Introduction
Historically, human cancer cell lines have been
widely used for studies on cancer biology or as preclinical models
to evaluate anticancer agents. However, these models may not
reflect the characteristics of the source tumor tissues, as they
are frequently passaged for long periods of time, which may alter
their genome sequence, gene expression profile and morphology. In
addition, almost all cell lines are cultured under monolayer
conditions or used as xenografts in mice, which is not physically
representative of tumor tissues (1,2).
Therefore, the clinical effects of anticancer agents are not
identical to the results of evaluations performed with cancer cell
lines. In fact, ~85% of preclinical agents entering oncology
clinical trials fail to demonstrate sufficient safety or efficacy
to gain regulatory approval (3–5).
Currently, patient-derived tumor xenograft models
(PDXs) are used as preclinical cancer models that better replicate
the diversity of human cancer biology (6–11).
Increasing evidence suggests that PDXs closely recapitulate human
cancer biology and may be used to predict patient drug responses
through direct comparisons between responses in patients and those
in corresponding xenografts; however, the evaluation of anticancer
agents using these models is difficult due to their low-throughput
nature and high associated cost (6–10).
Therefore, since cost-effective techniques are required, in
vitro systems, including patient-derived tumor organoid (PDO)
or spheroid models that accurately recapitulate tissue architecture
and function, have been developed recently. The establishment of
human tumor organoids has been recently reported for colon
(12–14), pancreatic (15), prostate (16), endometrial (17), and liver (18) tumors, among others. In particular,
Pauli et al (19) reported
56 PDOs that were established from bladder, breast, brain, colon,
lung, kidney, ovarian, pancreatic, prostate, stomach and uterine
cancers, among others, in addition to the development of
high-throughput screening (HTS) using these systems. In addition,
Kondo et al (20) developed
a cancer tissue-originated spheroid (CTOS) method based on the
principle of retaining cell-cell contact throughout cancer cell
preparation and culture; as such, CTOSs from various types of tumor
tissues (e.g. colon, lung and endometrium) have been established,
which have been used to evaluate anticancer agents (20–23).
These PDOs are promising models to facilitate a better
understanding of cancer biology and for the evaluation of drug
efficacy in vitro, prior to employing PDXs. However, reports
utilizing HTS and PDOs for the evaluation of anticancer agents have
been limited.
In this study, a novel series of 53 PDOs was
established from human tumor tissues, including those from the
lung, breast, ovary, uterus, digestive organs and peritoneum,
termed Fukushima (F)-PDOs, as presented in Table I. It was additionally confirmed that
F-PDOs were able to be cultured for a long period of time and that
they retained similar characteristics to those of source tumors
based on histological and comprehensive gene expression analyses.
In addition, the in vivo tumorigenesis of F-PDOs was tested
using a xenograft model. Thus, to evaluate anticancer agents, a
suitable HTS system using F-PDOs with multi-well plates was
developed.
 | Table I.Established F-PDOs. |
Table I.
Established F-PDOs.
| Tissue | Number of
F-PDOs |
|---|
| Lung | 15 |
| Breast | 2 |
| Ovary | 12 |
| Uterus | 18 |
| Digestive
organs | 3 |
| Peritoneal | 3 |
| Total | 53 |
Materials and methods
Compounds
A total of 61 anticancer agents were used in the
present study (Table II). Stock
solutions (10 mM) of the compounds were prepared in dimethyl
sulfoxide (DMSO) and stored at −80°C until use. The purity and
integrity of all compounds were measured using ultra performance
liquid chromatography-mass spectrometry (Waters Corporation,
Milford, MA, USA) as follows (the injection volume was 1 µl): A
Waters CORTECS C18 column (particle size, 1.6 µm; column size,
2.1×50 mm; Waters Corporation) was developed with aqueous
acetonitrile (MeCN) containing a 0.1% formic acid linear gradient
system (5–90% MeCN, 1.6 min; flow rate, 1 ml min−1) at
40°C, verifying the ultraviolet (UV) adsorption and mass of the
major UV peaks (Table II).
| Table II.Anticancer agents used in the present
study. |
Table II.
Anticancer agents used in the present
study.
| Compound | Target | Supplier | Purity, % |
|---|
| Nilotinib | ABL | Carbosynth | 100 |
| Ponatinib | ABL, Kit, Ret,
FGFR | LC
Laboratories | 100 |
| DCC-2036 | ABL, SRC, FLT3 | AdooQ
BioScience | 100 |
| GDC-0068 | AKT | MedChemExpress | 100 |
| Ceritinib | ALK | Chemietek | 100 |
| Crizotinib | ALK, HGFR | LC
Laboratories | 100 |
| Entrectinib | ALK, TrkA, B, and
C, ROS1 | MedChemExpress | 100 |
| Bicalutamide | Androgen | Enzo Life
Sciences | 100 |
| Pentostatin | Antimetabolite | Toront Research
Chemicals | 100 |
| Elesclomol | Apoptosis | Selleck
Chemicals | 100 |
|
Aminoglutethimide | Aromatase | MP Biomedicals | 100 |
| Obatoclax | Bcl | LC
Laboratories | 100 |
| Ibrutinib | Btk | MedChemExpress | 100 |
| Tacrolimus | Calcineurin | LC
Laboratories | 100 |
| PAC-1 | Caspase | AdooQ
BioScience | 100 |
| Dinaciclib | CDK | Cayman
Chemical | 100 |
| PHA-793887 | CDK | AdooQ
BioScience | 100 |
| Dexamethasone | Corticosteroid | Fujifilm Wako | 100 |
| Methotrexate | Dihydrofolate
reductase | Fujifilm Wako | 100 |
| Leflunomide | Dihydroorotate
dehydrogenase | TCI | 100 |
| Etoposide | DNA
topoisomerase | Fujifilm Wako | 100 |
| Melphalan | DNA alkylation | Fujifilm Wako | 100 |
| Temozolomide | DNA alkylation | Fujifilm Wako | 100 |
| Decitabine | DNA
demethylating | TCI | 100 |
| Fluorouracil | DNA synthesis | Fujifilm Wako | 100 |
| Mitomycin C | DNA synthesis | Fujifilm Wako | 100 |
| Carboplatin | DNA synthesis | TCI | 100 |
| Mycophenolic
acid | DNA synthesis | TCI | 100 |
| Erlotinib | EGFR | Carbosynth | 100 |
| Afatinib | EGFR, HER2 | Selleck
Chemicals | 100 |
| Lapatinib | EGFR, HER2 | LC
Laboratories | 100 |
| Tamoxifen | Estrogen | Fujifilm Wako | 100 |
| GSK126 | EZH2 | AdooQ
BioScience | 100 |
| AZD 4547 | FGFR | Active Biochem | 100 |
| Entinostat | HDAC | Carbosynth | 100 |
| Panobinostat | HDAC | Cayman
Chemical | 98.19 |
| Belinostat | HDAC | Selleck
Chemicals | 100 |
| PCI-34051 | HDAC | Selleck
Chemicals | 98.28 |
| Tubastatin A | HDAC | Selleck
Chemicals | 100 |
| Vismodegib | Hedgehog | LC
Laboratories | 100 |
| Varlitinib | HER2, EGFR | Selleck
Chemicals | 97.51 |
| Tivantinib | HGFR | MedChemExpress | 100 |
| Foretinib | HGFR, VEGFR, PDGFR,
Kit, FLT3, Tie, Ron | Selleck
Chemicals | 99.01 |
| Ganetespib | HSP90 | Selleck
Chemicals | 100 |
| Alvespimycin | HSP90 | Selleck
Chemicals | 100 |
| AGI-6780 | IDH2(R140Q) | MedChemExpress | 97.68 |
| BMS-754807 | IGF | Chemscene | 98.52 |
| OSI-906 | IGF | Chemietek | 100 |
| Ruxolitinib | JAK | Chemscene | 100 |
| AZD 6244 | MEK | LC
Laboratories | 100 |
| Binimetinib | MEK | Active Biochem | 100 |
| Rapamycin | mTOR | LC
Laboratories | 100 |
| Everolimus | mTOR | AdooQ
BioScience | 97.97 |
| MLN-4924 | NAE | AdooQ
