Introduction
Cardiac arrest is a leading cause of mortality and
disability worldwide (1).
Approximately half of the survivors of cardiac arrest experience
impairments in memory, attention and executive functioning
(2-4).
These cognitive deficits are considered to be primarily due to
global cerebral ischemia (GCI) following cardiac arrest, which may
induce delayed neuronal cell death in the vulnerable hippocampal
CA1 region (5-8).
Unfortunately, the treatment options for cardiac arrest-induced
neurological impairments are currently limited. A study by Harukuni
and Bhardwaj reported that therapeutic hypothermia may have a
neurological benefit in cardiac arrest (9), but a recent research failed to confirm
any significant benefit on survival or neurological outcome
(10). Thus, there is a clear need
to identify new potential therapies for reducing brain damage after
GCI.
Autophagy plays an important role in neuronal death
induced by ischemic brain injury (11-13).
Autophagy has been reported to protect neurons from apoptosis, both
in a cerebral ischemia model and an oxygen-glucose deprivation
mouse model (14,15). Another research demonstrated that
autophagy contributed to neuron survival by suppressing apoptosis
in a middle cerebral artery occlusion (MCAO) stroke rat model
(16). Additionally, Wang et
al demonstrated that deletion of β-arrestin-1, a vital
scaffolding protein that interacts with beclin-1 and Vps34 forming
a pro-autophagic complex in neurons, hampered autophagosome
formation and enhanced neuronal apoptosis (17).
Silent information regulator transcript-1 (SIRT1) is
a nicotinamide adenosine dinucleotide (NAD+)-dependent
deacetylase enzyme (18,19). SIRT1 regulates the activity of
downstream target genes through deacetylation (20,21).
Studies have demonstrated that SIRT1 can attenuate neuronal death
and improve cognitive function in mice, indicating that it may also
be a novel target for neuroprotection (22,23).
EX527 is a potent and selective inhibitor of SIRT1 that exerts
different effects in experimental models of diseases and disorders.
EX527 has been shown to exert beneficial effects on cerebral
ischemia-reperfusion injury and acute lung injury (24,25).
The Forkhead box O (FoxO) transcription factor family consists of
four members: FoxO1, FoxO3, FoxO4 and FoxO6(26), among which FoxO1 is widely expressed
in brain tissue and may be activated by SIRT1 (27,28).
It has been observed that the SIRT1/FoxO1 axis regulates autophagy
and promotes phagocytosis of abnormal organelles and proteins
(29). When SIRT1 was silenced,
autophagy was inhibited and neuron apoptosis was enhanced (30). These data revealed that the
SIRT1/FoxO1 axis is a key regulator of neuron autophagy.
It has been established that hypoxia
postconditioning (HPC) may trigger endogenous mechanisms to protect
the heart and brain from ischemia-reperfusion injury. Wang et
al demonstrated that HPC can significantly reduce the
ischemia-reperfusion injury of the heart (31). Similarly, Albrecht et al
observed that HPC reduced brain injury caused by perinatal asphyxia
(32). Additionally, Nguyen et
al reported that HPC reduced brain injury in a MCAO rat model,
which indicated a novel approach to the treatment of MCAO-induced
brain injury (33). Recent studies
have demonstrated that treatment with 8% oxygen for 120 min applied
1 day after ischemia effectively ameliorated neuronal death induced
by GCI in the hippocampal CA1 subregion (34,35).
The neuroprotective mechanism induced by HPC after GCI, which may
be associated with a variety of intracellular signal transduction
pathways, has been investigated, but has yet to be fully
elucidated.
The aim of the present study was to examine the
efficacy of HPC as a potential novel treatment for brain injury
following GCI and to explore the potential mediators through which
HPC activates the SIRT1/FoxO1 signaling pathway to promote the
survival of neurons in the hippocampal CA1 region after GCI. It was
also investigated whether the upregulation of autophagy induced by
HPC was caused by the activation of the SIRT1/FoxO1 signaling
pathway.
Materials and methods
Animals and groups
Adult male Sprague-Dawley rats (250-300 g body
weight, Animal Center Laboratory of North China University of
Science and Technology) were housed in an animal care facility
under laboratory conditions of 22-25˚C, 50-60% humidity and a 12-h
light/dark cycle. The rats were given free access to water and food
and were allowed to acclimate for 1 week prior to the experimental
procedures. All procedures used in the present study were approved
by the Institutional Animal Care and Use Committee of North China
University of Science and Technology (no. 2015-99), and were
conducted in accordance with the guidelines of the National
Institutes of Health of the United State of America for animal
research. In the present study, as improvement of GCI was an
endpoint, determining when the animals should be euthanized was
key. Following the study design, the rats were sacrificed under
anesthesia once they demonstrated inactivity, difficulty in
drinking or eating or abnormal behaviors. Before euthanasia,
surgical plane of anesthesia was confirmed by absence of the
toe-pinch reflex. Following euthanasia, death was confirmed by
checking for absence of heartbeat and respiration.
