclin B1, Bax and Bcl-2
after modulating ICT1 expression in HCC cells. ICT1 knockdown
reduced the levels of CDK1, cyclin B1 and Bcl-2 and increased Bax
expression in HepG2 cells (Fig. 6).
In turn, ICT1 overexpression led to upregulation of CDK1, cyclin B1
and Bcl-2 and suppression of Bax in Hep3B cells. Thus, ICT1
regulates the levels of CDK1, cyclin B1, Bax and Bcl-2 in HCC
cells.
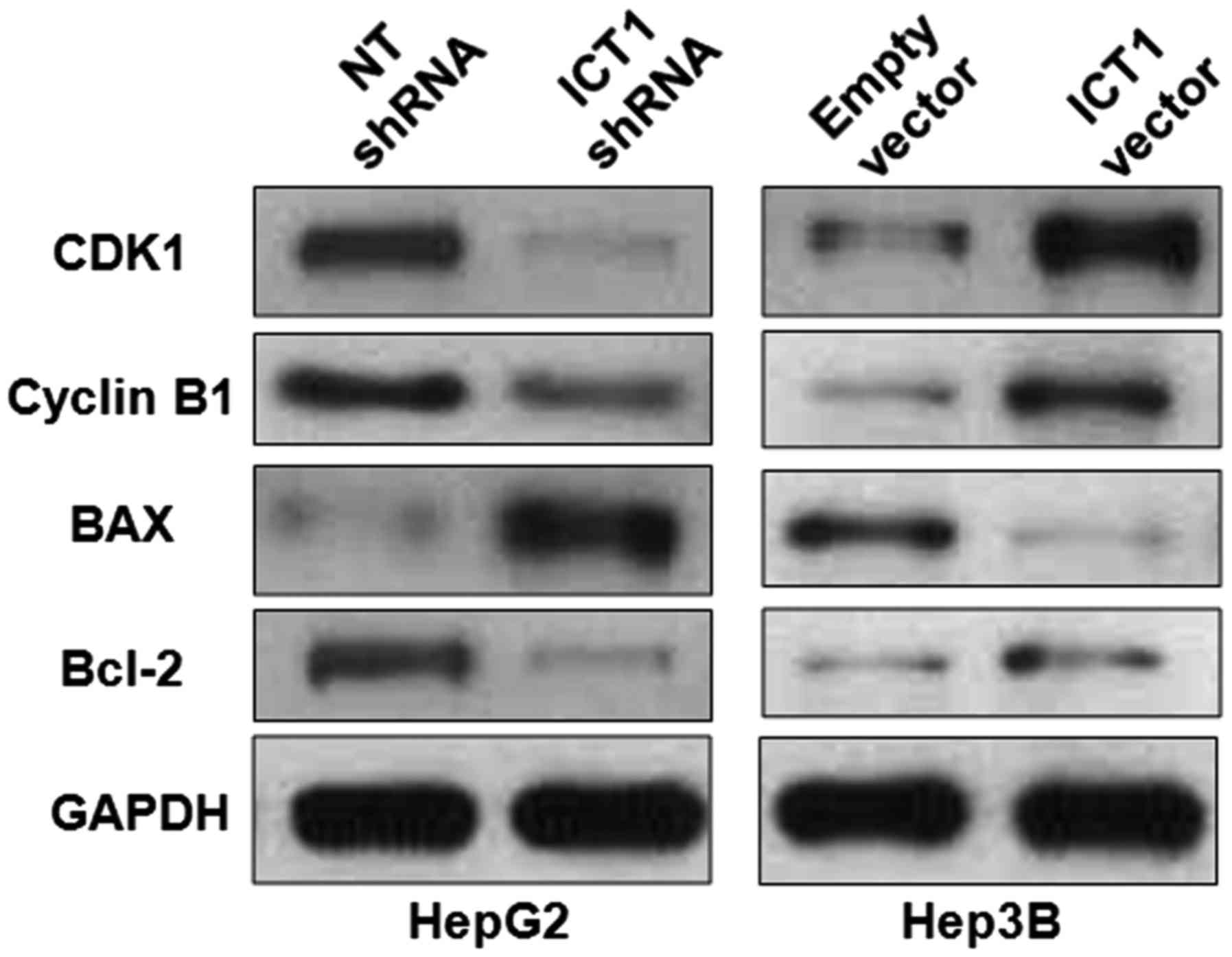 | Figure 6.ICT1 modulates the levels of CDK1,
cyclin B1, Bcl-2 and Bax in HCC cells. HepG2 cells that were
transfected with ICT1 shRNA and non-targeting (NT) shRNA,
respectively, were subjected to immunoblotting. ICT1 knockdown