BioScience | 100 |
| Olaparib | PARP | AdooQ
BioScience | 100 |
| Tandutinib | PDGFR, Kit,
FLT3 | LC
Laboratories | 100 |
| Idelalisib | PI3K | AdooQ
BioScience | 100 |
| NVP-BKM120 | PI3K | AdooQ
BioScience | 100 |
| GDC-0980 | PI3K, mTOR | MedChemExpress | 98.63 |
| Volasertib | PLK | Chemietek | 100 |
| Carfilzomib | Proteasome | Chemietek | 100 |
| Bortezomib | Proteasome | AdooQ
BioScience | 100 |
| Dabrafenib | BRAF | AdooQ
BioScience | 100 |
| Vemurafenib | BRAF | ChemScene | 100 |
| RO-4929097 | Secretase | AdooQ
BioScience | 100 |
| AZD 0530 | SRC, ABL | ChemScene | 100 |
| BX-795 | TBK, PDK, IKK | AdooQ
BioScience | 97.34 |
| Lenalidomide | Thalidomide | AdooQ
BioScience | 100 |
| Paclitaxel | Tubulin | TCI | 100 |
| Vindesine | Tubulin | Sigma-Aldrich | 100 |
| Nutlin-3 | Ubiquitin | KareBay
Biochem | 100 |
| Brivanib | VEGFR, FGFR | AdooQ
BioScience | 100 |
| Sunitinib | VEGFR, FGFR, PDGFR,
Kit, FLT3 | Cayman
Chemical | 96.85 |
| Regorafenib | VEGFR, Kit, Ret,
FGFR, PDGFR | MedChemExpress | 100 |
Establishment of F-PDOs
The present study was approved by the Institutional
Animal Care and Use Committee of Fukushima Medical University
(Fukushima, Japan). Solid tumor tissues obtained from surgical or
biopsy specimens, and ascites and pleural fluids, were acquired
from patients with cancer at Fukushima Medical University Hospital,
upon providing informed consent. Ascites and pleural fluids were
centrifuged at 400 × g for 3 min at room temperature to concentrate
the tumor tissues, and the supernatants were removed. Parts of the
solid tumor tissues, and their associated tissues from ascites and
pleural fluids, were used for comprehensive gene expression
analysis and histological analysis. Solid tumor tissues were also
cut up into small (~1 mm3) pieces with a scalpel. The
minced and concentrated tumor tissues were cultured as suspension
cultures for 3–6 months to establish the F-PDOs. For example,
REME16 cells were derived from the ascites of a patient with
adenocarcinoma of the endometrium. Ascites collected from the
patient was centrifuged at 400 × g for 3 min at room temperature
and the tumor tissues were concentrated. The tumor tissues were
cultured in 5 ml modified FBIM002 medium (Fukushima Translational
Research Project, Fukushima, Japan) supplemented with 1%
penicillin-streptomycin mixed solution (cat. no. 26253-84; Nacalai
Tesque, Inc., Kyoto, Japan) using ultra-low attachment 6-well
plates (cat. no. 3471; Corning Incorporated, Corning, NY, USA) at
37°C in a humidified incubator with 5% CO2. The 50–80%
of the volume of the medium was changed twice weekly, while
observing the condition of the cells. Subculture was performed when
the concentration of the cells was not increased and cellular
debris or single cells were increased in the culture medium. The
organoids were cultured under the same conditions for the following
60 days. During this time, they were observed under a microscope
(Leica DMC2900; Leica Microsystems GmbH, Wetzlar, Germany), to
identify alterations in cellular morphology. When REME16 cells had
been established as an F-PDO, the organoids were stored in liquid
nitrogen vapor phase. The cryopreserved REME16 cells were thawed
and cultured in FBIM002 medium, and it was confirmed that they were
capable of culture for >3 months.
F-PDOs were tested for pathogens [human
immunodeficiency virus (HIV), hepatitis C virus (HCV), hepatitis B
virus (HBV) and Treponema pallidum] with the StepOnePlus
Real-Time Polymerase Chain Reaction System (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) using HIV Real-TM Qual (cat.
no. R-V0-100FRT; Sacace Biotechnologies Srl, Como, Italy), HCV
Real-TM Qual (cat. no. V1-100FRT; Sacace Biotechnologies Srl), HBV
Real-TM Qual (cat. no. V5-100FRT; Sacace Biotechnologies Srl), and
Treponema pallidum Real-TM (cat. no. B20-100FRT; Sacace
Biotechnologies Srl), respectively, according to the manufacturer's
protocols. All F-PDOs were negative.
Cell culture
F-PDOs were cultured in 15 ml FBIM002 using
ultra-low attachment 75-cm2 flasks (cat. no. 3814;
Corning Incorporated) at 37°C in a humidified incubator with 5%
CO2. Since accurate cell numbers of F-PDOs were not able
to be determined using a cell counter, the cell pellet volume
following centrifugation of the cell suspension was visually
measured by comparing the F-PDO pellet in a 15-ml tube with tubes
marked at 50, 75, 100 and 150 µl volume. The 50–80% medium was
changed twice weekly. When F-PDOs reached their maximum saturation
density, the cells were passaged at a 1:2 ratio. In detail, F-PDO
suspensions were transferred from the flask to a 15-ml tube and
centrifuged at 200 × g for 2 min at room temperature. Following
removal of the supernatant, 10 ml medium was added to the pelleted
cells and gently mixed five times. A total of one-half of the cell
suspension was subsequently seeded into a flask containing 10 ml
fresh medium.
Cancer cell lines (AN3 CA, KLE, RL95-2 and SK-UT-1B)
were purchased from the American Type Culture Collection (Manassas,
VA, USA). The cells were cultured at 37°C in a humidified incubator
with 5% CO2. All cell culture media were supplemented
with fetal bovine serum (cat. no. 172012; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) and penicillin-streptomycin solution
(cat. no. 168-23191; Fujifilm Wako Pure Chemical, Ltd., Osaka,
Japan) at final concentrations of 10 and 1%, respectively. AN3 CA
and SK-UT-1B cells were maintained in Eagle's minimum essential
medium (cat. no. 051-07615; Fujifilm Wako Pure Chemical, Ltd.). KLE
cells were cultured in Dulbecco's modified Eagle's medium
(DMEM)/F-12, 4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
(HEPES) medium (cat. no. 11330; Thermo Fisher Scientific, Inc.).
RL95-2 cells were grown in DMEM/F-12, HEPES medium supplemented
with 5 µg/ml insulin (cat. no. 097-06474; Fujifilm Wako Pure
Chemical, Ltd.). Cell number and viability were automatically
measured using trypan blue dye exclusion with a Vi-Cell XR Cell
Viability Analyzer (Beckman Coulter, Inc., Brea, CA, USA),
according to the manufacturer's protocol.
Histological analysis
F-PDOs were fixed using 4% paraformaldehyde for 1 h
at room temperature, and washed with PBS. Subsequently, F-PDOs were
embedded using iPGell (cat. no. GSPG20-1; GenoStaff Co., Ltd.,
Tokyo, Japan). The blocks were embedded in paraffin and sections
(3-µm) were obtained for histological analysis. Hematoxylin and
eosin (HE) staining was performed using a DRS-Prisma-J0D (Sakura
Finetek Japan, Tokyo, Japan) automated slide stainer. Xenograft
tumor tissues were fixed using 10% neutral buffered formalin
solution at least 24 h at room temperature, and the following
steps, including paraffin embedding, paraffin sectioning and HE
staining, were performed under contract with BoZo Research Center,
Inc. (Tokyo, Japan). Paraffin-embedded tissues were sliced to 3-µm.
HE-stained samples were observed using an upright light microscope
(magnification, ×40; BX43; Olympus, Corporation, Tokyo, Japan).
Comprehensive gene expression
analysis
Comprehensive gene expression analysis was performed
according to previous reports (24–26).
