Introduction
The growth of solid tumors, as well as the
metastasis of aggressive cancers, depend on angiogenesis and
lymphangiogenesis that are triggered by chemical signals
originating from the tumor cells during the phase of rapid growth
(1). Generally angiogenesis, the
process of new blood vessel formation, is inactive in normal
vasculature except during the wound healing process, where
angiogenesis is transiently turned on in order to facilitate the
healing of wound tissue. However, it plays a central role in
regulating local tumor growth, invasion and distant metastasis of
various cancers including breast cancers (2). Among the known pro-angiogenic factors,
the dominant regulator of normal and pathological angiogenesis is
vascular endothelial growth factor (VEGF) and its receptors
(VEGFRs). Activation of the VEGFR2 signaling pathway leads to
phosphorylation of various downstream signal transduction proteins,
including phosphoinositide 3-kinase and protein kinase B
(PI3K-AKT), p38 mitogen-activated protein kinase (p38 MAPK), and
extracellular signal-regulated kinase (ERK), to promote the
pro-angiogenic effects, which include endothelial cell
proliferation, migration and tube formation resulting in the
creation of new vascular walls. Therefore, antagonizing VEGFR2 has
become one of the common modes of cancer therapy for its role in
inhibiting tumor growth and metastasis (3). However, current cancer therapies are
often discontinued due to considerable side-effects (4). Therefore, it is necessary to search
for new drugs that have the ability to attack specific tumor
targets to arrest growth or sensitize cancer cells to cytotoxic
chemotherapy to enhance therapeutic outcomes (4).
In this context, a novel small molecule code named
JFD was developed using molecular modeling (MODELLER 6v2) to
specifically antagonize VEGFR2 (5).
The model was refined further by energy minimization using DISCOVER
module of Insight II Accelrys Inc. (San Diego, CA, USA) (5). The JFD original with poor water
solubility was modified into a water soluble form (JFD-WS) to
enhance the bioavailability. Prior to the development of JFD-WS,
the parent compound JFD original was tested and confirmed for its
anti-angiogenic activity using HUVEC Matrigel assay (5,6).
Several in vitro studies have attempted to recreate the
complex sequence of angiogenic events using HUVEC cells. Therefore,
HUVECs are used as the cell based (in vitro) model to study
abnormal and tumor-associated angiogenesis (7). In addition to confirmation of the
anti-angiogenic properties, both the JFD original and JFD-WS were
shown to have antitumorigenic effects in a patient-derived
xenograft (PDX) implanted animal model (6). As seen with several anti-angiogenic
drugs, JFD-WS produces significant control of cancer growth when
used in combination with paclitaxel (Taxol). The cytotoxic compound
paclitaxel is classified as a taxane, an anti-microtubule agent
with a unique mechanism of action and potent cytotoxic activity
against several tumor types, including ovarian, breast, lung and
bladder (8,9). Taxol inhibits the cell cycle at the
G2-M phase transition points and subsequently induces apoptosis of
cancer cells (10). The two most
commonly described pathways of apoptosis are the death
receptor-mediated (extrinsic) and the mitochondria-mediated
(intrinsic) pathway. The extrinsic pathway is initiated by
activation of death receptors of the tumor necrosis factor (TNF)
superfamily that are expressed on the cell surface. The intrinsic
pathway is typically initiated subsequent to chemotherapy
treatment, growth factor withdrawal, hypoxia or via induction of
tumor-suppressor genes (11). Apart
from evaluating the pro-apoptotic signals, we also assessed the
tumor burden by measuring the levels of circulating tumor marker
MUC1 (CA 15-3), a transmembrane glycoprotein of the mucin family,
which is overexpressed in >90% of breast carcinomas (12). Due to overexpression and its
secretion into the blood circulation, MUC1 is considered as a tumor
marker (13).
Therefore, the main focus of the present study was
to determine the anti-angiogenic and pro-apoptotic effects of
JFD-WS alone and in combination with Taxol in GI-101A xenografts, a
PDX breast cancer mouse model. In addition, the chronic toxicity of
JFD-WS alone and in combination with Taxol was also studied. Taken
together, our data suggest that JFD-WS could be further advanced
towards the clinical translation of its use.
Materials and methods
Cell lines and reagents
For in vitro experiments, HUVECs were
purchased from Lonza (Walkersville, MD, USA) and maintained in
endothelial cell growth medium-2 (EGM-2) supplemented with
BulletKit containing VEGF and all the required growth factors.
Cells were incubated at 37°C in a humidified incubator with 95%
air, 5% CO2. For in vivo experiments, GI-101A
human breast carcinoma cells derived at the Rumbaugh-Goodwin
Institute for Cancer Research (Plantation, FL, USA) from a
57-year-old female patient with recurrent ductal adenocarcinoma
(stage IIIa; T3N2MX) who had not previously received any
chemotherapy or radiation therapy other than surgery were used in
the present study (14). GI-101A
and MCF-7 cells were maintained in RPMI-1640 and Dulbecco's
modified Eagle's medium (DMEM), respectively, supplemented with 10%
fetal bovine serum (FBS), 2 mM L-glutamine, 1.5 g/l sodium
bicarbonate and 1% penicillin/streptomycin. Cultures were incubated
under the same aforementioned conditions. Only single-cell
suspensions with >95% viability were used for injection into the
experimental mice. The antibodies against p53 (9282; 1:1,000 rabbit
polyclonal), Bcl-2 (D55G8; 1:1,000 rabbit monoclonal), Bax (D2E11;
1:1,000 rabbit monoclonal), APAF-1 (D7G4; 1:1,000 rabbit
monoclonal), cytochrome c (D18C7; 1:1,000 rabbit
monoclonal), PARP (46D11; 1:1,000 rabbit monoclonal),
phospho-VEGFR2 (19A10; 1:1,000 rabbit monoclonal) and cleaved
caspase-3 (9662; 1:1,000 rabbit polyclonal) were purchased from
Cell Signaling Technology (Danvers, MA, USA). Sunitinib was
purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). All other
chemicals used in this experiment were of research grade.
Cell proliferation assay
The proliferation of HUVECs was determined by
bromodeoxyuridine (BrdU) labeling assay. Briefly, HUVECs were
plated at 5×103 cells/well in 96-well plates and allowed
to attach for 24 h in EGM-2 BulletKit (100 µl/well). Cells were
exposed to different concentrations of JFD-WS (1–40 µM) and
incubated in the presence of 1X BrdU for 24 h, and then assayed
with BrdU Cell Proliferation kit (Cell Signaling Technology)
following the manufacturer's instructions.
Cytotoxicity assay using MCF-7 and
GI-101A cells
To further confirm the effect of JFD-WS on breast
cancer cell lines, cytotoxicity on GI-101A and MCF-7 cells was
assessed using trypan blue dye exclusion method. Briefly, the cells
were treated with different concentrations of JFD-WS (1–100 µM) for
24 h. Then, the cell death was determined by calculating the
relative percentage of live cells.
Cell migration assay
The effect of JFD-WS on cell migration was assessed
using both Transwell and scratch assays. For Transwell migration
assay, 6.5-mm Transwell inserts with an 8-µm polycarbonate membrane
(Corning Inc., Corning, NY, USA) were used. The upper chamber of
the Transwell contained HUVECs suspended with basal media exposed
to different concentrations of JFD-WS. The lower chamber was filled
with EGM-2 supplemented with 50 ng/ml VEGF-A as a chemoattractant.
Then, the Transwell plates were incubated for 24 h to allow for
migration of HUVECs across the porous membrane. Transwell filters
were fixed in 70% ethanol and stained with crystal violet and
examined under a Leica microscope (DMI3000 B; Leica Microsystems,
Inc., Buffalo, Grove, IL, USA). Migratory cells were detached from
the lower chamber and counted using Bio-Rad TC10™ automated cell
counter. For the scratch assay, a confluent monolayer of HUVECs was
grown on 24-well plates. A sterile 200-µl tip was used to scratch a
straight line and fresh medium with corresponding concentrations of
JFD-WS (0.1–10 µM) was added to the scratched monolayer. The drug
concentrations used in this assay were determined based on the
results obtained from the Transwell migration assay, in which we
observed >70% inhibition at 10 µM. Therefore, 10 µM JFD-WS was
used as a maximum concentration for the scratch assay. Images were
captured using Leica microscope (DMI3000 B) at 12 h post-scratch.
The cell migratory effect of JFD-WS was calculated as the
percentage of the cell migration after 12 h. Images were analyzed
using ImageJ software (http://rsbweb.nih.gov/ij/download.html).
Matrigel tube formation assay
The anti-angiogenesis ability of JFD-WS was
evaluated using the Chemicon in vitro angiogenesis assay kit
(ECM 625; Millipore, Billerica, MA, USA). Briefly, a 96-well plate
was pre-incubated with EC-Matrigel at 37°C for 1 h. Then, HUVECs
(1×104 cells) suspended in the EGM-2 medium were seeded
onto the Matrigel and treated with various concentrations of JFD-WS
(0.05–10 µM). After a 6-h incubation at 37°C, tube formation was
assessed by counting the capillary tube branch points in 5 randomly
selected fields for each well using Leica microscope (DMI3000 B).
The capillary network formation was scored according to the method
of Sridhar et al (5).
Sunitinib was used as a positive control.
Inhibition of growth factor-stimulated
VEGFR2 phosphorylation in vitro
The ability of JFD-WS to inhibit VEGFR2
phosphorylation was determined using western blotting. HUVECs were
treated with JFD-WS (2 µM) in the presence and absence of VEGF (50
ng/ml). After 24 h of treatment, the cells were lysed on ice;
proteins were extracted and analyzed by western blotting. Protein
sample extracted from sunitinib-treated cells was included as a
positive control.
Phospho-VEGFR2 sandwich ELISA
assay
Endogenous levels of phospho-VEGFR2 (Tyr1175) were
determined using PathScan® Phospho-VEGFR2 (Tyr1175)
Sandwich ELISA kit (Cell Signaling Technology). Briefly, the
protein samples were diluted to the equal concentration; 100 µl of
each diluted cell lysate was added to the mouse anti-VEGFR2
antibody-coated microwells and allowed to incubate overnight. Then,
the microwells were incubated with rabbit anti-pVEGFR2 (Y1175)
detection antibody followed by horseradish peroxidase (HRP)-linked
secondary antibody and then developed with
3,3,5,5′-tetramethylbenzidine (TMB) substrate. Absorbance was read
at 450 nm after adding the stop solution, and the results were
recorded using a VersaMax microplate reader (Molecular Devices,
Sunnyvale, CA, USA). Sunitinib was used as a positive control.