reduced the levels of CDK1, cyclin B1 and Bcl-2, and increased Bax
expression in HepG2 cells. Furthermore, Hep3B cells were
transfected with ICT1 vector and empty vector, respectively. ICT1
overexpression increased the levels of CDK1, cyclin B1 and Bcl-2,
and reduced Bax expression in Hep3B cells. |
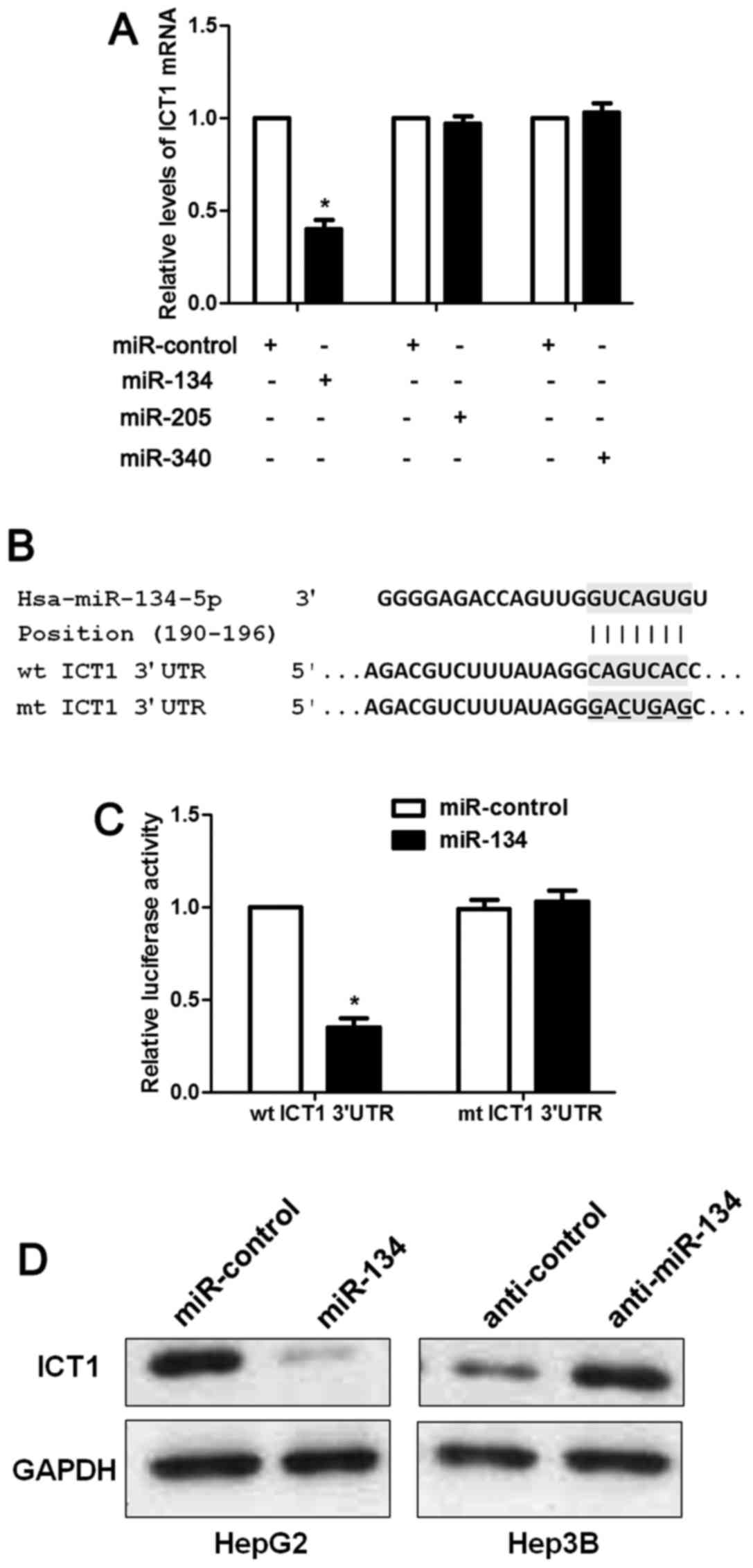
ICT1 is a direct target of miR-134 in
HCC cells
The mechanism involved in ICT1 overexpresion in HCC
has rarely been investigated. Increasing evidence indicates that
miRNAs play a critical role in regulating the expression of
oncogenes and tumor suppressors in human cancers (12–14).