All rats were randomly divided into five groups
(n=12/group) as follows: i) Sham; ii) GCI; iii) GCI + HPC; iv) GCI
+ HPC + DMSO; and v) GCI + HPC + EX527 groups. EX527 is a potent
and selective inhibitor of SIRT1 that exerts different effects in
experimental models of diseases and disorders (36). One day prior to the surgery, rats in
the GCI + HPC + DMSO and GCI + HPC + EX527 groups underwent lateral
ventricle catheterization, after which time 50 µl DMSO or EX527 (1
mmol/ml) were slowly injected into the lateral ventricle (2
µl/min), leaving the needle in for 5 min. With the exception of the
sham group, all rats underwent the GCI operation. All rats were
weighed daily. One day after the surgery, there were 12 rats in the
sham group, 10 in the GCI, 10 in the GCI + HPC, 11 in the GCI + HPC
+ DMSO and 10 in the GCI + HPC + EX527 group. A total of 7 rats
were excluded from the study due to death after the operation. The
duration of the experiment was 2 weeks.
GCI model
The four-vessel occlusion rat model of GCI was
constructed as previously described (36). The rats were fasted for 10 h and
were not allowed to drink water for 4 h before the operation.
Briefly, after anesthetizing rats with 40 mg/kg sodium
pentobarbital (Diamondback Drugs) by intraperitoneal injection,
both vertebral arteries were permanently electrocauterized with an
electrocautery needle. Subsequently, the bilateral common carotid
arteries (CCAs) were exposed and isolated. After 24 h, the CCAs
were exposed and occluded with aneurysm clips for 12 min to induce
ischemia. Successful cerebral ischemia was ensured by monitoring
the loss of righting reflex, dilated pupils and unresponsiveness to
light during GCI. Following the procedure, arterial blood flow was
confirmed before the wound was closed with tissue glue. The body
temperature of the animals was maintained within the range of
36.5-37.5˚C during surgery with a baking lamp. For sham-operated
animals, all procedures were performed exactly as for ischemic
animals, except that the CCAs were not occluded. After modeling, no
rats exhibited behavioral defects.
Prior to surgery, healthy animals were observed and
assessed twice daily (8:00 a.m. and 8:00 p.m.). During the
anesthesia recovery period, the animals were observed continuously
until they were able to move, drink and eat independently. After
surgery, the animals were observed and assessed every 4 h to
determine whether painkiller administration was required if they
were suffering from pain and distress.
HPC protocols
After GCI surgery, the rats were placed into a
sealed plastic chamber (BioSpherix, Ltd.), through which air
containing 8% O2 and 92% N2 (normal air
composition ratio: 21% O2 and 78% N2) flowed
continuously via a Pro-Ox oxygen controller for 2 h per day for 3
days. The gas mixture flow was maintained at 200 ml/min and no more
than 4 rats were placed into the chamber at any given time
(37). Sham group rats were placed
in the chamber with no hypoxia applied.
Morris water maze (MWM) tests
MWM testing was performed in a circular pool
(diameter, 180 cm; height, 60 cm; depth, 35 cm) filled with opaque
water by stirring in milk (temperature, 26±1˚C) to evaluate
cognitive function in rats. Briefly, the rats were placed in the
water facing the wall at random in one of the four quadrants (I,
II, III and IV), located equidistant from one another around the
rim of the pool. For each trial, the rat was given a maximum of 90
sec to locate the hidden platform; any rat that failed the mission
within 90 sec would be guided to the hidden platform and allowed to
stay on the platform for 15 sec before the training was terminated.
The procedure was repeated from each of the four start locations,
within a 4-h interval. The escape latency, representing the average
time required to reach the submerged platform, was recorded. The
training tests were administered on days 1, 3, 5 and 7 after GCI.
Additionally, probe trials were performed 4 h after the last
training session. During the probe trial, the platform was removed
from the tank and each rat was allowed to swim freely for 90 sec.