Chorioallantoic membrane (CAM)
angiogenesis assay
The CAM assay was performed according to the method
described by Tamilarasan et al (15). Fertilized chicken eggs were
incubated in an automated digital egg incubator at 37°C and 67%
relative humidity (RH). On the fourth day, the incubated eggs were
broken and gently plated on Petri dishes under sterile conditions.
JFD-WS at 5 and 10 µM concentrations was added to a paper disc
containing 50 ng/ml VEGF and incubated for 12 and 24 h. Images were
captured using a digital camera (Canon PowerShot A810).
Angiogenesis was quantified by counting the number of branching
blood vessels.
Treatment with PDX-implanted athymic
nude mice
Eight- to 10-week-old in-house bred female athymic
nude mice, weighing ~25 g, were used for tumor implantation. All
animal care and experiments were performed in accordance with the
guidelines and approval of the Institutional Animal Care and Use
Committee (IACUC) of Nova Southeastern University. Briefly,
3.0×106 cells were subcutaneously injected into the
right flank of athymic nude mice. The tumor-bearing mice were
divided randomly into 4 groups: group I was the untreated control;
group II was treated with JFD-WS (100 mg/kg); group III was treated
with Taxol (10 mg/kg); and group IV was treated with the
combination of JFD-WS (100 mg/kg) and Taxol (10 mg/kg). The
experimental mice were treated once every 2 days for a period of 22
days. The tumor volume (V) was calculated according to the formula:
V = ½ × (L × W2) where L, is the length and W, is the
width. All the animals in the control and experimental groups were
sacrificed at the end of the treatment. Blood samples were
collected and plasma was separated for MUC1 analysis. The tumor
tissues from control and experimental animals were harvested for
DNA isolation and protein extraction.
Plasma MUC1 levels
Enzyme-linked immunosorbent assay (ELISA) was used
for measuring the levels of MUC1 following the manufacturer
protocol (Sigma-Aldrich, St. Louis, MO, USA). Briefly, the plasma
collected from the control and experimental animals was added to
microwells coated with anti-MUC1 antibody and allowed to incubate
for 2 h at 37°C. Then, the microwells were incubated with rabbit
anti-MUC1 detection antibody followed by HRP-conjugated secondary
antibody. The color for the measurement was developed using TMB
substrate. The absorbance was read at 450 nm after adding the stop
solution, and the results were recorded using a microplate reader
(Molecular Devices).
Western blot analysis
Tumor tissues (50 mg) were homogenized, and the
tissue lysates were subjected to western blot analysis. Briefly, 25
µg of total protein was subjected to electrophoresis on 10–12%
polyacrylamide gel, and then transferred onto a nitrocellulose
membrane. After blocking with 5% non-fat dry milk solution, the
membranes were probed with antibodies for p53, Bcl-2, Bax, APAF-1,
PARP, cytochrome c and cleaved caspase-3 (1:1,000 dilution).
The protein bands were visualized using the LumiGLO
chemiluminescence substrate system (KPL Biosolutions, Milford, MA,
USA) after incubation of the blotted membrane with HRP-conjugated
secondary antibody (anti-rabbit A6154; 1:10,000; Sigma, St. Louis,
MO, USA). As a loading control, membranes were stripped and
reprobed with β-actin (A5441; 1:5,000 dilution; Sigma). The protein
band intensity was quantified using ImageJ software (http://rsbweb.nih.gov/ij/download.html).
DNA fragmentation assay
DNA fragmentation was determined by gel
electrophoresis. For this experiment, tumor tissue (~25 mg) was
collected to extract DNA using DNeasy® Blood and Tissue
kit (Qiagen, Valencia, CA, USA). DNA extracts (50 ng) mixed with
peqGreen were loaded onto a 1.5% agarose gel, and then
electrophoresis was performed at 50 V for 2 h. The gel also