Briefly, synthetic polynucleotides (80-mers) representing 14,400
human transcripts (MicroDiagnostic, Inc., Tokyo, Japan) were
arrayed using a custom arrayer. Total RNA was extracted from the
cells using ISOGEN (Nippon Gene Co., Ltd., Tokyo, Japan) and 5 µg
was labeled using SuperScript II (Invitrogen; Thermo Fisher
Scientific, Inc.) and cyanine 5-dUTP (PerkinElmer, Inc., Waltham,
MA, USA) for samples, or cyanine 3-dUTP (PerkinElmer, Inc.) for
human common reference RNA, which was prepared by mixing equal
amounts of total RNA extracted from 22 cell lines. Hybridization
was performed with a labeling and hybridization kit
(MicroDiagnostic, Inc.). Signals were measured with a GenePix 4000B
scanner (Molecular Devices, LLC, Sunnyvale, CA, USA).
The signals were converted into primary expression
ratios (ratio of cyanine-5 intensity of each sample to cyanine-3
intensity of the human common reference RNA). Each ratio was
normalized through multiplication using normalization factors and
Gene Pix Pro 3.0 software (Molecular Devices, LLC). The primary
expression ratios were converted to log2 values
(designated as log ratios). Spots that exhibited fluorescence
intensities below the detection limit were assigned a log ratio
value of 0 and were not included in the signal calculations of the
averages and subtracted log ratios. The data were processed using
Microsoft Office Excel 2016, version 16.0.4266.1001 (Microsoft
Corporation, Redmond, WA, USA) and the ExpressionView_Pro Version
3.3.8.1 (MicroDiagnostic, Inc.). Cluster analysis was performed
using the unweighted pair group method with arithmetic mean
hierarchical clustering method (24).
Assessment of tumorigenesis using a
xenograft model
These experiments were performed with the approval
of the Institutional Animal Care and Use Committee of Fukushima
Medical University. Tumorigenesis of F-PDOs was examined using
immunodeficient NOG
(NOD.Cg-Prkdcscidll2rgtm1Sug/ShiJic)
mice (27). A total of three male
NOG mice (6–8 weeks old; 20–22 g) were obtained from the Central
Institute for Experimental Animals (Kawasaki, Japan). All mice were
housed in plastic cages (136×208×115 mm) within a safety rack (CLEA
Japan, Inc., Tokyo, Japan) in a pathogen-free state, at a
temperature of 22±2°C with 55±5% humidity, and a 12-h light/12-h
dark cycle. The plastic cages, bedding and filter caps for these
mice were sterilized either in an autoclave or via gas
sterilization. The mice were allowed ad libitum access to
commercial diet sterilized by gamma irradiation at 30 kGy (CE-2;
CLEA Japan, Inc.) and ultra-filtered membrane water. REME9, REME11
and REME16 (34–70 mg) lines were suspended in 0.1 ml Hanks'
balanced salt solution without magnesium and calcium (cat. no.
085-09355; Fujifilm Wako Pure Chemical, Ltd.), and were injected
subcutaneously into the back of the NOG mice using a 1-ml syringe
with a 26 G needle (28). Tumor
sizes were estimated by performing two-dimensional caliper
measurements once per week; the formula for estimating the volume
of the ellipsoid tumor was L × W2/2, where L is the
length of the major axis and W is the width of the tumor (29).
Growth inhibition assays using
multi-well plates
The growth inhibitory activity of anticancer agents
against F-PDOs was assayed by measuring the amount of 5′ adenosine
triphosphate (ATP) in the cells using the CellTiter-Glo 3D Cell
Viability Assay (Promega Corporation, Madison, WI, USA). F-PDOs
were cultured in flasks until sufficient numbers of cells were
present for the assay. A total of 1 day prior to seeding, the
F-PDOs were transferred from a 75-cm2 flask to a 15-ml
tube and centrifuged at 200 × g for 2 min at room temperature to
measure the cell pellet volume. Subsequently, the cell pellet was
suspended with 15 ml fresh medium. The suspension was transferred
to a 75-cm2 flask and cultured in an incubator. After 24
h, the F-PDO was minced using a CellPet FT (JTEC Corporation,
Osaka, Japan) set with a filter holder containing a 70-µm mesh
filter. The F-PDO suspension was diluted 10- or 20-fold and seeded
into 96-well round-bottomed, ultra-low attachment microplates (cat.
no. 7007; Corning Incorporated) with 150 µl medium using a
Multidrop Combi dispenser (Thermo Fisher Scientific, Inc.). To
monitor apoptosis activity, CellEvent™ Caspase-3/7 Green Detection
Reagent (cat. no. C10423; Thermo Fisher Scientific, Inc.) was added
to each well. F-PODs were incubated for 24 h and then treated with
0.1-µl solutions containing the agent at final concentrations
ranging between 10 µM and 1.5 nM (nine serial dilutions) using an
ADS-348-8 Multistage-dispense station (Biotec Co., Ltd., Tokyo,
Japan). DMSO was used as the vehicle control at a maximum
concentration of 0.1%. The dynamics of apoptosis activity were
monitored using an IncuCyte ZOOM live cell imaging system (Essen
BioScience, Ann Arbor, MI, USA) and IncuCyte ZOOM version 2016B
(Essen BioScience). The plates were placed in an IncuCyte for 6
days to monitor caspase-3/7 activation through the capture of
green-filter images (λex, 440–480 nm; λem, 504–544 nm) of the cells
every 6 h. After 144 h, 40 µl CellTiter-Glo 3D reagent solution
(Promega Corporation) was added to each well. The plates were mixed
using a mixer and incubated for 10 min at 30°C. Luminescence was
measured using an EnSpire plate reader (PerkinElmer, Inc.). Cell
viability was calculated by dividing the amount of ATP in the test
wells by that in the vehicle control wells, with the background
subtracted. The growth rate over 6 days was calculated by dividing
the amount of ATP in the wells without anticancer agents by that in
the vehicle control wells 24 h after seeding.
HTS using 384-well plates was conducted as performed
for the 96-well plates, except for the volume used for seeding and
the agent concentration range. A total of 40 µl F-PDO suspension
was seeded in 384-well round-bottomed, ultra-low attachment
spheroid microplates (cat. no. 4516; Corning Incorporated) using
with a dispenser. At 24 h after seeding, F-PDOs were treated with
0.04-µl solutions of agent at final concentrations ranging between
20 µM and 1.0 nM (ten serial dilutions) using an Echo 555 (Labcyte,
Inc., San Jose, CA, USA). After 144 h, 10 µl CellTiter-Glo 3D
reagent solution was added to the medium and the luminescence was
measured.
The half-maximal inhibitory concentration
(IC50) and area under the curve (AUC) values were
calculated from the dose-response curves and analyzed using Morphit
version 5.0 (The Edge Software Consultancy, Ltd., Guildford, UK).
The data represent the mean ± standard deviation of triplicate
experiments. The Z' factor, a dimensionless parameter that ranges
between 1 (infinite separation) and <0, was defined as Z' = 1 -
(3σc+ + 3σc-) / |µc+ - µc-|, where σc+, σc-, µc+ and µc- are the
standard deviations (σ) and averages (µ) of the high (c+) and low
(c-) controls (30).
Results
Establishment of F-PDOs
The present study attempted to establish organoids
from lung, breast, ovarian, uterine and digestive organ tumor
tissues using an independently developed method, which had the
following characteristics: (i) Tumor tissues were cut without
tissue-dissolving enzymes, for example trypsin or collagenase; (ii)
F-PDOs were cultured in suspension without extracellular matrix;
and (iii) F-PDOs expand for long periods of time in culture, based
on the principle of retaining the histological architecture of
their source tissue. Thus, during long periods of culture, almost
all stromal cells were removed except for cancer cells, and
heterogeneous tumor organoids were cultured. F-PDOs formed cell
clusters that exhibited various heterogeneous morphologies.
Furthermore, F-PDOs were able to be cultured for >6 months and
cryopreserved for future use. At present, 53 F-PDOs have been
established from several tumor tissues (Table I). The present study describes the
characteristics of three F-PDOs (REME9, 11 and 16) established from
endometrial cancer tissues, in addition to HTS using these
F-PDOs.