The number of platform crossings (the number of times passing
through the previous location of the platform) was used to evaluate
the level of spatial reference memory. Each rat was placed in the
pool at the same random start location. All behavioral tracks of
the trials were recorded and analyzed using video tracking software
(Huaibeizhenghua Biological Equipment Co., Ltd.).
Brain tissue and hematoxylin and eosin
(H&E) staining
Rats were administered an overdose of 4% sodium
pentobarbital (200 mg/kg) by intraperitoneal injection, and were
then transcardially perfused with normal saline (0.9%) followed by
4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.3). The
brain was then dissected, postfixed with 4% paraformaldehyde at 4˚C
for 3 days, embedded in paraffin, cut into 5-µm sections and
stained with H&E. The population of necrotic neurons in the CA1
subfield was counted using a light microscope (BX53; Olympus
Corporation) at x400 magnification by two independent investigators
who were blinded to the experimental conditions. The mean number of
apoptotic neurons was obtained by counting 3 sections per brain and
5 representative fields were randomly selected per section.
Immunohistochemistry staining
Coronal brain sections were prepared as described
above. Immunofluorescence staining was performed following a
standard protocol as described previously (34). Briefly, the sections were first
treated with 3% hydrogen peroxide for 30 min, followed by 5% normal
serum for 1 h, and they were then incubated overnight at 4˚C with
primary antibodies, including LC3-II (1:300; cat. no. 2275; Cell
Signaling Technology, Inc.). Immunopositive cells in which the
reaction product was present in the cytoplasmic or nuclear border
were quantified under a light microscope at x400 magnification. The
number of immunopositive cells was counted in a total of 4
non-repeated random fields (0.037 mm2/field x4=0.148
mm2 in total) in the CA1 subregions. Data were
quantified bilaterally in 4 sections from each rat and assessed in
a double-blinded manner.
Sample preparation and western blot
analysis
As described previously (38), both hippocampi were rapidly removed
from animals under deep anesthesia. The hippocampal CA1 regions
were quickly microdissected on an ice pad and immediately frozen in
liquid nitrogen. The rat hippocampus was homogenized in RIPA lysis
buffer (cat. no. P0013B; Beyotime Institute of Biotechnology)
containing PMSF, followed by centrifugation at 12,000 x g for 10
min at 4˚C. Protein concentrations were determined using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology). A total of 30 µg of protein per lane was separated
by 12% SDS-PAGE and transferred onto PVDF membranes. The membranes
were blocked with 3% BSA (Sigma-Aldrich; Merck KGaA) in
Tris-buffered saline at room temperature for 30 min and incubated
overnight at 4˚C with primary antibodies against mammalian target
(LC3-II; 1:200; cat. no. 3868; Cell Signaling Technology, Inc.),
SIRT1 (1:500; cat. no. 9475; Cell Signaling Technology, Inc.),
FoxO1 (1:500; cat. no. 2880; Cell Signaling Technology, Inc.) or
actin (1:1,000, cat. no. 4967; Cell Signaling Technology, Inc.),
followed by incubation with horseradish-peroxidase conjugated
anti-rabbit IgG (1:2,000; cat. no. 5127; Cell Signaling Technology,
Inc.) for 1 h at room temperature. Protein signals were detected
using an enhanced chemiluminescence system (EMD Millipore) and
quantified using Quantity-One software, version 4.6.3 (Bio-Rad
Laboratories, Inc.). Actin was used as a protein-loading
control.
Statistical analysis
IBM SPSS 22.0 (IBM Corp.) and GraphPad Prism 8.0
(GraphPad Software, Inc.) were used to analyze the data. All data
are expressed as mean ± standard deviation and differences between
three or more groups were analyzed by one-way ANOVA. Repeated
measures ANOVA was used to compare the weight loss and cognitive
results. Tukey's post hoc test was applied where there was a
significant difference. P<0.05 was considered to indicate
statistically significant differences.