contained a DNA ladder for comparing the sizes of the DNA
fragments.
Evaluation of chronic toxicity with
JFD-WS
To test the potential toxicity of JFD-WS, 8- to
10-week-old in-house bred BALB/c mice (weight, 22–30 g) were used.
All animals were maintained under sterile 12 h light-dark
controlled conditions. They also had free access to autoclaved
water and a conventional mouse diet throughout the experiment. The
experimental animals were divided into 4 groups (6 in each group).
The first group was the control that was not treated with any drug;
the second group was injected with JFD-WS (100 mg/kg body weight);
the third group was injected with Taxol (10 mg/kg body weight) and
the fourth group was injected with JFD-WS (100 mg/kg body weight)
and Taxol (10 mg/kg body weight). The injections were given
intraperitoneally (i.p.) once every 2 days for the duration of 30
days. All the animals were monitored twice a day for harmful
side-effects such as allergy or ulceration, anorexia, and other
relevant symptoms. At the end of the treatment period, blood
samples were collected for performing blood chemistry and
hematological analysis.
Pharmacokinetic studies
All pharmacokinetic parameters were evaluated using
BALB/c mice. Briefly, JFD-WS was dissolved in sterile water and
given i.p. at a concentration of 100 mg/kg body weight. The plasma
samples (100 µl) were collected at different time points (2.5–1,440
min) while 20 µl urine samples were collected at 15–1,440 min.
Then, JFD-WS was extracted using simple liquid extraction procedure
with HPLC grade acetonitrile (ACN). Final extracts were analyzed by
reverse-phase high-performance liquid chromatography (Hitachi 2000,
Monroe, GA, USA). The stationary phase consisted of Hitachi Lachrom
C18 column (15 × 4.6 mm; 5 µm particle size), whereas the mobile
phase consisted of 100% ACN.
Statistical analysis
The data presented in the present study represent
mean ± standard deviation (SD) values from at least 4 independent
experiments. Statistical analyses were performed using a one-way
analysis of variance (ANOVA) and the differences between means were
tested by unpaired Student's t-test using SPSS software (SPSS,
Inc., Chicago, IL, USA). The value of P<0.05 was considered as
statistically significant.
Results
Effect of JFD-WS on HUVEC
proliferation and migration
JFD-WS was designed to be easily dissolved in
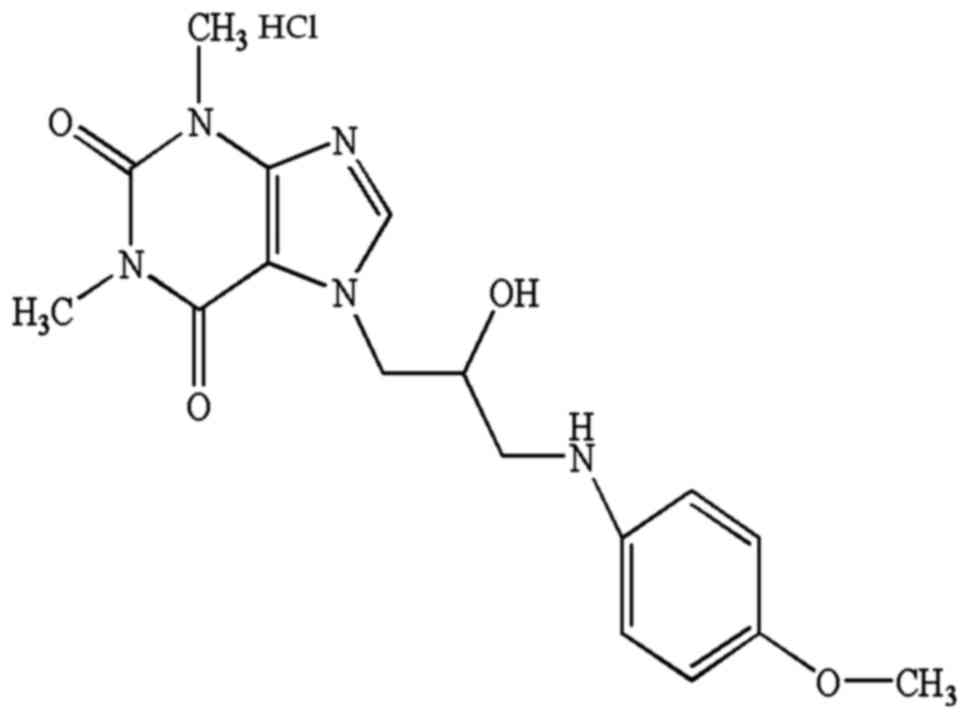
aqueous solutions. Its chemical structure is shown in Fig. 1. We initially sought to confirm the
inhibitory effects of JFD-WS on endothelial cell (EC)
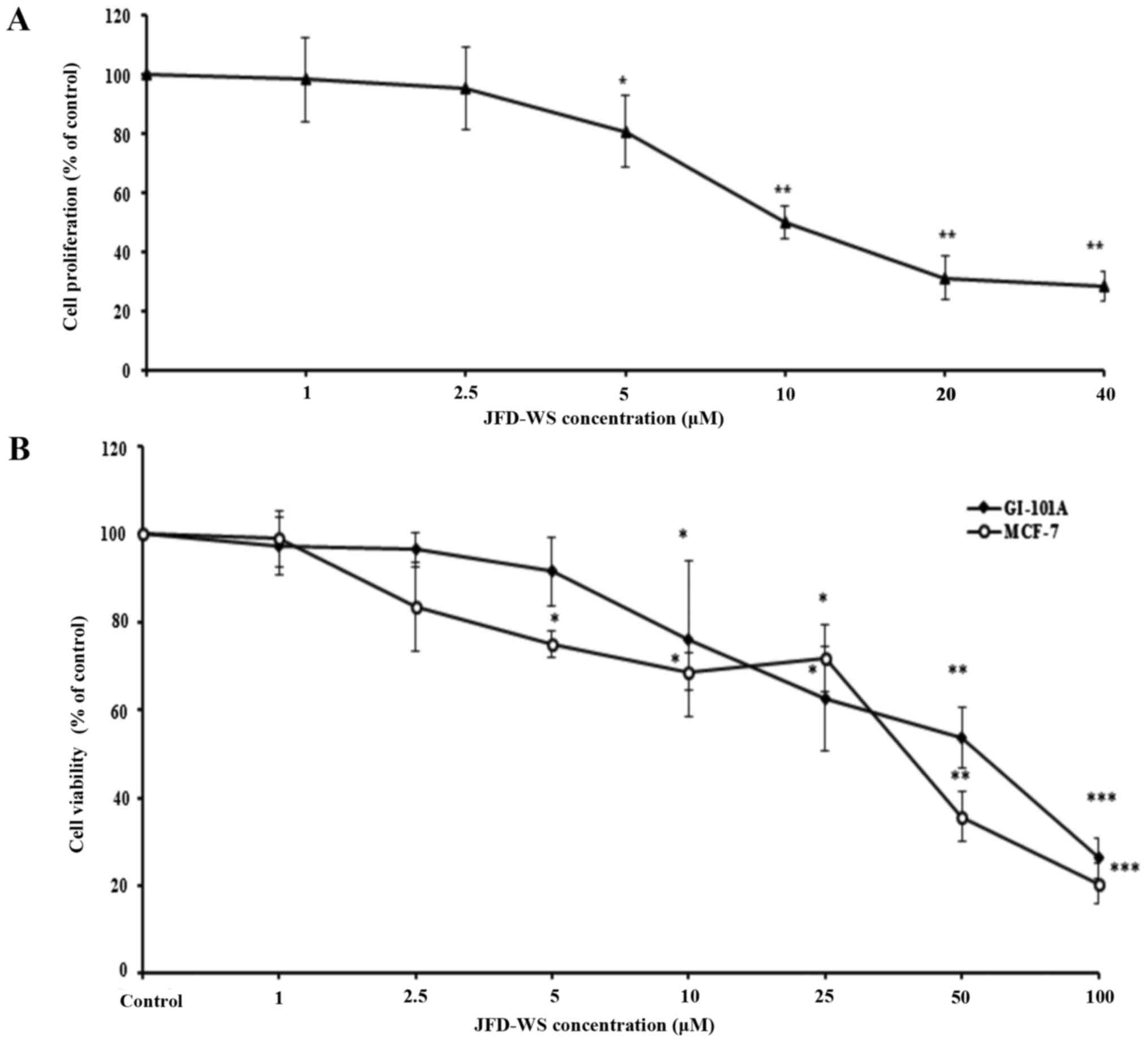
proliferation. As shown in Fig. 2A,
the proliferation of HUVECs stimulated by VEGF was significantly
decreased after JFD-WS treatment at a concentration ranging from 5
to 40 µM. In contrast, trypan blue assay results exhibited
significant dose-dependent cell viability reduction of MCF-7 and
GI-101A cells with IC50 values of 36.5 and 55.5 µM
respectively, after a 24-h treatment with JFD-WS (Fig. 2B). To further confirm the
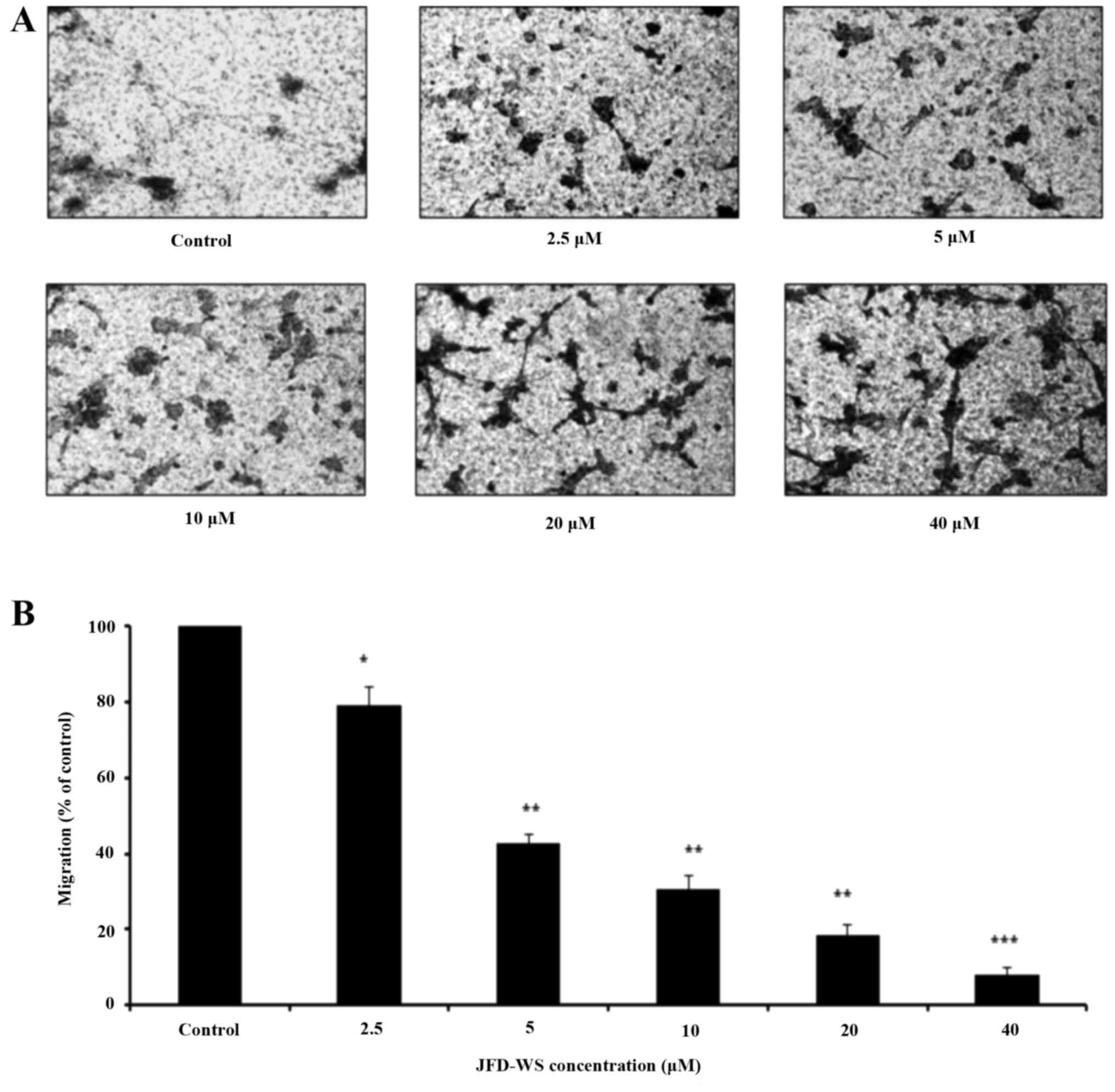
anti-angiogenic property of JFD-WS, we explored the effect of
JFD-WS on the migration of HUVECs using Transwell migration assay.
Our results showed that JFD-WS significantly inhibited the
migration ability of HUVECs in a concentration-dependent manner
(Fig. 3A), which is well correlated
with the fewer number of migrated JFD-WS-treated cells in the lower
chamber (Fig. 3B). After 24 h, ~90%
of HUVECs were trapped in the upper compartment when treated with
40 µM JFD-WS as compared to the untreated cells, indicating a
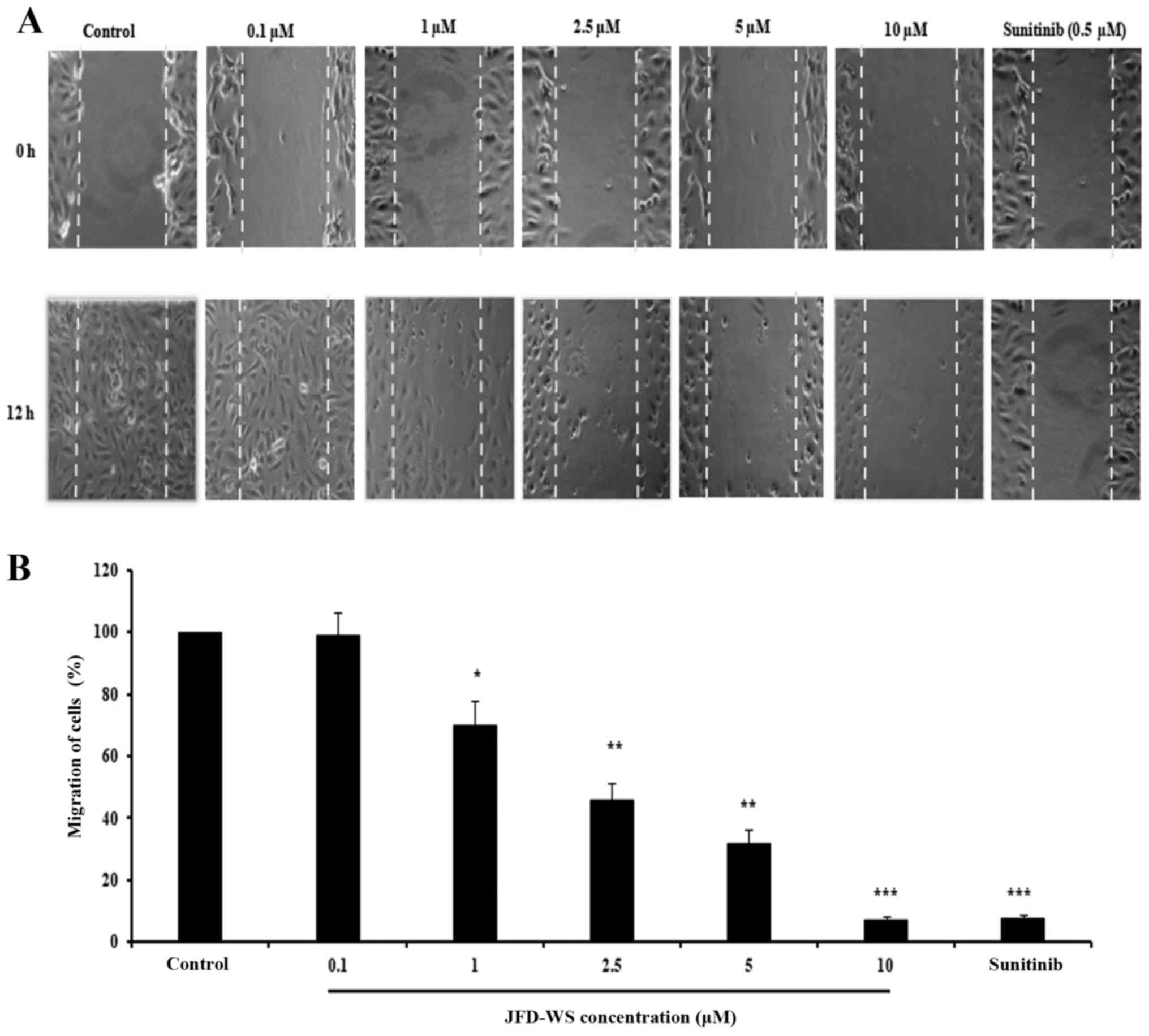
potential anti-migratory effect of JFD-WS (Fig. 3A). Similarly, JFD-WS exhibited
consistent inhibitory effects on cell migration when assayed by
scratch assay (Fig. 4A and B).