Characterization of F-PDOs
To investigate whether the REME9, 11 and 16 lines
had similar characteristics to their source tumor tissues,
histological and comprehensive gene expression analyses were
performed. REME9 was from endometrial cancer tissue, which was
pathologically diagnosed as endometrioid adenocarcinoma with
squamous differentiation. REME11 and 16 were from clear cell
adenocarcinoma specimens. REME9 was a mixture of cells with
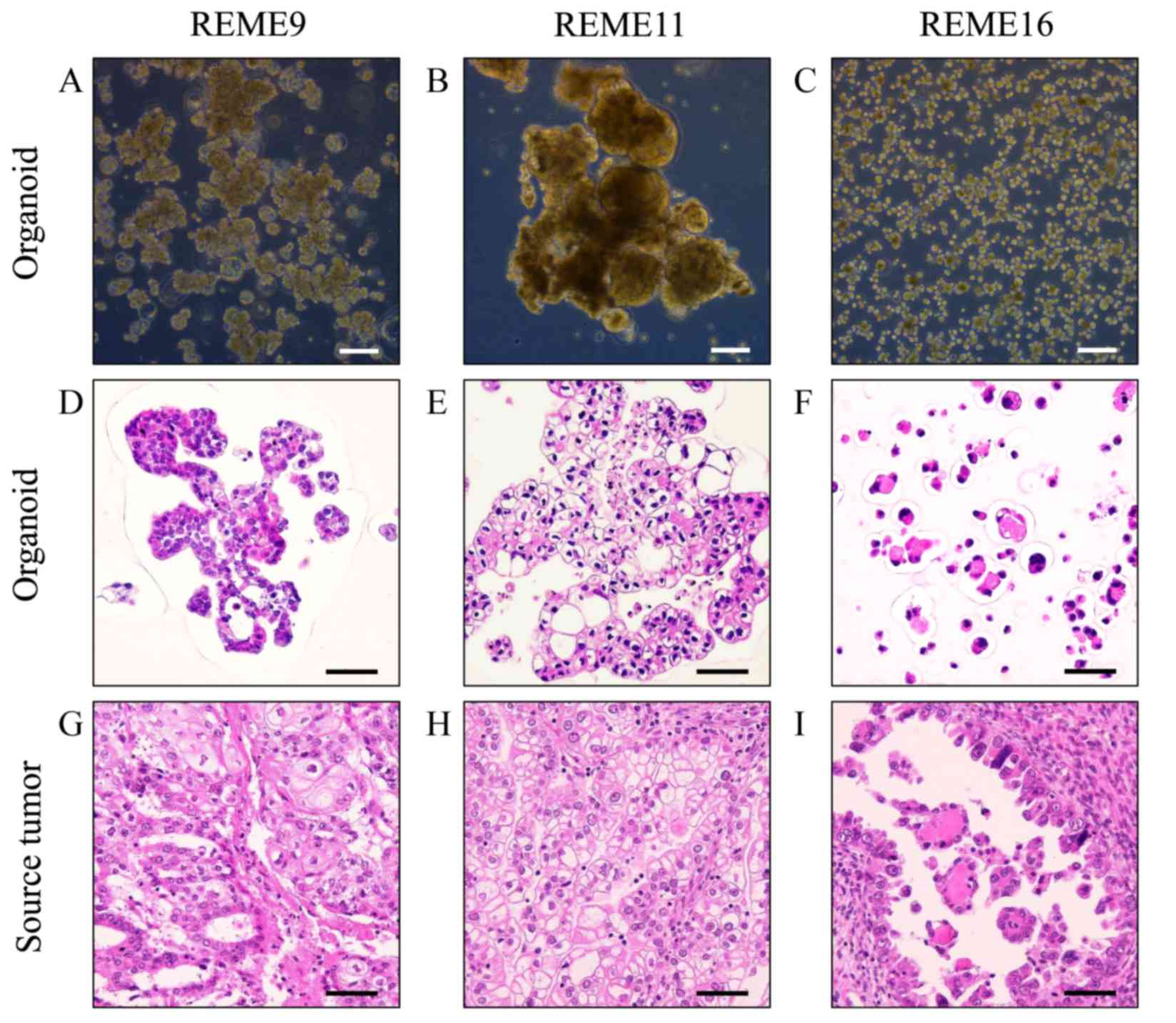
different morphological features (Fig.
1A). The majority of REME9 cells appeared as big cell clusters
that were ~100–500 µm in diameter. Certain organoids were ~100 µm
and included hollow cells. Cell debris existed in the culture.
REME11 grew as large cell clusters of 300–1,000 µm, which were
composed of round and elliptical cells of 100–200 µm (Fig. 1B). These cell clusters frequently
merged to form clusters that were >1,000 µm in diameter. REME16
comprised primarily round cells that were <50 µm (Fig. 1C). Although the cells sometimes
formed clusters, they were rarely >100 µm, unlike REME9 and 11.
The doubling times of REME9, 11 and 16 were ~5, 8 and 6 days,
respectively.
Subsequently, REME9, 11 and 16, and their source
tissues, were stained with HE for histological evaluation.
HE-stained images of REME9, 11, and 16 (Fig. 1D-F) were similar to those of their
source tissues (Fig. 1G-I). REME9
was characterized by its resemblance to endometrioid adenocarcinoma
in accordance with the source tissue (Fig. 1D and G). REME11 and its source
tissue clearly possessed characteristics of clear cell
adenocarcinoma, specifically the presence of clear cytoplasm
(Fig. 1E and H). Images of REME16
and its source tissue illustrated a glandular structure with
eosin-positive material at the center of the spheroid (Fig. 1F and I).
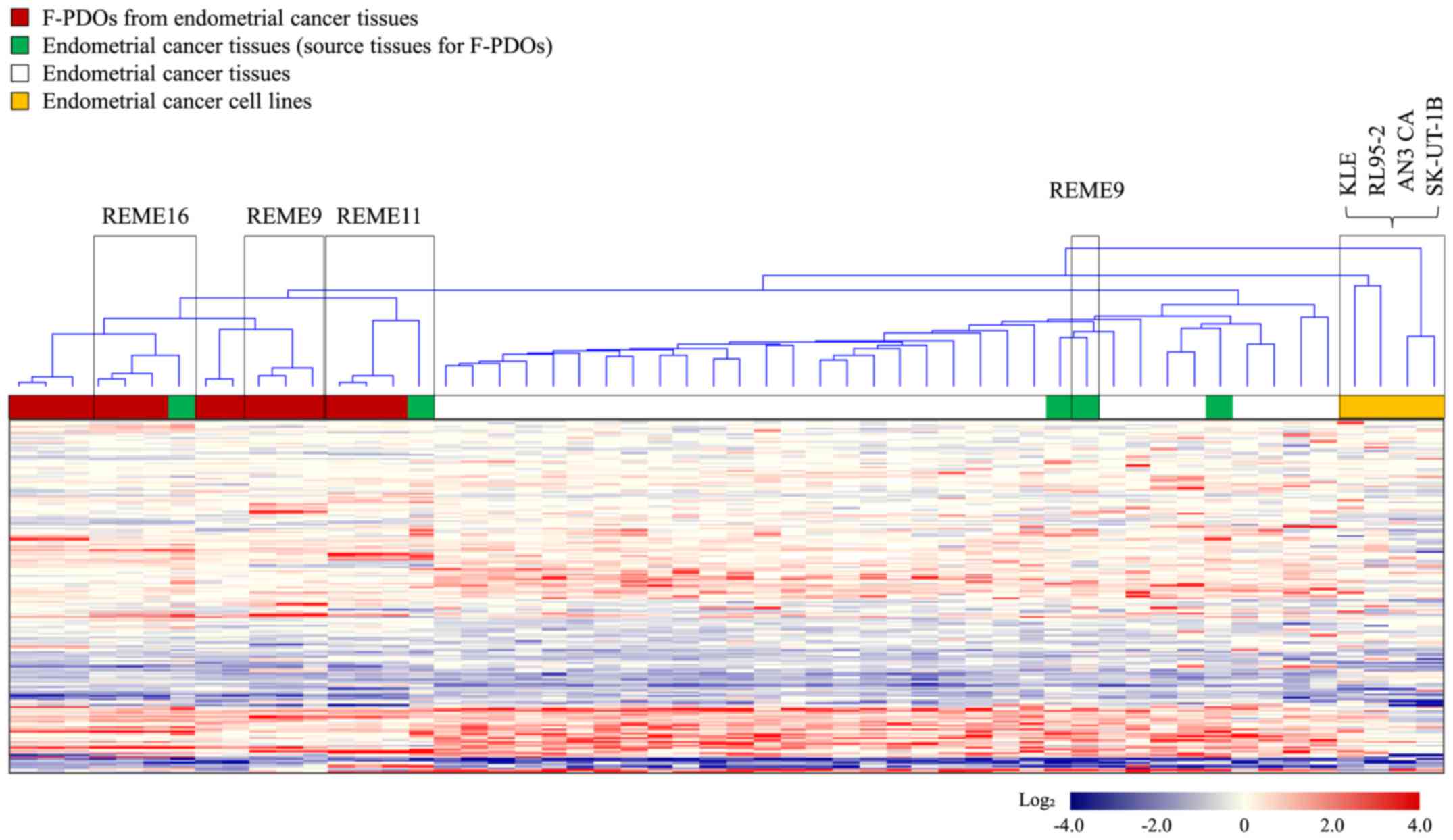
Gene expression profiles were obtained from five
F-PDOs from endometrial tumor tissues, source endometrial tumor
tissues, various other endometrial tumor tissues (31 samples), and