Thus, we used two publicly available databases (TargetScan and
miRanda) to search for the potential miRNAs involved in regulation
of ICT1 in HCC. Five miRNAs including miR-134-5p, miR-340-5p,
miR-205-5p, miR-543 and miR-758-3p were recognized as candidates.
miR-758-3p is rarely investigated and miR-543 is reported to be
overexpressed in HCC (15). Thus,
we checked the regulatory effects of miR-134, miR-340, miR-205 on
ICT1 mRNA expression. Our data indicated that only miR-134
overexpression significantly reduced the level of ICT1 mRNA in
HepG2 cells (P<0.05; Fig. 7A).
As suggested by Fig. 7B, the
complementary sequence of miR-134 was found in the 3′-UTR of ICT1
mRNA. Notably, luciferase reporter assays demonstrated that miR-134
overexpression reduced the luciferase activity of wt but not mt
3′-UTR of ICT1 (P<0.05; Fig.
7C). Next, we found that miR-134 inversely regulated the levels
of ICT1 protein in HepG2 and Hep3B cells (Fig. 7D). Hence, these results disclose
that miR-134 inversely regulates ICT1 abundance in HCC cells.
Discussion
The expression status and role of ICT1 in human
cancers is limited. Thus, further study is necessary to investigate
the clinical significance and role of ICT1 in HCC. In the present
study, the expressions of ICT1 in the HCC tissues were prominently
higher than those in the matched tumor-adjacent tissues.
Furthermore, our data suggested that positive expression of ICT1
was prominently associated with large tumor size and advanced TNM
tumor stage. Additionally, we supported the first evidence that
ICT1-positive expression conferred a obvious poor prognosis of HCC
patients. Thus, ICT1 potentially functions as a prognostic marker
in HCC.
ICT1 has been reported to promote proliferation,
cell cycle progression and inhibit apoptosis in CRC, lung, prostate
and breast cancer (5–10). Then, we disclosed the biological
function of ICT1 in HCC. We demonstrated that ICT1 knockdown
inhibited proliferation and cell cycle progression and induced
apoptosis of HepG2 cells. While, ICT1 restoration showed opposite
effects on these cellular behaviors of Hep3B cells in vitro.
In addition, ICT1 knockdown reduced the tumor growth of HCC in nude
mice. These data reveal that ICT1 functions as an oncogene by
regulating proliferation, cell cycle progression and apoptosis in
HCC cells.
Previous studies report that ICT1 regulate cell
growth of breast, CRC, prostate and lung cancer by modulating cell
cycle and apoptosis-associated proteins including CDK1, cyclin B1,
Bax and Bcl-2 (7–10). CDK1 and cyclin B1 are key regulators
of cell cycle in HCC cells and restrained activation of cyclin
B1-CDK1 kinase leads to G2/M arrest (16,17).
Otherwise, Bcl-2/Bax axis plays an essential role in modulating
apoptosis of HCC cells (18,19).
In the present study, we revealed that ICT1 regulates the levels of
CDK1, cyclin B1, Bax and Bcl-2 in HCC cells. These data indicate
that ICT1 promotes growth of HCC probably by regulating these cell
cycle and apoptosis-associated proteins. Furthermore, ICT1 was
identified as a direct target of miR-134, which was previously
reported to exert a dramatically suppressive effect on HCC
malignancy (20,21). Thus, miR-134 regulation of ICT1
promotes malignant phenotypes of HCC cells probably via modulating
CDK1, cyclin B1, Bax and Bcl-2 expression.
In conclusion, we show that ICT1 acts as an oncogene
in HCC. First, our results demonstrate that ICT1 expression was
upregulated in HCC tissues and cell lines. Then, our clinical data
suggest that ICT1 may be used as a novel prognostic marker for HCC
patients. Moreover, miR-134 regulation of ICT1 facilitates
proliferation, cell cycle progression and suppresses apoptosis
probably via modulating CDK1, cyclin B1, Bax and Bcl-2 expression
in HCC cells. Taken together, our results verify that ICT1 may
serve as a potential target for cancer therapeutics in HCC.
Acknowledgements
The present study was supported by the Scientific
Research Project of Shaanxi Provincial Department of Education
(2013JK0791).