Inhibition of HUVEC tube formation and
VEGFR2 phosphorylation
Beyond endothelial cell proliferation and migration,
angiogenesis is dependent on the ability of ECs to organize and
form cellular networks. Therefore, the ability of JFD-WS to inhibit
angiogenesis was evaluated using capillary tube formation assay. As
shown in Fig. 5A, HUVECs were
seeded on Matrigel and treated with increasing concentrations of
JFD-WS. Untreated HUVECs formed capillary-like structures within 6
h. However, JFD-WS strongly inhibited the tube formation of HUVECs
in a concentration-dependent manner (0.05–10 µM). Almost the
maximum destruction of tube network was observed when HUVECs were
incubated with JFD-WS at 10 µM. Then, we investigated the effects
of JFD-WS on phospho-VEGFR2 protein expression. As shown in
Fig. 5B, there was a significant
reduction in VEGF-stimulated phospho-VEGFR2 levels in HUVECs
treated with JFD-WS at a 2 µM concentration. In addition, the
effect of JFD-WS on VEGF-induced tyrosine phosphorylation activity
of VEGFR2 was measured using PathScan Sandwich ELISA kit. Results
in Fig. 5C, show that the
phosphorylation activity of VEGFR2 was increased by the exogenously
added VEGF. However, the VEGF-induced phosphorylation activity of
VEGFR2 was significantly attenuated in cells treated with
JFD-WS.
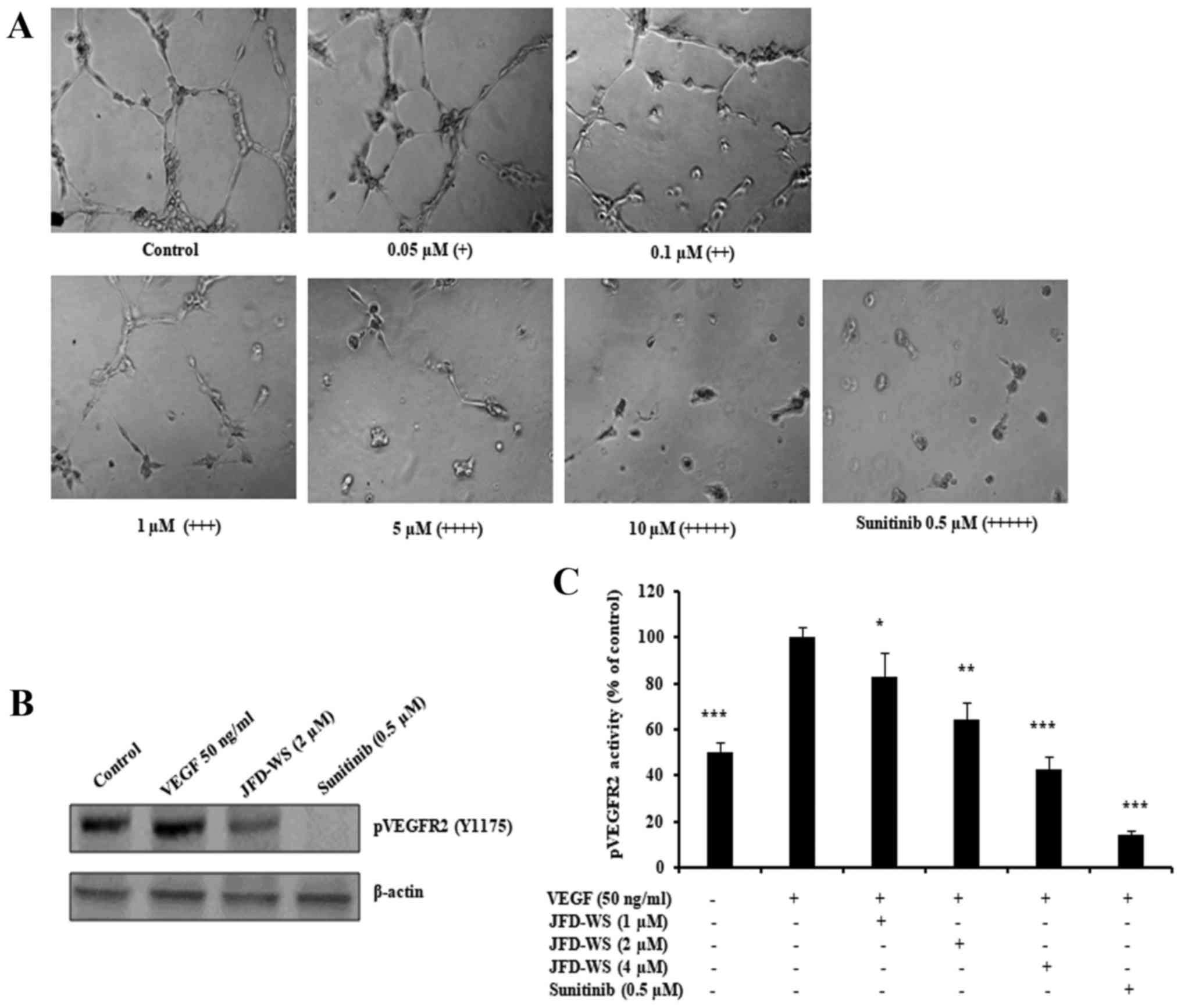 | Figure 5.JFD-WS inhibits HUVEC tube formation.
(A) HUVECs were seeded on a Matrigel layer and treated with
different concentrations of JFD-WS (0.05–10 µM). HUVEC tube
formation was assessed 6 h later. The score is based on the extent
of cellular networks: (+++++), individual cells well separated;
(++++), cells begin to migrate and align; (+++), cells lineup but
do not sprout; (++), visible sprouting; and (+), closed polygons
begin to form. (B) JFD-WS inhibited VEGF-induced phosphorylation of
VEGFR2 in HUVECs. (C) The phosphorylation activity of VEGFR2 in the
Tyr1175 phosphorylation site was measured after treatment with
JFD-WS for 24 h using the PathScan Phospho-VEGFR2 Sandwich ELISA
kit. The VEGF stimulated bar was compared with the basal level,
which shows a significant elevation in the VEGFR2 phosphorylation.
However, JFD-WS treatment showed a significant inhibition of
pVEGFR2 levels caused by VEGF stimulation. Therefore, the VEGF
stimulated effect was used as 100% for comparison. The error bar
represents the SD for the results obtained from at least 3
independent experiments. Sunitinib (0.5 µM) was used as a positive
control (*P≤0.05, **P≤0.01, ***P≤0.005). |
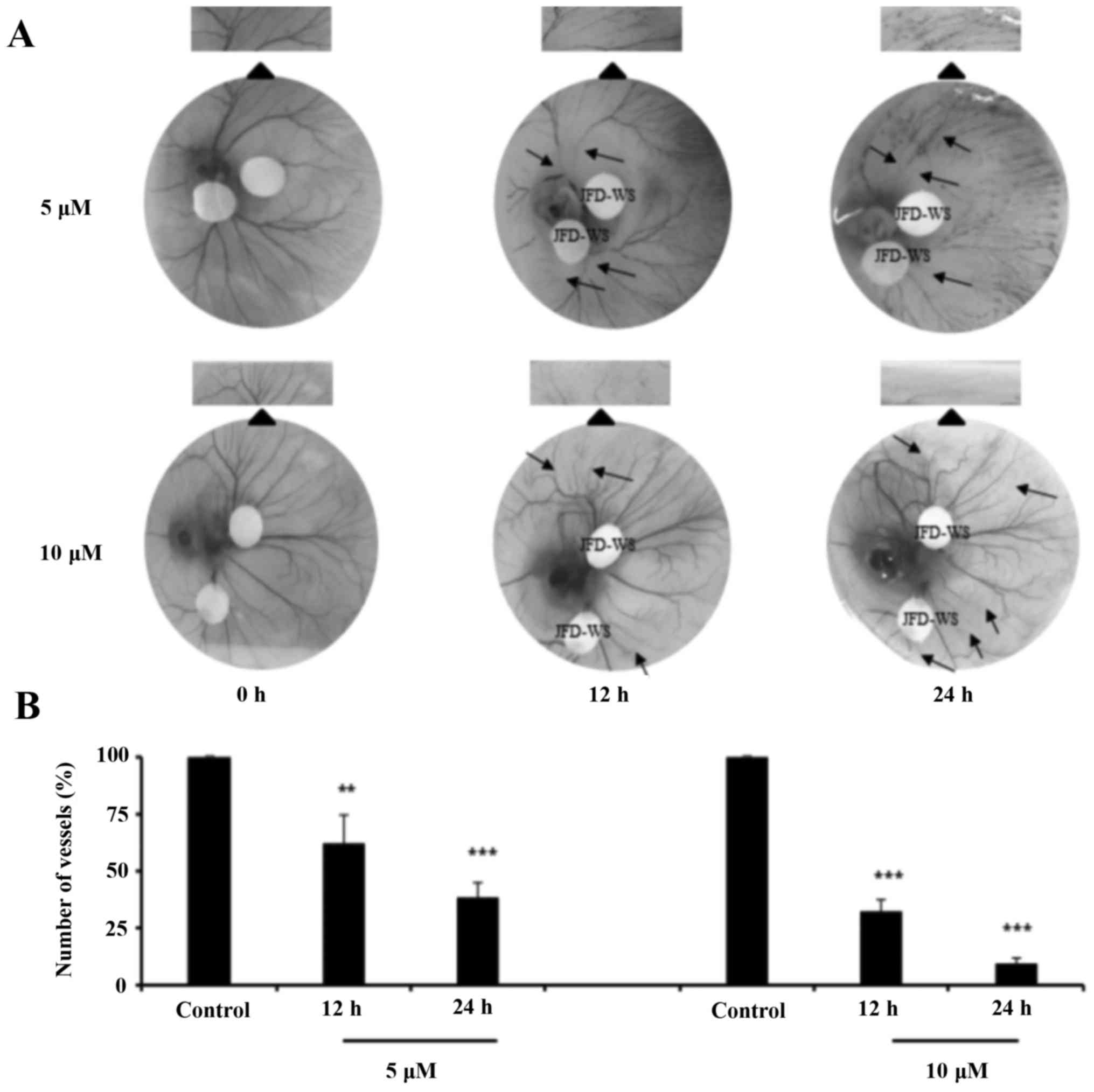
Effects of JFD-WS on angiogenesis
using CAM
To confirm the anti-angiogenic effects of JFD-WS,
CAM assay was performed with 5 and 10 µM concentrations of JFD-WS.