five cell lines (AN3 CA, KLE, RL95-2 and SK-UT-1B) derived from
endometrial cancer. Furthermore, samples used for this analysis
included different generations of the same F-PDO, in addition to
two different types of endometrial tumor-derived F-PDOs in addition
to REME9, 11 and 16. Cluster analysis of gene expression profiles
was performed with the resultant 2,579 genes, which indicated
variation in gene expression among all samples, as presented in
Fig. 2. This analysis resulted in
the presence of two groups: The first group consisted of F-PDOs and
endometrial tumors, and the second group comprised cancer cell
lines. The profiles of F-PDOs were completely different from those
of the cell lines, although similar to those of the endometrial
tumors. The differences between the profiles of F-PDOs and those of
endometrial tumors may be due to the fact that stromal cells were
removed from their source tissues during F-PDO culture. These
results indicated that F-PDOs possessed characteristics of the
source tissues, although they were not similar to endometrial cell
lines.
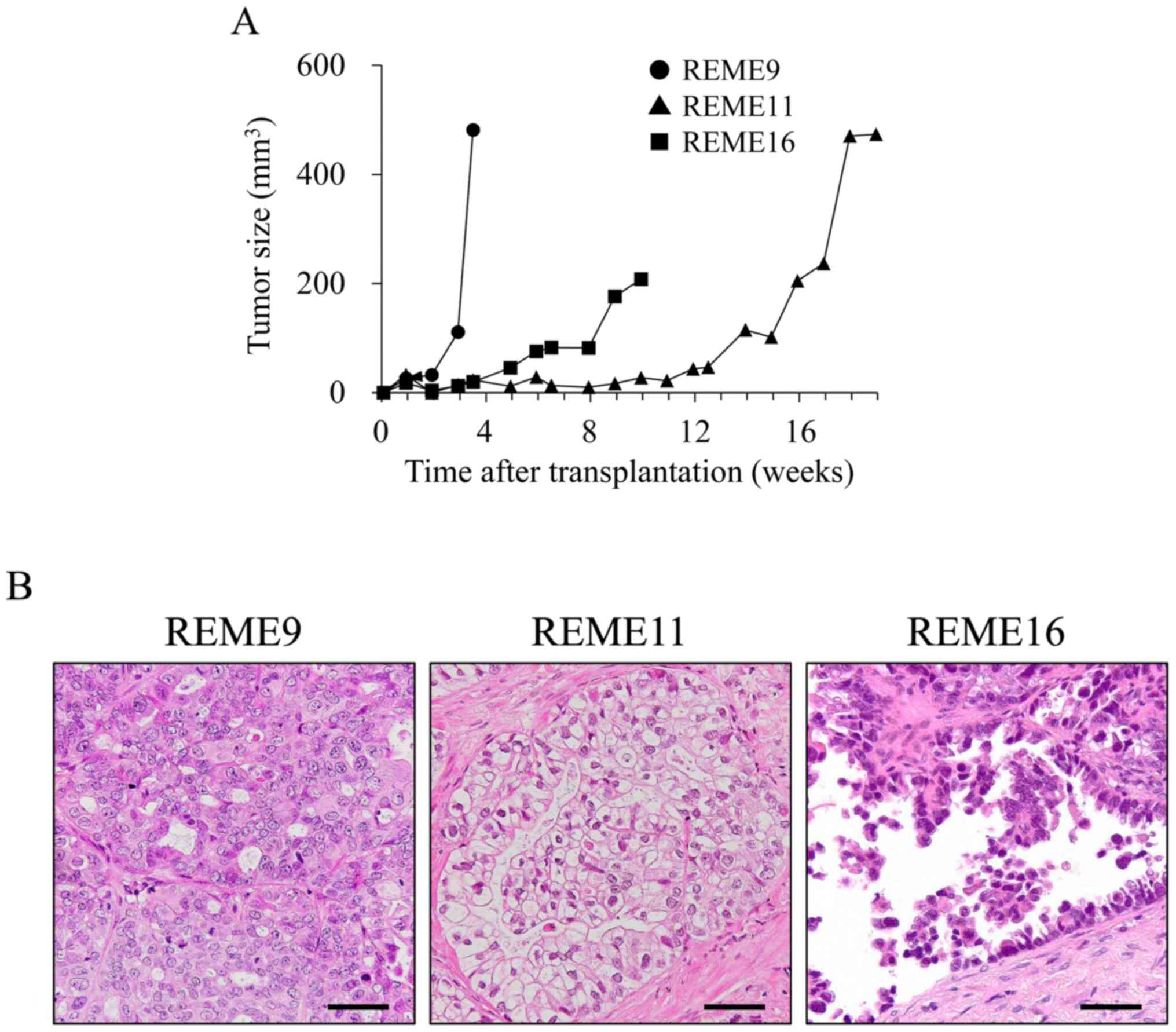
To verify in vivo tumorigenesis, REME9, 11
and 16 F-PDOs were xenografted subcutaneously into NOG mice. All
F-PDOs were confirmed to engraft in NOG mice 6 days
post-inoculation, and thus demonstrated in vivo
tumorigenesis (representative data presented in Fig. 3A). However, growth rates among the
three F-PDOs were different. The growth rate of REME9 was highest,
as the tumor size was 481.6 mm3 24 days post-grafting.
The growth rate of REME11 was very slow, requiring 120 days to
reach a tumor size of 400 mm3. The tumor size of REME16
increased to 200 mm3 within 70 days. The tissues from
REME9, 11 and 16 xenografts were stained with HE, and
histologically observed. Resulting images showed tissue
characteristics that were identical to those of their source F-PDOs
and tissues (Figs. 1 and 3B). Furthermore, the gene expression
profiles of REME9, 11 and 16 xenografts were similar to those of
their source F-PDOs and tissues (data not shown).