References
|
1
|
Njei B, Rotman Y, Ditah I and Lim JK:
Emerging trends in hepatocellular carcinoma incidence and
mortality. Hepatology. 61:191–199. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yin G, Liu Z, Wang Y, Dou C, Li C, Yang W,
Yao Y, Liu Q and Tu K: BCORL1 is an independent prognostic marker
and contributes to cell migration and invasion in human
hepatocellular carcinoma. BMC Cancer. 16:1032016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anwar SL and Lehmann U: MicroRNAs:
Emerging novel clinical biomarkers for hepatocellular carcinomas. J
Clin Med. 4:1631–1650. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
van Belzen N, Diesveld MP, van der Made
AC, Nozawa Y, Dinjens WN, Vlietstra R, Trapman J and Bosman FT:
Identification of mRNAs that show modulated expression during colon
carcinoma cell differentiation. Eur J Biochem. 234:843–848. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Belzen N, Dinjens WN, Eussen BH and
Bosman FT: Expression of differentiation-related genes in
colorectal cancer: Possible implications for prognosis. Histol
Histopathol. 13:1233–1242. 1998.PubMed/NCBI
|
|
6
|
Huang P, Cao K and Zhao H: Screening of
critical genes in lung adenocarcinoma via network analysis of gene
expression profile. Pathol Oncol Res. 20:853–858. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang Z, Xu D, Gao Y, Liu Y, Ren J, Yao Y,
Yin L, Chen J, Gan S and Cui X: Immature colon carcinoma transcript
1 is essential for prostate cancer cell viability and
proliferation. Cancer Biother Radiopharm. 30:278–284. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lao X, Feng Q, He G, Ji M, Zhu D, Xu P,
Tang W, Xu J and Qin X: Immature colon carcinoma transcript-1
(ICT1) expression correlates with unfavorable prognosis and
survival in patients with colorectal cancer. Ann Surg Oncol.
23:3924–3933. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, He J, Zhang S, Yang Q, Wang B, Liu
Z and Wu X: Knockdown of immature colon carcinoma transcript 1
inhibits proliferation and promotes apoptosis of non-small cell
lung cancer cells. Technol Cancer Res Treat. 16:559–569. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang C, Liang C, Feng W, Xia X, Chen F,
Qiao E, Zhang X, Chen D, Ling Z and Yang H: ICT1 knockdown inhibits
breast cancer cell growth via induction of cell cycle arrest and
apoptosis. Int J Mol Med. 39:1037–1045. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Goh BK, Teo JY, Chan CY, Lee SY, Jeyaraj
P, Cheow PC, Chow PK, Ooi LL and Chung AY: Importance of tumor size
as a prognostic factor after partial liver resection for solitary
hepatocellular carcinoma: Implications on the current AJCC staging
system. J Surg Oncol. 113:89–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Garzon R, Calin GA and Croce CM: MicroRNAs
in cancer. Annu Rev Med. 60:167–179. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jansson MD and Lund AH: MicroRNA and
cancer. Mol Oncol. 6:590–610. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin S and Gregory RI: MicroRNA biogenesis
pathways in cancer. Nat Rev Cancer. 15:321–333. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yu L, Zhou L, Cheng Y, Sun L, Fan J, Liang
J, Guo M, Liu N and Zhu L: MicroRNA-543 acts as an oncogene by
targeting PAQR3 in hepatocellular carcinoma. Am J Cancer Res.
4:897–906. 2014.PubMed/NCBI
|
|
16
|
Huang Z, Zhou X, He Y, Ke X, Wen Y, Zou F
and Chen X: Hyperthermia enhances 17-DMAG efficacy in
hepatocellular carcinoma cells with aggravated DNA damage and
impaired G2/M transition. Sci Rep. 6:380722016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cheng P, Li Y, Yang L, Wen Y, Shi W, Mao
Y, Chen P, Lv H, Tang Q and Wei Y: Hepatitis B virus X protein
(HBx) induces G2/M arrest and apoptosis through sustained
activation of cyclin B1-CDK1 kinase. Oncol Rep. 22:1101–1107.
2009.PubMed/NCBI
|
|
18
|
Gai X, Tu K, Li C, Lu Z, Roberts LR and
Zheng X: Histone acetyltransferase PCAF accelerates apoptosis by
repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell
Death Dis. 6:e17122015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ou X, Lu Y, Liao L, Li D, Liu L, Liu H and
Xu H: Nitidine chloride induces apoptosis in human hepatocellular
carcinoma cells through a pathway involving p53, p21, Bax and
Bcl-2. Oncol Rep. 33:1264–1274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yin C, Wang PQ, Xu WP, Yang Y, Zhang Q,
Ning BF, Zhang PP, Zhou WP, Xie WF, Chen WS, et al: Hepatocyte
nuclear factor-4α reverses malignancy of hepatocellular carcinoma
through regulating miR-134 in the DLK1-DIO3 region. Hepatology.
58:1964–1976. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zha R, Guo W, Zhang Z, Qiu Z, Wang Q, Ding
J, Huang S, Chen T, Gu J, Yao M, et al: Genome-wide screening
identified that miR-134 acts as a metastasis suppressor by
targeting integrin β1 in hepatocellular carcinoma. PLoS One.
9:e876652014. View Article : Google Scholar : PubMed/NCBI
|