The number of blood vessels formed in the chorioallantoic membrane
was significantly suppressed in both 5 and 10 µM JFD-WS treated
groups as compared with the controls (Fig. 6A). The number of newly formed
vessels was suppressed by ~40% (P<0.01) and 70% (P<0.001)
after 12 and 24 h treatment of 5 µM JFD-WS, respectively. However,
the inhibition of blood vessel formation by JFD-WS was
significantly higher at 10 µM after 12 h (70%; P<0.001) and 24 h
(94%; P<0.001) compared to lower concentrations (Fig. 6B).
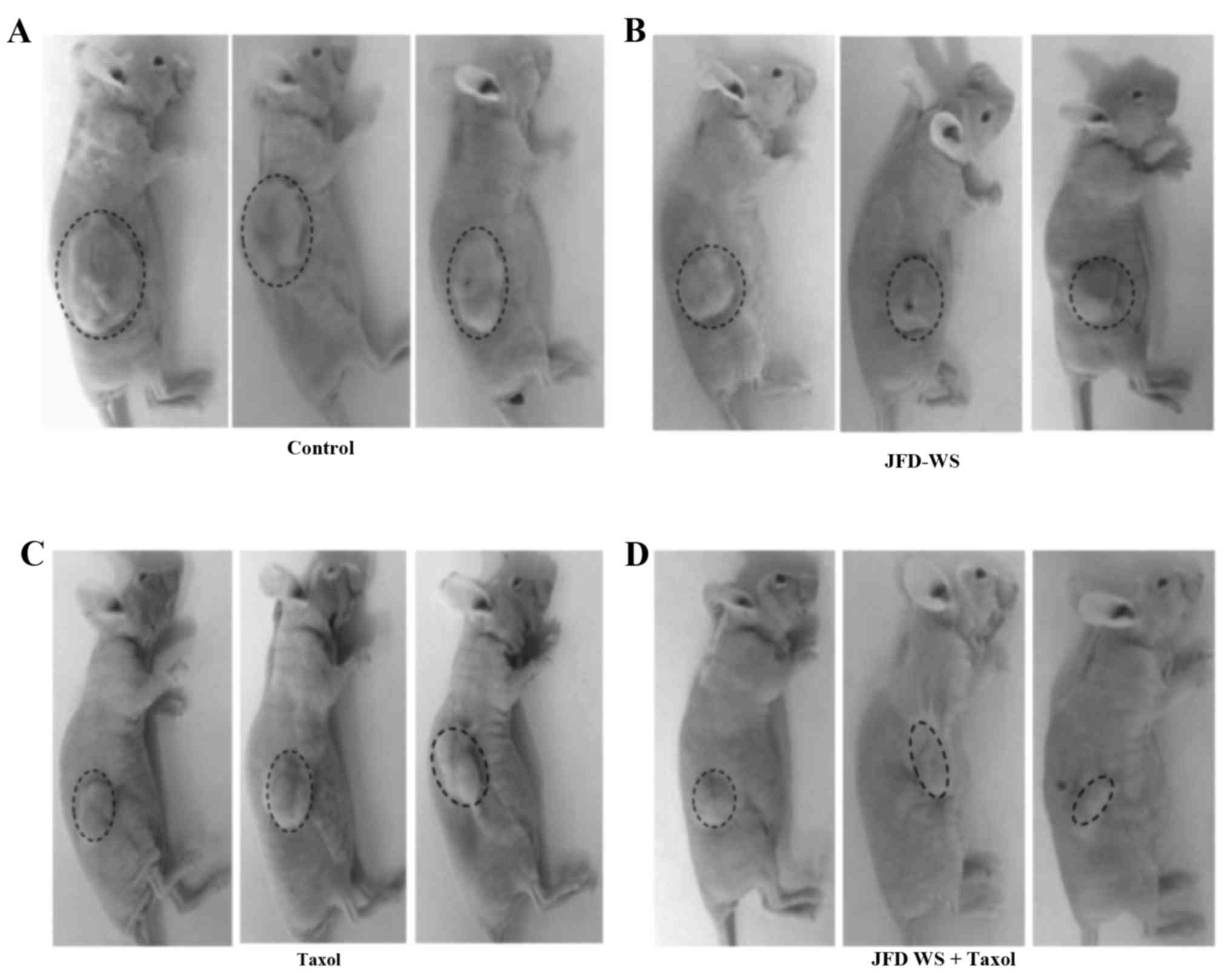
Antitumor efficacy of JFD-WS in a
GI-101A breast xenograft mouse model
The effect of JFD-WS on growth and metastasis of
breast cancer was examined using immunocompromised nude mice
implanted with GI-101A breast xenografts. To assess the
effectiveness of JFD-WS, tumor volume and body weight of
tumor-bearing animals were observed every 3 days until the end of
the experiment. Mice implanted with GI-101A tumors showed 40 and
50% suppression of tumor growth after treatment with 100 mg/kg
JFD-WS and 10 mg/kg Taxol, respectively, for 22 days (Fig. 7A-E). The doses of JFD-WS used in the
present study were based on our initial experiments in which, we
used 50 mg/kg body weight (data not shown) and 100 mg/kg body
weight JFD-WS. However, better pharmacological (Fig. 7A-E) effects were observed in animals
treated with 100 mg/kg body weight JFD-WS. Notably, the inhibitory
effect of JFD-WS monotherapy was comparable to Taxol at the
specified dose with no signs of toxicity. In contrast, 100 mg/kg
dose of Taxol produced >50% mortality in the experimental groups
(data not shown). However, JFD-WS combination with Taxol treatment
caused 85% suppression of tumor growth (Fig. 7D and E) confirming the additive
effects of JFD-WS with no observed toxicity symptoms. Thus, JFD-WS
combination with Taxol was well-tolerated, and there were no
significant changes in food intake or body weight during the
experimental period (Fig. 7F).
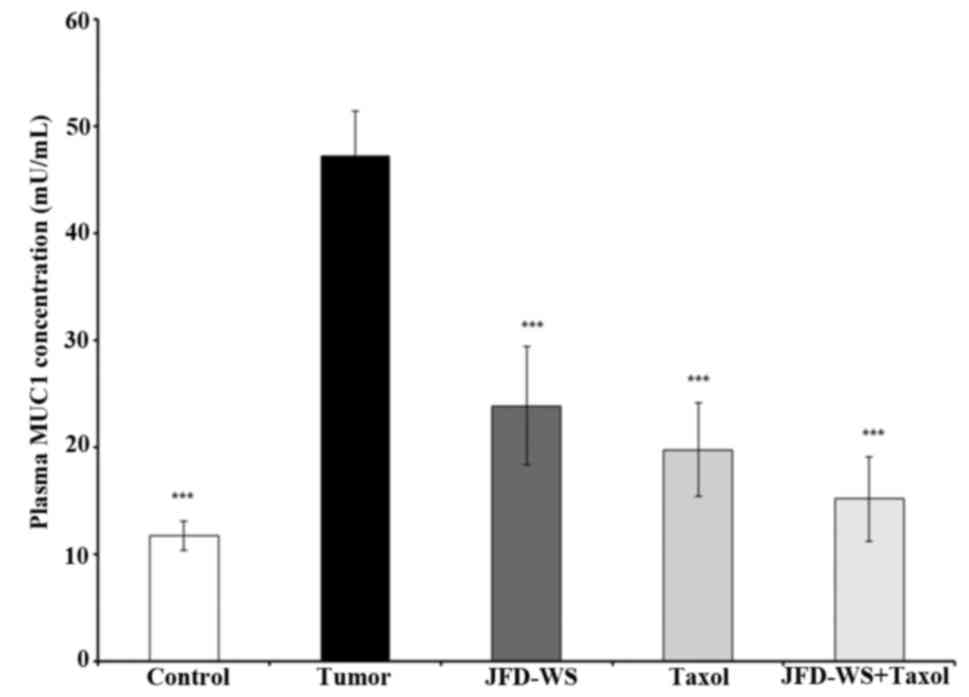
Effect of JFD-WS on plasma MUC1
levels
Since MUC1 is currently used as a marker of
responsive therapy and as a prognostic indicator for survival, the
effect of JFD-WS on the plasma levels of MUC1 in control mice with
no tumor burden and tumor-bearing mice with and without treatment
were analyzed using ELISA. As shown in Fig. 8, the level of MUC1 in tumor-bearing
animals was nearly 80% higher when compared to the control animals.
However, the level of MUC1 in JFD-WS and Taxol-treated animals
showed 50 and 60% decrease respectively, after 22 days of
treatment. Notably, an additional 9% decrease was observed when
JFD-WS was treated in combination with Taxol. The effect of the
combination treatment was found to be higher than the individual
drug treatments.
JFD-WS increases p53 protein
expression
To determine whether induction of apoptosis is
preceded by activation of the cell cycle controlling component p53,
the effect of JFD-WS on the expression levels of p53 protein in the
xenograft tumors was examined. A marked induction of p53 protein
level (Fig. 9A) was observed in
JFD-WS-treated tumors, nearing ~10-fold increase, while the p53
protein level in the combination treatment group was nearly 11-fold
as compared with the control tumors (untreated). These results
indicate that in addition to the anti-angiogenic effect, JFD-WS has
the ability to induce apoptosis. The results also suggest that the
induction of apoptosis in the xenografts may occur through
induction of the intrinsic pathway consequent to the activation of
p53-dependent pathways.