Development of a cell growth
inhibition assay using F-PDOs
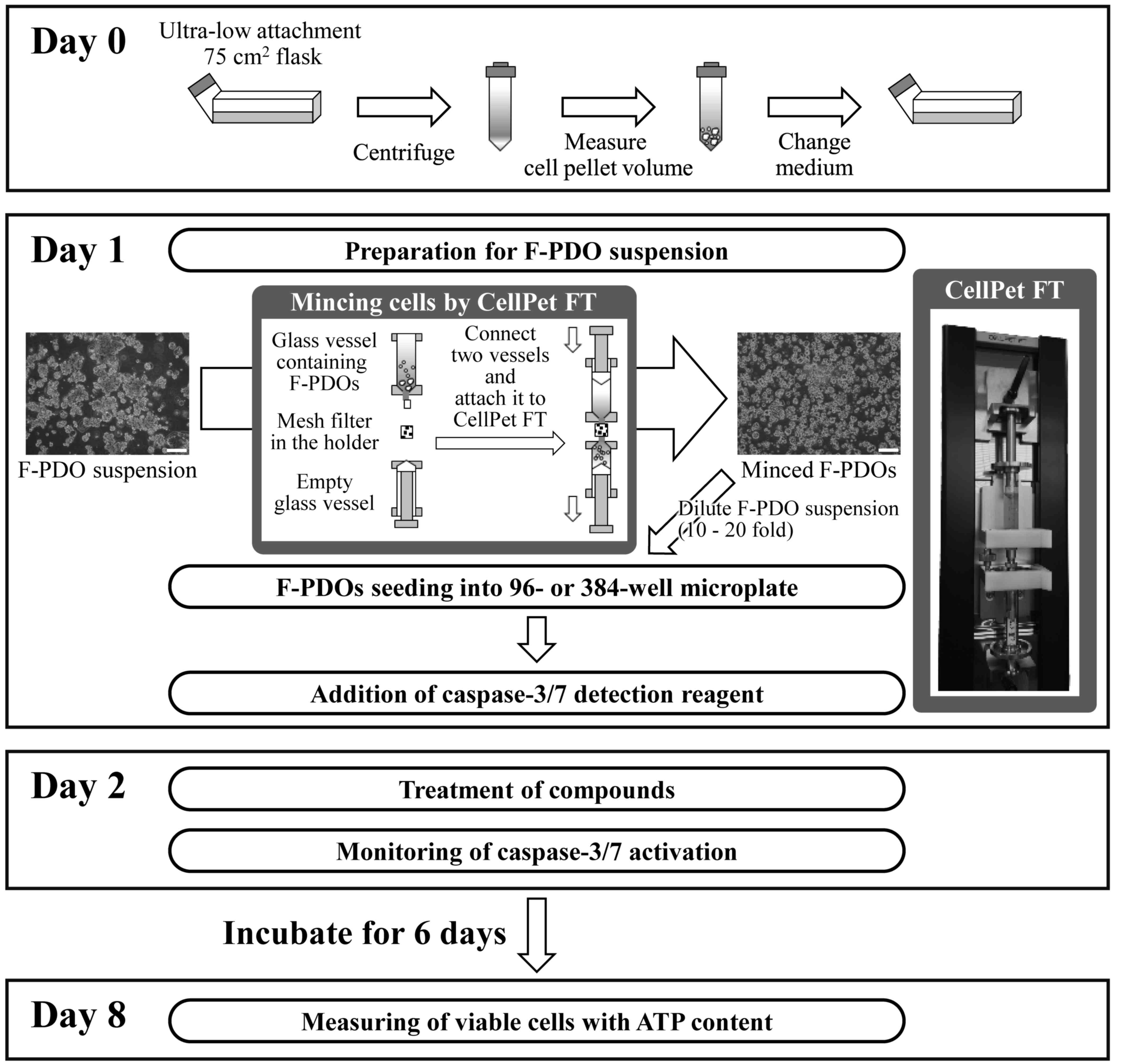
To evaluate anticancer agents using F-PDOs, the
present study aimed to develop an HTS assay using 96- or 384-well
microplates. In preliminary experiments, it was difficult to obtain
high precision data with multi-well plates, as the majority of
F-PDOs form large cell clusters that cannot be dispensed equally
into each well. Therefore, CellPet FT with a 70-µm mesh filter was
used to mince the F-PDOs, which were subsequently seeded into
microplates as presented in Fig. 4.
F-PDOs were incubated for 7 days following seeding. To count viable
cells, the ATP content was measured. HTS performance was evaluated
by computing the coefficients of variation (CV) and the Z'-factor.
The Z'-factor has been widely accepted for the evaluation of assay
quality and performance (30), and
an assay is suitable for HTS when this value is >0.5. The growth
rate of F-PDOs in the plates was 2.31–5.16 fold (Table III). The control data points in
the 96-well plate assay exhibited little variability, with CV
values of 1.12, 3.08 and 0.74% and with a calculated Z'-factor of
0.97, 0.91 and 0.98, for REME9, 11 and 16, respectively (Table III). These results suggested that
this assay had excellent performance for HTS. The CV and Z' values
for REME9 and 16 in 384-well plates were also good; specifically,
the CV values were 5.38 and 4.44% and the Z'-factor values were
0.84 and 0.87, for REME9 and 16, respectively (Table III). However, REME11 was
unsuitable for 384-well plate assays, since the CV and Z'-factor
values were 21.65% and 0.35, respectively. Consequently, HTS was
performed with F-PDOs using 96- or 384-well plates to evaluate
anticancer agents.
| Table III.High-throughput screening performance
using Fukushima patient-derived organoids and 96- or 384-well
microplates. |
Table III.
High-throughput screening performance
using Fukushima patient-derived organoids and 96- or 384-well
microplates.
| Plate size | Factor | REME9 | REME11 | REME16 |
|---|
| 96-well | CV, % | 1.12 | 3.08 | 0.74 |
|
| Z'-factor | 0.97 | 0.91 | 0.98 |
|
| Growth rate | 2.97 | 2.31 | 2.77 |
| 384-well | CV, % | 5.38 | 21.65 | 4.44 |
|
| Z'-factor | 0.84 | 0.35 | 0.87 |
|
| Growth rate | 5.16 | 3.56 | 4.01 |
Evaluation of anticancer agents using F-PDOs. To
investigate the sensitivity of F-PDOs to anticancer agents using
our HTS system, growth inhibition and apoptosis were assessed using
REME9, 11 and 16 lines treated with three representative anticancer
agents, specifically paclitaxel, carboplatin and mitomycin C, in
96-well plates. Paclitaxel and carboplatin are used as the standard
clinical treatments for endometrial cancer. F-PDOs were treated
with the drugs 24 h post-seeding and subsequently incubated for 6
days. In addition to counting the viable cells, apoptosis was
monitored and analyzed in parallel over time using IncuCyte and
CellEvent™ Caspase-3/7 Green Detection Reagent (Fig. 4). The data points for viable cells
and caspase-3/7 activity exhibited little variation (Fig. 5).