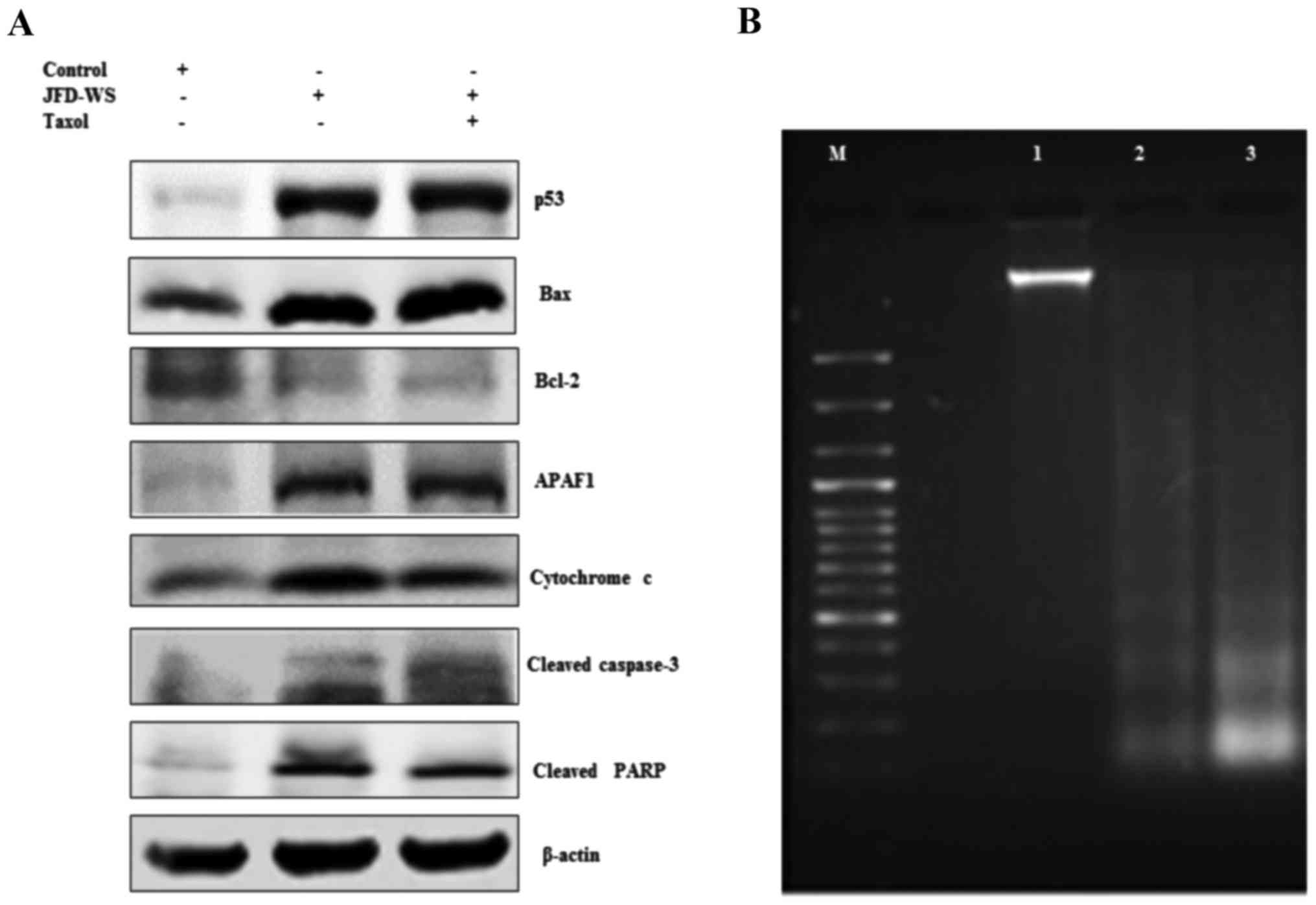 | Figure 9.JFD-WS alters apoptotic signals in
the xenograft tumors. (A) Expression of p53, Bax, Bcl-2, APAF-1,
cytochrome c, cleaved caspase-3 and cleaved PARP proteins
extracted from tumors of untreated control and treated groups were
analyzed by western blotting. The protein extracts (25 µg of
protein) were separated by SDS-PAGE gel (7.5–12%). After
electro-blotting, the separated proteins were probed with the
corresponding antibody. Significant alteration in the expression
pattern of pro- and anti-apoptotic proteins was observed in
treatment groups as compared to untreated control tumor. β-actin
was used as a loading control. (B) Demonstration of apoptosis by
DNA fragmentation. Lane M, 100 bp DNA ladder; lane 1, DNA extracted
from control tumors; lane 2, DNA extracted from JFD-WS-treated
tumors and lane 3, DNA extracted from JFD-WS + Taxol-treated
tumors. The DNA was separated by electrophoresis using 1.5% agarose
gel and visualized using UVP image analyzer. |
Effects of JFD-WS on Bcl-2 and Bax
protein expression levels
Alteration in the levels of both Bcl-2 and Bax
proteins have been shown to be associated with the anti- and
pro-apoptotic functions, respectively. We explored the impact of
JFD-WS treatment on the expression of Bcl-2 and Bax. As shown in
Fig. 9A, JFD-WS monotherapy and
also the combination treatment decreased the Bcl-2 protein level
significantly as compared to the levels of the control group. In
addition, the expression level of Bax was increased by 4-fold in
the group treated with JFD-WS alone. Likewise, in the JFD-WS/Taxol
combination group, Bax level was increased by 5-fold as compared to
the control.
Cytochrome c release and expression of
APAF-1
The release of cytochrome c from mitochondria
is critical for the initiation of APAF-1 mediated caspase
activation which is shown to subsequently induce apoptosis in a
multitude of experimental models. Therefore, we assessed the
induction of cytochrome c release from mitochondria and the
expression of APAF-1 in JFD-WS-treated and untreated groups.
However, the increase in the cytochrome c level observed in
the combination treatment group was only slightly higher when
compared to treatment with JFD-WS alone. In consistent with these
intracellular alterations, the immunoblotting analysis revealed a
markedly increase in the expression of APAF-1 in both JFD-WS and
combination treated groups as compared with the control (Fig. 9A).
Activation of caspase-3, PARP
cleavage, and DNA fragmentation
Accumulated evidence from the literature indicates
that caspases play a pivotal role in the terminal, execution phase
of apoptosis (17). It is known
that the activated caspase-3 receives apoptotic signals from
mitochondria and transmits to PARP to act at the DNA level. To
ascertain whether caspase-3 and PARP are involved in JFD-WS-induced
cell death, the expression levels of active caspase-3 and PARP
cleavage were examined. As shown in Fig. 9A, JFD-WS treatment alone was able to
cause a significant increase in cleaved caspase-3 levels in the
xenograft tumor leading to cleavage of PARP (Fig. 9A), which further evidenced the
induction of apoptosis in the xenograft tumors subsequent to the
anti-angiogenic effects elicited by JFD-WS. To confirm the final
execution of apoptosis, the genomic DNA was isolated from tumors of
the control and treated animals to determine DNA fragmentation.
Electrophoretic separation of the genomic DNA revealed the
fragmentation of DNA in the xenograft tumor tissues treated with
JFD-WS as well as the JFD-WS + Taxol combination (Fig. 9B).
Safety profile of JFD-WS
Treatment of BALB/c mice with JFD-WS for 30 days did
not cause any adverse effects as revealed by the analysis of
pathological and hematological parameters. Furthermore, no abnormal
clinical signs or behavior were detected in either of the groups.
Hematological observations of all treated mice, including total
blood count, red blood cells (RBC), white blood cells (WBC),
neutrophils, monocytes, lymphocytes, platelet counts and hemoglobin
levels were within normal limits as the control group. There were
no significant differences noted between control and treated groups
for the hematological parameters measured (Table I). Conversely, it can be interpreted
that the injection of JFD-WS at a dose of 100 mg/kg neither showed
significant changes in serum biochemical parameters such as
albumin, total protein, globulin, urea, sodium and creatinine
levels when compared to control group nor revealed an abnormality
in the functions of the vital organs (Table II). However, significant difference
in the total bilirubin was observed in the Taxol and JFD + Taxol
combination groups when compared with the control (Table II). Consistent with biomarker
analyses, there were no observable changes in body weight, food
intake, behavior and lethargy and gastrointestinal toxicity, in the
combination treatment.
 | Table I.Hematological parameters after 30
days treatment with JFD-WS. |
Table I.
Hematological parameters after 30
days treatment with JFD-WS.
| Parameters | Control | JFD-WS | Taxol | JFD-WS + Taxol |
|---|
| Red blood cell
count | 9.54±0.58 | 9.60±0.47 | 9.91±0.34 | 9.14±1.20 |
| White blood cell
count | 3.07±0.65 | 3.87±1.6 | 3.57±0.20 |
5.47±0.55a |
| Hemoglobin | 13.17±0.29 | 13.33±0.40 | 12.13±0.81 | 12±1.90 |
| Hematocrit | 44.67±2.08 | 45±2.65 | 44±4.58 | 40±8 |
| MCV | 46.67±2.52 | 47±2 | 48±3.61 | 43.7±3.51 |
| MCH | 13.67±1.15 | 14±0 | 13.33±0.58 | 13±0 |
| MCHC | 29.67±0.58 | 29.33±1.15 | 27.33±2.08 | 30±1 |
| Segmented
neutrophils | 0.53±0.15 | 0.49±0.41 | 1.2±0.77 |
1.08±0.26a |
| Lymphocytes | 2.15±0.51 | 1.83±0.41 | 2.31±0.31 | 2.92±0.73 |
| Monocytes | 0.17±0.03 | 0.17±0.01 | 0.14±0.04 | 0.20±0.05 |
| Eosinophils | 0.15±0.01 | 0.16±0.05 | 0.16±0.04 | 0.19±0.09 |
| Basophils | 0.06±0.01 | 0.05±0.02 | 0.07±0.01 | 0.07±0.03 |
| RBC morphology | Normal | Normal | Normal | Normal |
| Platelet
morphology | Normal | Normal | Normal | Normal |
| WBC morphology | Normal | Normal | Normal | Normal |
| Table II.Blood clinical chemistry values after
administration of JFD-WS on BALB/c mice. |
Table II.
Blood clinical chemistry values after
administration of JFD-WS on BALB/c mice.
| Parameters | Control | JFD-WS | Taxol | JFD-WS + Taxol |
|---|
| Glucose | 111.70±22.3 | 106.30±12.70 | 106.70±20.20 | 98.3±10.20 |
| BUN | 21±2.65 | 15±1 | 20.33±1.15 | 19.70±2.89 |
| CREA | 0.20±0.00 | 0.20±0.00 | 0.17±0.06 | 0.20±0.00 |
| BUN/crea ratio | 105±13.20 | 75±5 | 106±15.70 | 98.3±14.4 |
| Amylase | 2799±513 | 2930±523 | 2529±78.6 | 2416±311 |
| Calcium | 7.93±0.37 | 8.60±0.00 | 8.77±0.38 | 7.90±2.25 |
| Phosphorus | 9.9±0.69 | 9.63±3.47 | 7.6±1.31 | 6.65±0.21 |
| Total protein | 6.17±0.21 | 5.57±0.40 | 5.33±1.31 | 5.77±0.40 |
| Albumin | 3.57±0.06 | 3.23±0.25 | 2.90±0.20 | 3.30±0.17 |
| AST | 294±53.80 | 251±69.70 | 281±65.80 | 319±87 |
| ALT | 54.67±8.08 | 55.67±13.10 | 79.67±39.30 | 89.30±25.70 |
| CPK | 2429±483 | 2403±554 | 2756±675 | 2281±765 |
| Total
bilirubin | 0.77±0.15 | 0.52±0.08 |
0.33±0.12a |
0.47±0.21a |
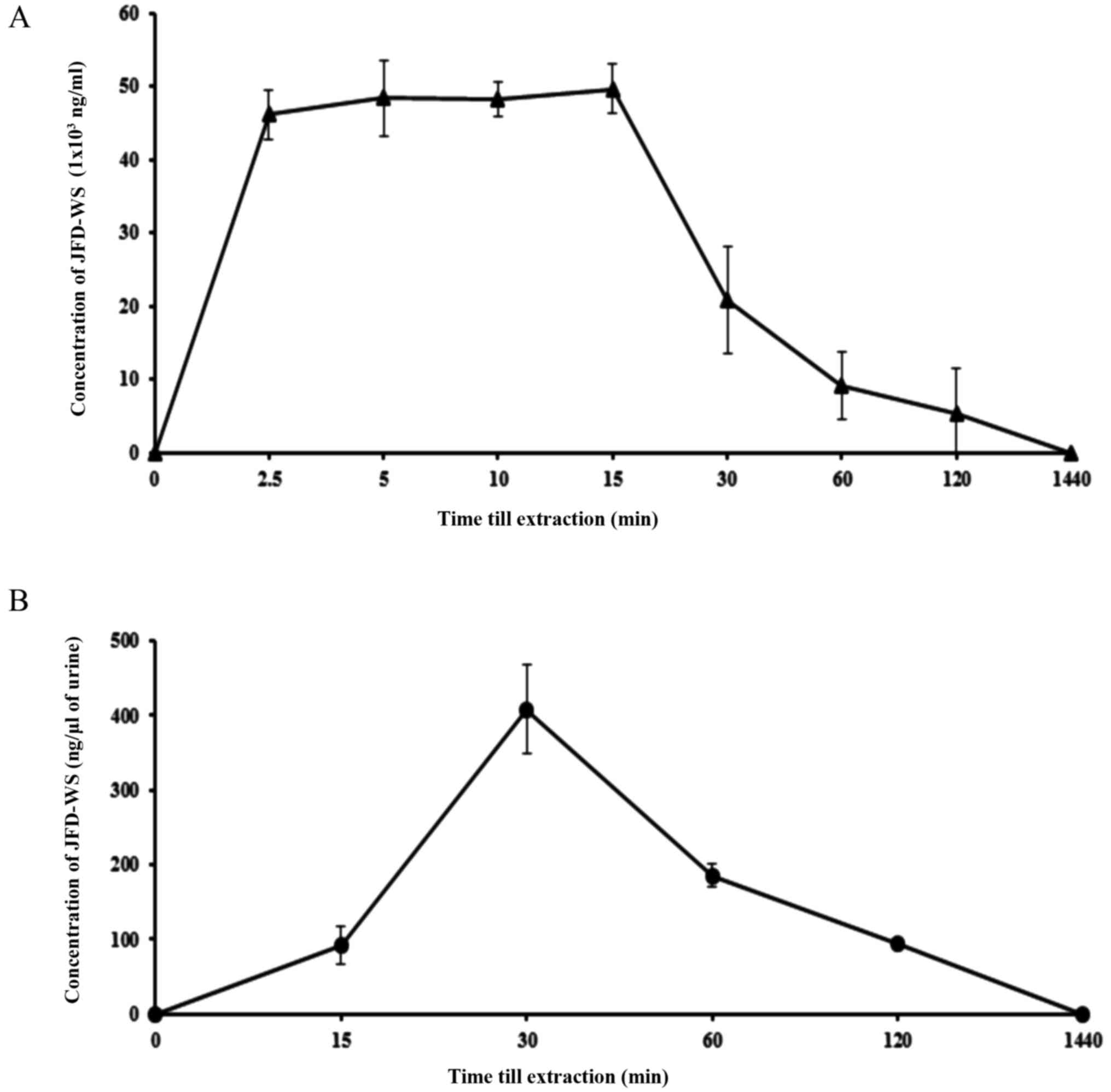
Pharmacokinetics of JFD-WS in
mice
As mentioned previously, original JFD was modified
into JFD-WS to increase its bioavailability. To test this, we
investigated the pharmacokinetic (PK) profile of JFD-WS in BALB/c
mice (Table III). The plasma
profile was established by measuring the plasma concentrations of
JFD-WS at different time points after i.p. administration of the
drugs at the dose of 100 mg/kg body weight (Fig. 10A). JFD-WS reached a maximum plasma
concentration after 15 min of i.p administration when compared to
oral administration (data not shown). Since JFD-WS is highly water
soluble, urine elimination was expected to be the major route of
elimination. As expected, JFD-WS reached maximum urine elimination
after 30 min of i.p. injection (Fig.
10B). Following i.p. injection, JFD-WS is largely distributed
in extravascular regions as indicated by its volume of
distribution. The results of PK studies confirm that i.p.
administration allows for quick distribution of JFD-WS to tumor
sites while high clearance rate could contribute to the lower
toxicity of this compound.
| Table III.Summary of pharmacokinetic
parameters. |
Table III.
Summary of pharmacokinetic
parameters.
| Pharmacokinetic
parameters | JFD-WS (100 mg/kg
body weight) |
|---|
|
Cmax | 49.69 µg/ml |
| T½ | 34 min |
| Ke | 1.24
h−1 |
| Vd | 0.25 l |
| Cl | 0.084 l/h |
Discussion
Overexpression and aberrant activation of receptor
tyrosine kinases (RTKs) such as EGFR and VEGFR are associated with
increased proliferation rates, angiogenesis and metastasis and
reduced apoptosis (18). Our group
has been engaged in the design, screening and testing for novel
VEGFR2 inhibitors such as JFD (5,6). This
molecule belongs to a class of organic compound which is analogous
to the purine moiety
(7-[2-hydroxy-3-(4-methoxyanilino)propyl]-1,3-dimethyl-3,7-dihydro-1H-purine-2,6-dione).
The original form of JFD was modified to its water soluble form to
increase bioavailability while preserving its pharmacological
activities. Various studies directed towards the development of
anti-angiogenic agents have used HUVECs as the cell based model to
study physiological and pathological processes of angiogenesis
(7). In addition, it provides an
optimal model system for the study of the regulation of endothelial
cells and their response to anti-angiogenic molecules (7). Recent results from our laboratory
confirm the anti-angiogenic activity of JFD-WS by decreasing the
level of VEGFR2 phosphorylation in human endothelial cells, through
specific binding to VEGFR2 (19).
As a result of VEGFR2 antagonism, JFD-WS specifically inhibits
angiogenesis and some of the related events that include
endothelial cell proliferation, migration, survival and vascular
network formation (16).
Conversely, when we extended these analyses to the in vivo
systems, our results showed that new blood vessel formation on the
CAM was significantly inhibited by JFD-WS even at sub-lethal
concentrations, providing critical evidence to the ability of
JFD-WS to inhibit angiogenesis.
Since many of the anti-angiogenic agents produce
cytostatic effects (20), we
evaluated the effects of JFD-WS alone and also in combination with
Taxol on GI-101A xenograft tumor implanted athymic nude mice. As
outlined in the results section, beside the anti-angiogenic effect
in vitro, JFD-WS also exerted cytoreductive activity on
GI-101A xenograft implanted animals which were confirmed by the
reduction in tumor volume (Fig.
7E). The mechanism of action of JFD-WS is similar to various
anti-angiogenic inhibitors, which are known to inhibit tumor growth
by blocking angiogenesis in the xenograft tumors (19). Inhibition of tumor growth under
in vivo condition can be attributed primarily to the
anti-angiogenenic activity of JFD-WS. However, JFD-WS shows direct
antiproliferative effects also on MCF-7 and GI-101A cells.
Therefore, we are suspecting that combination of both these effects
may contribute to the tumor-suppressing ability of JFD-WS.
Induction of apoptosis is a therapeutic approach that is primarily
intended to impede rapid tumor growth. With this purpose in the
forefront, some of the earlier studies have shown that several
VEGFR2 inhibitors including sorafenib can induce apoptosis in the
HUVECs (21). However, in our
experiments, subsequent to the onset of anti-angiogenic effect by
JFD-WS, there appears to be the induction of apoptosis in the PDX
tumors, which was confirmed by DNA fragmentation. Treatment with
JFD-WS, both as a single agent and also in combination with Taxol,
increased the expression of tumor suppressor protein p53 in the
xenograft tumors with a subsequent increase in the pro-apoptotic
protein Bax. The increase in Bax levels was accompanied by
significant decrease in the levels of the anti-apoptotic protein
Bcl-2. In addition to the increase in the expression of Bax protein
levels, a significant increase in the level of APAF-1 with a
concomitant increase in the cytochrome c level was observed
in the tumor samples of JFD-WS-treated animals as compared to the
untreated controls, evidencing the activation of the apoptotic
pathway.
In response to activation of apoptotic signals,
cytochrome c is released from mitochondria to the cytosol,
and it eventually binds to APAF-1 to form a procaspase-9 activating
heptameric protein known as apoptosomes. Such apoptosomes are
required for the activation of caspase-9 through autocatalysis,
which subsequently generates the caspase-3 from procaspase-3
(22). Activation of caspase-3
eventually triggers the caspase-activated DNase, which enters the
nucleus and causes DNA cleavage that was clearly evidenced from the
present study (Fig. 9A).
Subsequently, the active caspase-3 cleaves the downstream substrate
PARP which is responsible for the morphological and biochemical
changes that constitute the final stages of apoptosis (23,24).
PARP cleavage, which is indicated by the generation of an 85-kDa
fragment, has been reported to disable/prevent the DNA repair
process which eventually leads to cell death (25).
Several studies have shown active cell death due to
apoptosis following anti-angiogenic therapy. However, the actual
cause of apoptotic cell death subsequent to anti-angiogenic therapy
remains as an area with the limited amount of knowledge and
understanding. Although, several possibilities can be considered
including direct effects coming from anti-angiogenic agents,
inhibition of VEGFR2 phosphorylation by a small molecule is
reported to induce apoptosis by inhibiting the PI3K/AKT pathway
(26). In addition, decreased
phosphorylation of AKT was shown to increase the p53 expression
level and trigger the intrinsic pathway of apoptosis (25). Experimental evidence also suggests
that p53 can negatively regulate VEGF expression and may decrease
angiogenesis and vessel permeability, thereby inducing and
sustaining the dormancy of the experimental tumors in
micro-metastasis stages by elevating the incidence of apoptosis in
tumor cells (27,28). Results observed in the present study
with increased expression of p53 (Fig.