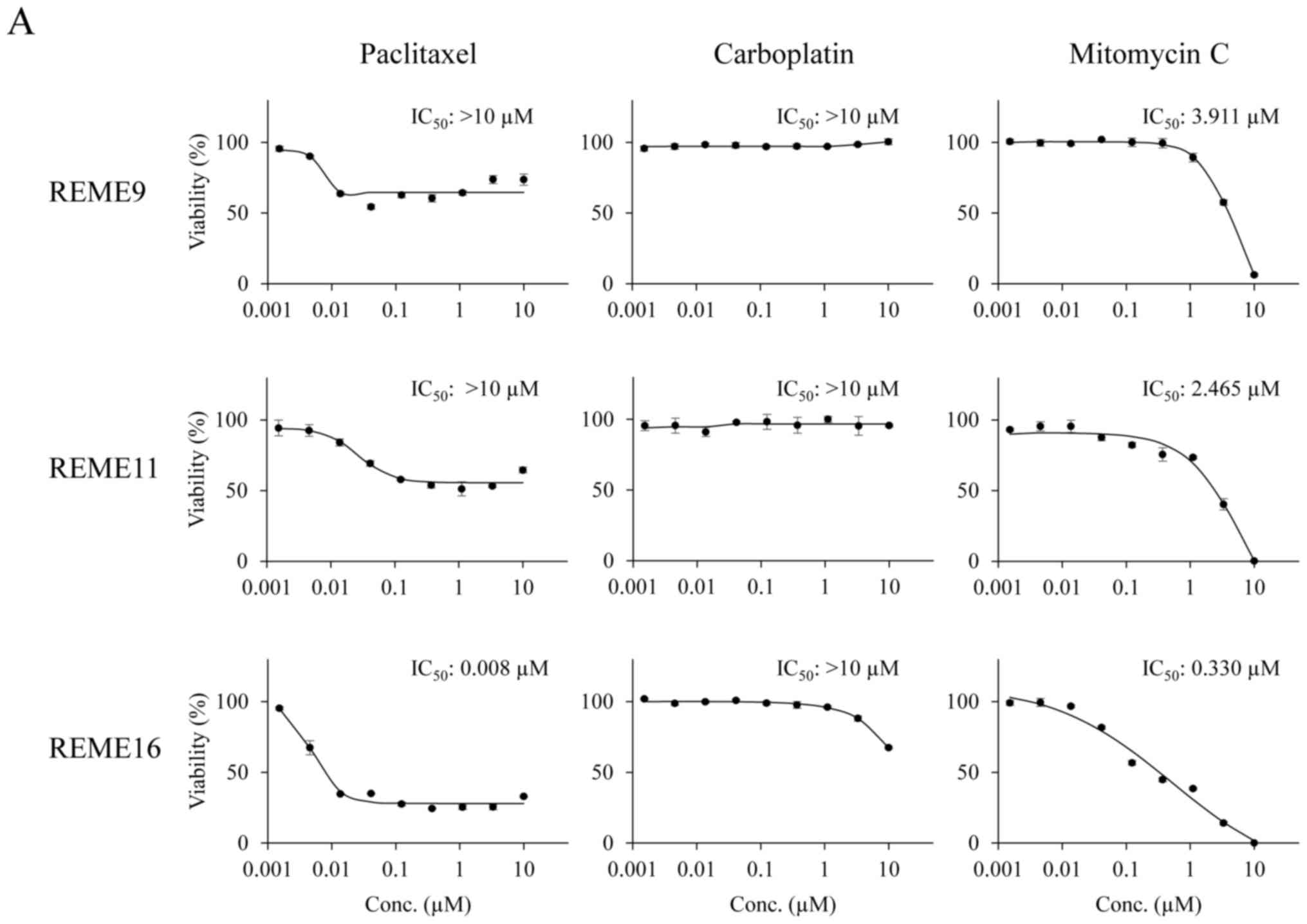 | Figure 5.Response of F-PDOs to clinically used
chemotherapeutics. (A) Dose-response curve of F-PDOs to anticancer
agents. The F-PDOs were minced, seeded in 96-well plates, and
treated with nine different concentrations (between 10 µM and 1.5
nM) of paclitaxel, carboplatin or mitomycin C for 6 days. The data
represent the mean ± standard deviation of triplicate experiments.
(B) Caspase-3/7 levels in F-PDOs treated with paclitaxel,
carboplatin or mitomycin C. The time course quantification of
caspase-3/7 fluorescence in F-PDOs treated with 10 µM each agent is
presented. The graphs represent the time-course of fluorescence
intensities, representing caspase-3/7 activity, between 6 and 144 h
post-drug treatment. The data represent the mean ± standard
deviation of triplicate experiments. For each F-PDO, images were
captured at 0 h or the time when the fluorescence intensity reached
its maximum following the drug treatment, as indicated by the
arrows. Scale bar, 400 µm. F-PDO, Fukushima patient-derived
organoid. |
The IC50 values of paclitaxel,
carboplatin and mitomycin C in each F-PDO are presented in Fig. 5A. Dose-response curves demonstrated
that REME9 and 11 lines were more resistant to all drugs than
REME16. In particular, the IC50 values of paclitaxel and
carboplatin, the standard therapeutics for endometrial cancer, were
>10 µM for REME9 and 11, whereas the IC50 value of
paclitaxel was 0.008 µM for REME16. In addition, 10 µM carboplatin
marginally affected the viability of REME16 cells. By contrast,
mitomycin C inhibited the proliferation of all three F-PDOs in a
dose-dependent manner and completely suppressed cell survival at a
concentration of 10 µM (Fig. 5A).
These results indicated that REME9 and 11 lines have high
resistance (IC50 >10 µM) to the standard therapeutics
used for endometrial cancer (paclitaxel and carboplatin).
To determine whether F-PDO cell death induced by
paclitaxel and mitomycin C was due to apoptosis, the enzymatic
activity of caspase-3/7 was monitored over time. Mitomycin C, which
inhibited the proliferation of F-PDOs, markedly activated
caspase-3/7, suggesting that this agent induces apoptosis at a
concentration of 10 µM (Fig. 5B).
By contrast, despite the fact that paclitaxel inhibited the growth
of REME16 cells, it only marginally activated caspase-3/7, similar
to that observed for REME9 and 11 (Fig.
5B). Thus, it is possible that the growth inhibition by
paclitaxel is not due to an apoptotic effect.
To compare the sensitivity profiles to anticancer
agents between F-PDOs (REME9 and 16) and endometrial cancer cell
lines (AN3 and RL95-2), 61 anticancer agents (Table II) that are used clinically or in
clinical devolvement were evaluated, using the 384-well HTS assay.
The overall sensitivity of REME9 and 16 to anticancer agents was
lower compared with that of the cell lines (Fig. 6). The sensitivities of REME9 and 16
were very similar, but the profiles of F-PDOs were not similar to
those of the cell lines. For example, paclitaxel weakly inhibited
the growth of REME9 and 16 cells (Fig.
5A), although it induced a strong growth inhibitory effect on
the cell lines (Fig. 6). Thus, the
evaluation of anticancer agents using F-PDOs, which possess the
characteristics of tumor tissues, may yield more precise
information regarding drug efficacy compared with conventional
methods. Furthermore, bortezomib, carfilzomib, dinaciclib and
panobinostat exerted marked inhibitory effects (AUC values <50)
against REME9 and 16. Thus, these anticancer agents may be
candidates for the development of novel therapeutic regimens for
endometrial cancer.
Discussion
We have established F-PDOs from a number of tumor
tissues and validated their characteristics relative to those of
their source tumor tissue. Furthermore, genome analysis of F-PDOs,
using next generation sequencers, is now in progress to understand
their genetic characteristics. Gene expression data for the F-PDOs
from xenograft mice were additionally obtained. The gene expression
profiles of F-PDOs between culture and xenograft samples were
virtually identical (data not shown). Thus, the characteristics of
F-PDOs in xenograft mice are maintained in culture, and are similar
to those of their source tissues. The primary difference between
F-PDOs and other tumor organoids is the method of establishment.
For this method, source tumors are not disrupted using enzymes, for
example trypsin and collagenase; rather, they are minced by
physical cutting, unlike previously reported methods (12–23).
In addition, these enzymes are not even used during passaging for
long-term culturing or assays. Therefore, it was hypothesized that
the maintenance of F-PDOs retains the conditions of the
heterogeneous source tumors. The characteristics of F-PDOs
recapitulate the histological architecture and expression profile
of the source tumor tissues, even following long-term expansion or
grafting in immunodeficient mice. Thus, the F-PDOs were stably and
continuously used to evaluate anticancer agents.
The characteristics of F-PDOs are unsuitable for HTS
using a 96-well or 384-well formats to evaluate compounds, as these
structures exhibit various and non-uniform sizes, in addition to
forming large clusters in culture. To solve this problem, CellPet
FT was used, which is able to mince cell spheroids without damaging
them using a mesh filter. This results in uniform cell sizes, and
when this was applied, the development of a precision HTS platform
using F-PDOs was successful. This protocol, using HTS, is
appropriate for screening anticancer agents from a large compound
library. In addition, it may be possible to assess the efficacy of
compounds identified by HTS using mice engrafted with F-PDOs used
for the original assay.
The present study evaluated the standard
chemotherapeutics (paclitaxel and carboplatin) that are used for
endometrial cancer using REME9, which was derived from a patient
who did not respond to paclitaxel and carboplatin. Accordingly, the
inhibitory effects of paclitaxel and carboplatin against this cell
line were weak (Fig. 5A). These
results suggested that F-PDOs may reflect the clinical status of
source tumors in terms of response to drugs. Thus, this assay
system may facilitate the evaluation of anticancer agents under
conditions that reflect clinical conditions more accurately
compared with conventional methods, and may aid the discovery of
markers to predict the pharmacological effects of anticancer
agents.
In conclusion, the results of the present study
demonstrated that F-PDOs are superior to conventional cell lines
for identifying potential novel therapeutic targets, thus
presenting opportunities for drug testing and advances in
personalized medicine approaches.
Acknowledgements
Not applicable.
Funding
The present study was supported by grants for
Translational Research Programs from Fukushima Prefecture.
Availability of data and materials
The datasets used during the present study are
available from the corresponding author upon reasonable
request.
Authors' contributions
MT, SW, KK and KF conceived and designed the
experiments. HT, AH, HH, GH, NT, MR, GM, YY, EI, JII, YD, SS and TW
performed the experiments. HT, AH, HH, GH, SW and MT analyzed the
data and wrote the paper. MT revised the paper.
Ethics approval and consent to
participate
These experiments were performed with the approval
of the Institutional Animal Care and Use Committee of Fukushima
Medical University (approval nos. 27011, 29035; Fukushima,
Japan).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sharma SV, Haber DA and Settleman J: Cell
line-based platforms to evaluate the therapeutic efficacy of
candidate anticancer agents. Nat Rev Cancer. 10:241–253. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shamir ER and Ewald AJ: Three-dimensional
organotypic culture: Experimental models of mammalian biology and
disease. Nat Rev Mol Cell Biol. 15:647–664. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Arrowsmith J and Miller P: Trial watch:
Phase II and phase III attrition rates 2011–2012. Nat Rev Drug
Discov. 12:5692013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Arrowsmith J: Trial watch: Phase II
failures: 2008–2010. Nat Rev Drug Discov. 10:328–329. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
DiMasi JA, Reichert JM, Feldman L and
Malins A: Clinical approval success rates for investigational
cancer drugs. Clin Pharmacol Ther. 94:329–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tentler JJ, Tan AC, Weekes CD, Jimeno A,
Leong S, Pitts TM, Arcaroli JJ, Messersmith WA and Eckhardt SG:
Patient-derived tumour xenografts as models for oncology drug
development. Nat Rev Clin Oncol. 9:338–350. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Siolas D and Hannon GJ: Patient-derived
tumor xenografts: Transforming clinical samples into mouse models.
Cancer Res. 73:5315–5319. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosfjord E, Lucas J, Li G and Gerber HP:
Advances in patient-derived tumor xenografts: From target
identification to predicting clinical response rates in oncology.
Biochem Pharmacol. 91:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hidalgo M, Amant F, Biankin AV, Budinská
E, Byrne AT, Caldas C, Clarke RB, de Jong S, Jonkers J, Mælandsmo
GM, et al: Patient-derived xenograft models: An emerging platform
for translational cancer research. Cancer Discov. 4:998–1013. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gao H, Korn JM, Ferretti S, Monahan JE,
Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al:
High-throughput screening using patient-derived tumor xenografts to
predict clinical trial drug response. Nat Med. 21:1318–1325. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Weeber F, Ooft SN, Dijkstra KK and Voest
EE: Tumor organoids as a pre-clinical cancer model for drug
discovery. Cell Chem Biol. 24:1092–1100. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Crespo M, Vilar E, Tsai SY, Chang K, Amin
S, Srinivasan T, Zhang T, Pipalia NH, Chen HJ, Witherspoon M, et
al: Colonic organoids derived from human induced pluripotent stem
cells for modeling colorectal cancer and drug testing. Nat Med.
23:878–884. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sato T, Stange DE, Ferrante M, Vries RG,
Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J,
Siersema PD, et al: Long-term expansion of epithelial organoids
from human colon, adenoma, adenocarcinoma, and Barrett's
epithelium. Gastroenterology. 141:1762–1772. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van de Wetering M, Francies HE, Francis
JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J,
Taylor-Weiner A, Kester L, et al: Prospective derivation of a
living organoid biobank of colorectal cancer patients. Cell.
161:933–945. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boj SF, Hwang CI, Baker LA, Chio II, Engle
DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al:
Organoid models of human and mouse ductal pancreatic cancer. Cell.
160:324–338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gao D, Vela I, Sboner A, Iaquinta PJ,
Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora
VK, et al: Organoid cultures derived from patients with advanced
prostate cancer. Cell. 159:176–187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Girda E, Huang EC, Leiserowitz GS and
Smith LH: The use of endometrial cancer patient-derived organoid
culture for drug sensitivity testing is feasible. Int J Gynecol
Cancer. 27:1701–1707. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Broutier L, Mastrogiovanni G, Verstegen
MM, Francies HE, Gavarró LM, Bradshaw CR, Allen GE, Arnes-Benito R,
Sidorova O, Gaspersz MP, et al: Human primary liver cancer-derived
organoid cultures for disease modeling and drug screening. Nat Med.
23:1424–1435. 2017. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pauli C, Hopkins BD, Prandi D, Shaw R,
Fedrizzi T, Sboner A, Sailer V, Augello M, Puca L, Rosati R, et al:
Personalized in vitro and in vivo cancer models to guide precision
medicine. Cancer Discov. 7:462–477. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kondo J, Endo H, Okuyama H, Ishikawa O,
Iishi H, Tsujii M, Ohue M and Inoue M: Retaining cell-cell contact
enables preparation and culture of spheroids composed of pure
primary cancer cells from colorectal cancer. Proc Natl Acad Sci
USA. 108:6235–6240. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Endo H, Okami J, Okuyama H, Kumagai T,
Uchida J, Kondo J, Takehara T, Nishizawa Y, Imamura F, Higashiyama
M, et al: Spheroid culture of primary lung cancer cells with
neuregulin 1/HER3 pathway activation. J Thorac Oncol. 8:131–139.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kiyohara Y, Yoshino K, Kubota S, Okuyama
H, Endo H, Ueda Y, Kimura T, Kimura T, Kamiura S and Inoue M: Drug
screening and grouping by sensitivity with a panel of primary
cultured cancer spheroids derived from endometrial cancer. Cancer
Sci. 107:452–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yoshida T, Okuyama H, Endo H and Inoue M:
Spheroid cultures of primary urothelial cancer cells: Cancer
tissue-originated spheroid (CTOS) method. Methods Mol Biol.
1655:145–153. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miura A, Honma R, Togashi T, Yanagisawa Y,
Ito E, Imai J, Isogai T, Goshima N, Watanabe S and Nomura N:
Differential responses of normal human coronary artery endothelial
cells against multiple cytokines comparatively assessed by gene
expression profiles. FEBS Lett. 580:6871–6879. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Okabe N, Ezaki J, Yamaura T, Muto S, Osugi
J, Tamura H, Imai J, Ito E, Yanagisawa Y, Honma R, et al: FAM83B is
a novel biomarker for diagnosis and prognosis of lung squamous cell
carcinoma. Int J Oncol. 46:999–1006. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Higa A, Hoshi H, Yanagisawa Y, Ito E,
Morisawa G, Imai JI, Watanabe S and Takagi M: Evaluation system for
arrhythmogenic potential of drugs using human-induced pluripotent
stem cell-derived cardiomyocytes and gene expression analysis. J
Toxicol Sci. 42:755–761. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ito M, Hiramatsu H, Kobayashi K, Suzue K,
Kawahata M, Hioki K, Ueyama Y, Koyanagi Y, Sugamura K, Tsuji K, et
al: NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse
model for engraftment of human cells. Blood. 100:3175–3182. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chijiwa T, Kawai K, Noguchi A, Sato H,
Hayashi A, Cho H, Shiozawa M, Kishida T, Morinaga S, Yokose T, et
al: Establishment of patient-derived cancer xenografts in
immunodeficient NOG mice. Int J Oncol. 47:61–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pearson AT, Finkel KA, Warner KA, Nör F,
Tice D, Martins MD, Jackson TL and Nör JE: Patient-derived
xenograft (PDX) tumors increase growth rate with time. Oncotarget.
7:7993–8005. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhang JH, Chung TD and Oldenburg KR: A
simple statistical parameter for use in evaluation and validation
of high throughput screening assays. J Biomol Screen. 4:67–73.
1999. View Article : Google Scholar : PubMed/NCBI
|