9A) following drug treatment tilting the balance towards
apoptosis, is in agreement with the study from Fontanini et
al (29) who showed the similar
influence of the p53 tumor-suppressor gene in non-small-cell lung
carcinoma (NSCLC).
Inhibition of VEGF/VEGFR2 with an increase in the
active caspase-9 and caspase-3-mediated apoptosis in
haemangioma-derived endothelial cells (HaemEC) has been previously
reported (30). The decreased
expression of anti-apoptotic protein Bcl-2 seen in the tumor
tissues (Fig. 9A) with the
concomitant increase in Bax level can easily shift the balance
towards programmed cell death. Our results are similar to the
inhibition of Bcl-2 by small molecules or antisense
oligonucleotides that led to multiple effects on the tumor,
including strong anti-angiogenic effect as well as restoration of
sensitivity to antineoplastic agents and thereby inducing apoptosis
(31). Substantial alteration in
the expression level of anti-apoptotic protein Bcl-2 in JFD-WS
treated mice that coincided with the decreased tumor volume, is
unique and confirms the ability for JFD-WS to induce cancer cell
death through multiple mechanisms.
Typically anti-angiogenic action of VEGFR2
inhibitors has been shown to induce apoptotic cell death of
endothelial cells (ECs) that are part of the tumor vasculature
(18). The apoptotic cell death of
the ECs was primarily responsible for the reduced density of the
tumor and regression of tumor growth. However, the present study
shows significant initiation of pro-apoptotic signals in
JFD-WS-treated tumor xenografts. In addition, both the JFD-WS and
Taxol as monotherapy and in combination showed a significant
reduction in the plasma level of tumor biomarker MUC1 further
indicating the reduction in the tumor burden of the experimental
animals subsequent to JFD-WS treatments.
Preliminary pharmacokinetic studies in mice confirm
that JFD-WS is largely distributed to extravascular regions within
a shorter duration of time after i.p injection, which enables
JFD-WS to control tumor growth. However, being target selective,
high urine clearance rate of JDF-WS contributed to the lower
toxicity of this compound. In addition, hematological and serum
biochemistry analysis of the mice treated with JFD-WS revealed no
pathological changes. However, some of the VEGFR2 inhibitors that
are already in clinical use have been found to inflict significant
toxicities in the gastrointestinal tract and liver after single
doses of treatment (21). Although,
the effective dose of JFD-WS required for inhibition of tumor
growth is slightly higher, the excellent safety profile of JFD-WS
indicates that it has the potential to be a safer and effective
anti-angiogenic drug. In conclusion, our anti-angiogenic drug
JFD-WS, whether used as a single agent or in combination with Taxol
produced strong antitumor effects by inducing apoptosis of cancer
cells in GI-101A breast xenograft tumor implanted athymic nude
mice. Elevating p53 levels and inducing apoptosis is a unique
property, and typically not seen with other anti-angiogenic drugs.
Therefore, our JFD-WS may be one of its type of anti-angiogenic
inhibitors with dual abilities.
Acknowledgements
The authors would like to thank the Royal Dames of
Cancer Research, Inc. (Ft. Lauderdale, FL, USA) for their financial
support in conducting this research. The authors would also like to
acknowledge the research grant from the Community Foundation of
Broward, Ft. Lauderdale (Florida, USA) which partially supported
the present study.
References
|
1
|
Folkman J: Role of angiogenesis in tumor
growth and metastasis. Semin Oncol. 29 Suppl 16:15–18. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ivy SP, Wick JY and Kaufman BM: An
overview of small-molecule inhibitors of VEGFR signaling. Nat Rev
Clin Oncol. 6:569–579. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamba T and McDonald DM: Mechanisms of
adverse effects of anti-VEGF therapy for cancer. Br J Cancer.
96:1788–1795. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sridhar J, Akula N, Sivanesan D,
Narasimhan M, Rathinavelu A and Pattabiraman N: Identification of
novel angiogenesis inhibitors. Bioorg Med Chem Lett. 15:4125–4129.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dhandayuthapani S and Rathinavelu A: JFD,
a novel small molecule for inhibiting vascular endothelial growth
factor receptor-mediated angiogenesis. Proceedings of the 105th
Annual Meeting of the American Association for Cancer Research;
2014 Apr 5–9; San Diego, CA Philadelphia (PA): AACR; Cancer Res. 74
Suppl 19:Abstract nr1021. 2014;
|
|
7
|
Auerbach R, Akhtar N, Lewis RL and
Shinners BL: Angiogenesis assays: Problems and pitfalls. Cancer
Metastasis Rev. 19:167–172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mekhail TM and Markman M: Paclitaxel in
cancer therapy. Expert Opin Pharmacother. 3:755–766. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ramaswamy B and Puhalla S: Docetaxel: A
tubulin-stabilizing agent approved for the management of several
solid tumors. Drugs Today. 42:265–279. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dorr RT: Pharmacology of the taxanes.
Pharmacotherapy. 17:96S–104S. 1997.PubMed/NCBI
|
|
11
|
Hollstein M, Hergenhahn M, Yang Q, Bartsch
H, Wang ZQ and Hainaut P: New approaches to understanding p53 gene
tumor mutation spectra. Mutat Res. 431:199–209. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Senapati S, Das S and Batra SK:
Mucin-interacting proteins: From function to therapeutics. Trends
Biochem Sci. 35:236–245. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tampellini M, Berruti A, Gerbino A, Buniva
T, Torta M, Gorzegno G, Faggiuolo R, Cannone R, Farris A,
Destefanis M, et al: Relationship between CA 15-3 serum levels and
disease extent in predicting overall survival of breast cancer
patients with newly diagnosed metastatic disease. Br J Cancer.
75:698–702. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Morrissey JJ and Raney S: A metastatic
breast tumor cell line, GI-101A, is estrogen receptor positive and
responsive to estrogen but resistant to tamoxifen. Cell Biol Int.
22:413–419. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tamilarasan KP, Kolluru GK, Rajaram M,
Indhumathy M, Saranya R and Chatterjee S: Thalidomide attenuates
nitric oxide mediated angiogenesis by blocking migration of
endothelial cells. BMC Cell Biol. 7:172006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lamalice L, Le Boeuf F and Huot J:
Endothelial cell migration during angiogenesis. Circ Res.
100:782–794. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Budihardjo I, Oliver H, Lutter M, Luo X
and Wang X: Biochemical pathways of caspase activation during
apoptosis. Annu Rev Cell Dev Biol. 15:269–290. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jimeno A and Hidalgo M: Pharmacogenomics
of epidermal growth factor receptor (EGFR) tyrosine kinase
inhibitors. Biochim Biophys Acta. 1766:217–229. 2006.PubMed/NCBI
|
|
19
|
Kanagasabai T, Alvarez J, Bhalani M,
Dhandayuthapani S and Rathinavelu A: The in vivo activity of a
novel anti-angiogenic compound, JFD-WS, in human breast
adenocarcinoma xenograft implanted athymic nude mice. Proceedings
of the 106th Annual Meeting of the American Association for Cancer
Research; 2015 Apr 18–22; Philadelphia (PA): AACR; Cancer Res. 75
Suppl 15:Abstract nr13802015;
|
|
20
|
Gotink KJ and Verheul HM: Anti-angiogenic
tyrosine kinase inhibitors: What is their mechanism of action?
Angiogenesis. 13:1–14. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang F, Brown C, Buettner R, Hedvat M,
Starr R, Scuto A, Schroeder A, Jensen M and Jove R: Sorafenib
induces growth arrest and apoptosis of human glioblastoma cells
through the dephosphorylation of signal transducers and activators
of transcription 3. Mol Cancer Ther. 9:953–962. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Susin SA, Zamzami N, Castedo M, Daugas E,
Wang HG, Geley S, Fassy F, Reed JC and Kroemer G: The central
executioner of apoptosis: Multiple connections between protease
activation and mitochondria in Fas/APO-1/CD95- and ceramide-induced
apoptosis. J Exp Med. 186:25–37. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zamzami N, Marchetti P, Castedo M, Zanin
C, Vayssière JL, Petit PX and Kroemer G: Reduction in mitochondrial
potential constitutes an early irreversible step of programmed
lymphocyte death in vivo. J Exp Med. 181:1661–1672. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marchetti P, Castedo M, Susin SA, Zamzami
N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M and Kroemer
G: Mitochondrial permeability transition is a central coordinating
event of apoptosis. J Exp Med. 184:1155–1160. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kroemer G and Reed JC: Mitochondrial
control of cell death. Nat Med. 6:513–519. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gottlieb TM, Leal JF, Seger R, Taya Y and
Oren M: Cross-talk between Akt, p53 and Mdm2: Possible implications
for the regulation of apoptosis. Oncogene. 21:1299–1303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mukhopadhyay D, Tsiokas L and Sukhatme VP:
Wild-type p53 and v-Src exert opposing influences on human vascular
endothelial growth factor gene expression. Cancer Res.
55:6161–6165. 1995.PubMed/NCBI
|
|
28
|
Bouvet M, Ellis LM, Nishizaki M, Fujiwara
T, Liu W, Bucana CD, Fang B, Lee JJ and Roth JA:
Adenovirus-mediated wild-type p53 gene transfer down-regulates
vascular endothelial growth factor expression and inhibits
angiogenesis in human colon cancer. Cancer Res. 58:2288–2292.
1998.PubMed/NCBI
|
|
29
|
Fontanini G, Boldrini L, Vignati S, Chinè
S, Basolo F, Silvestri V, Lucchi M, Mussi A, Angeletti CA and
Bevilacqua G: Bcl2 and p53 regulate vascular endothelial growth
factor (VEGF)-mediated angiogenesis in non-small cell lung
carcinoma. Eur J Cancer. 34:718–723. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kumar P, Miller AI and Polverini PJ: p38
MAPK mediates gamma-irradiation-induced endothelial cell apoptosis,
and vascular endothelial growth factor protects endothelial cells
through the phosphoinositide 3-kinase-Akt-Bcl-2 pathway. J Biol
Chem. 279:43352–43360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Milella M, Estrov Z, Kornblau SM, Carter
BZ, Konopleva M, Tari A, Schober WD, Harris D, Leysath CE,
Lopez-Berestein G, et al: Synergistic induction of apoptosis by
simultaneous disruption of the Bcl-2 and MEK/MAPK pathways in acute
myelogenous leukemia. Blood. 99:3461–3464. 2002. View Article : Google Scholar : PubMed/NCBI
|