Results
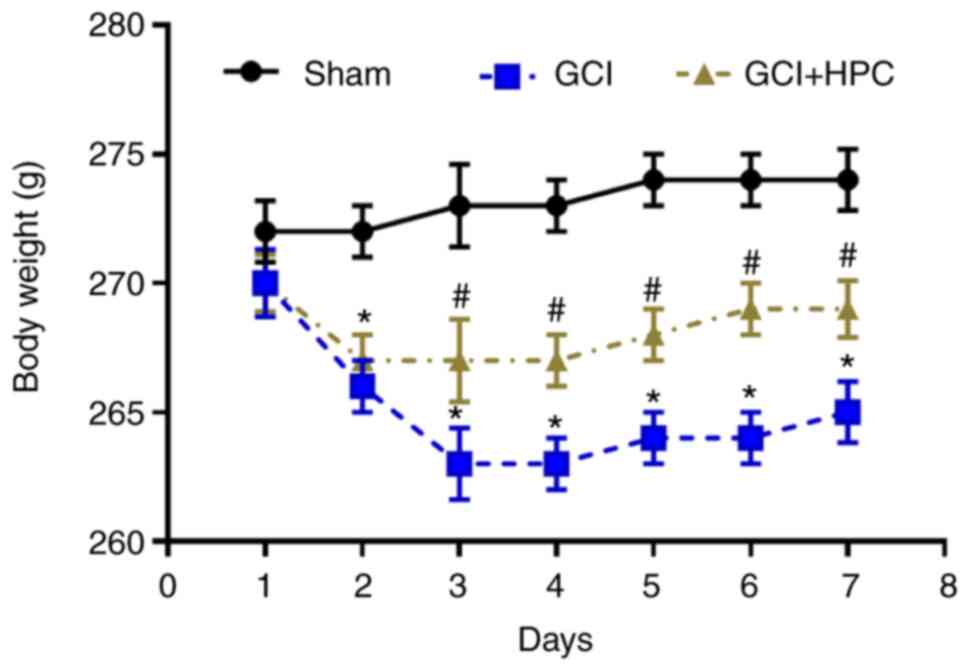
HPC ameliorates GCI induced weight
loss in rats
Rats in the GCI group exhibited more notable weight
loss compared with those in the sham group (P<0.05); however,
HPC attenuated this tendency in rats with GCI at 2 days after
surgery (P<0.05; Fig. 1).
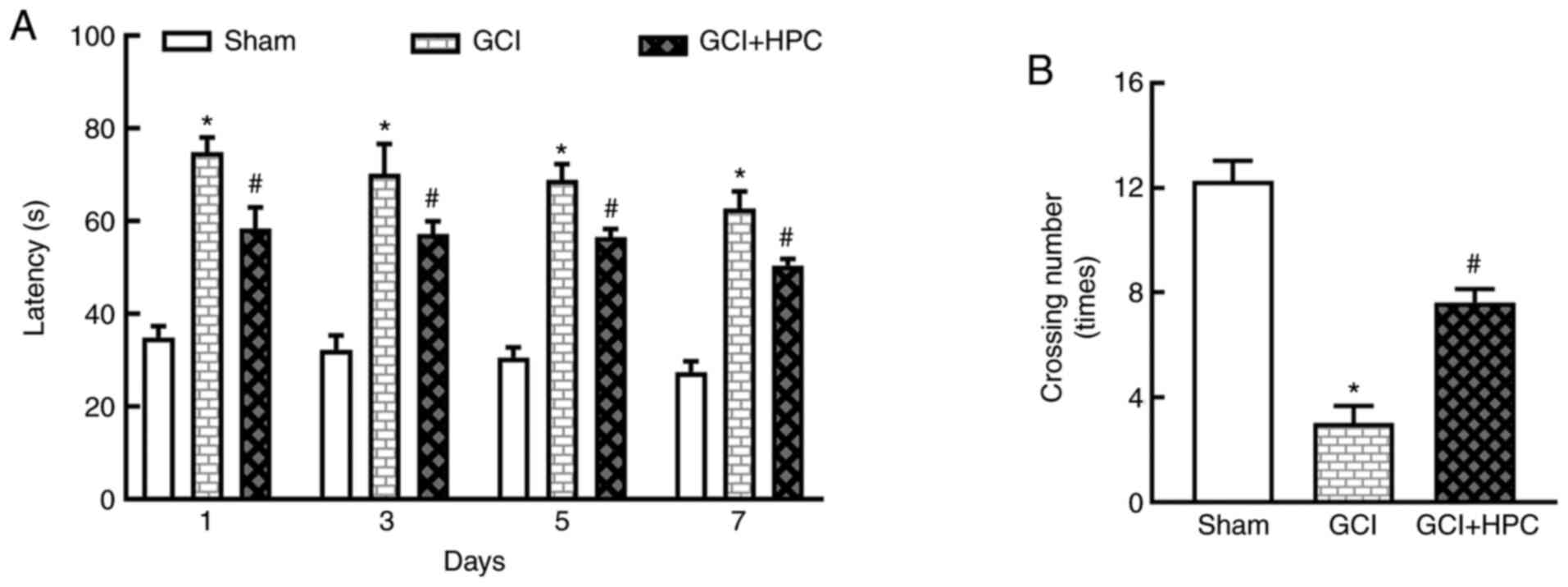
HPC ameliorates GCI induced cognitive
function decline in rats
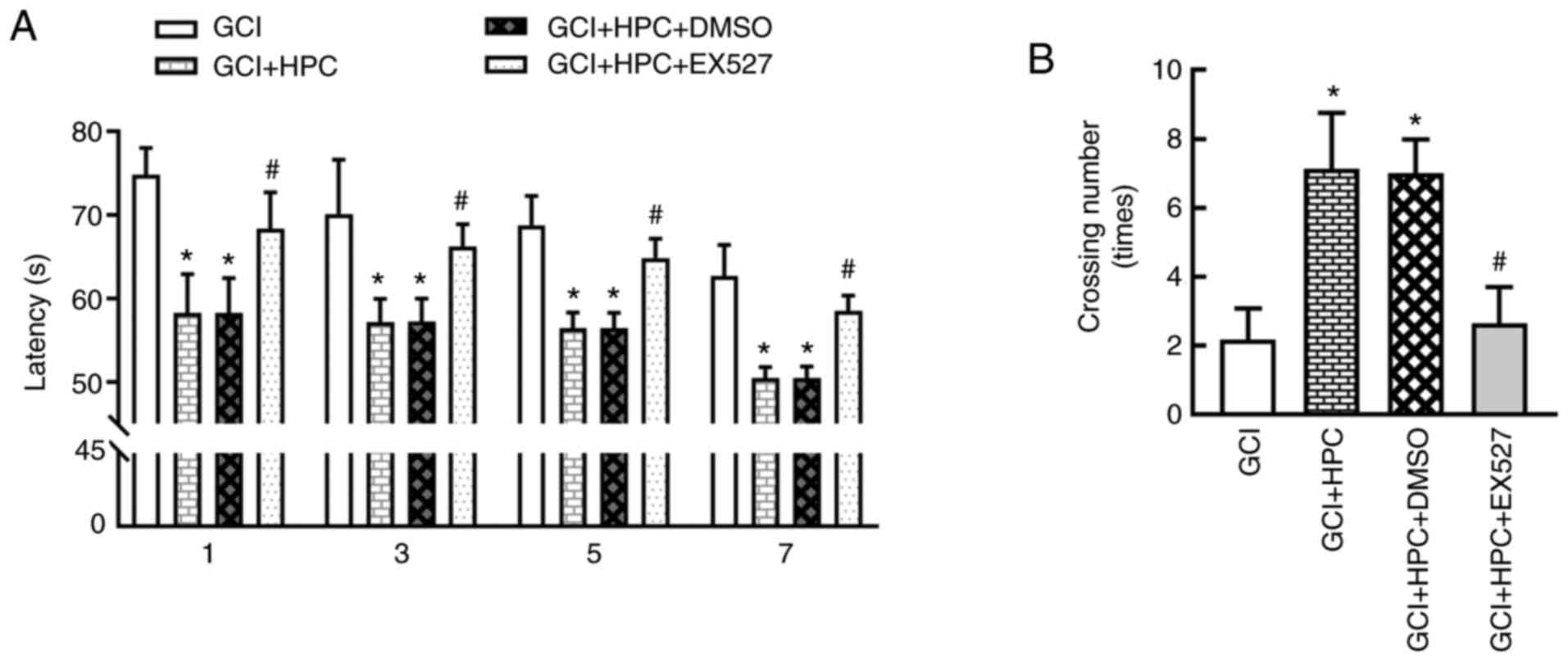
Cognitive function was determined using the MWM test
and statistical analysis was performed using repeated measures
ANOVA. During training, significant differences in performance were
observed. Rats in the GCI group exhibited a longer escape latency
compared with those in the sham group (P<0.05), and the escape
latency was shortened by treatment with HPC (P<0.05; Fig. 2A). Repeated measures ANOVA revealed
that the total effect was not statistically significant (F=2.874,
P=0.1666); however, the groups effect was significant (F=918.3,
P=0.000). In the probe test, the number of platform crossings was
significantly lower in the GCI group compared with that in the sham
group (P<0.05). HPC increased the number of platform crossings
compared with the GCI group (P<0.05; Fig. 2B). These results indicated that HPC
ameliorated the impairment of cognitive function induced by
ischemia.
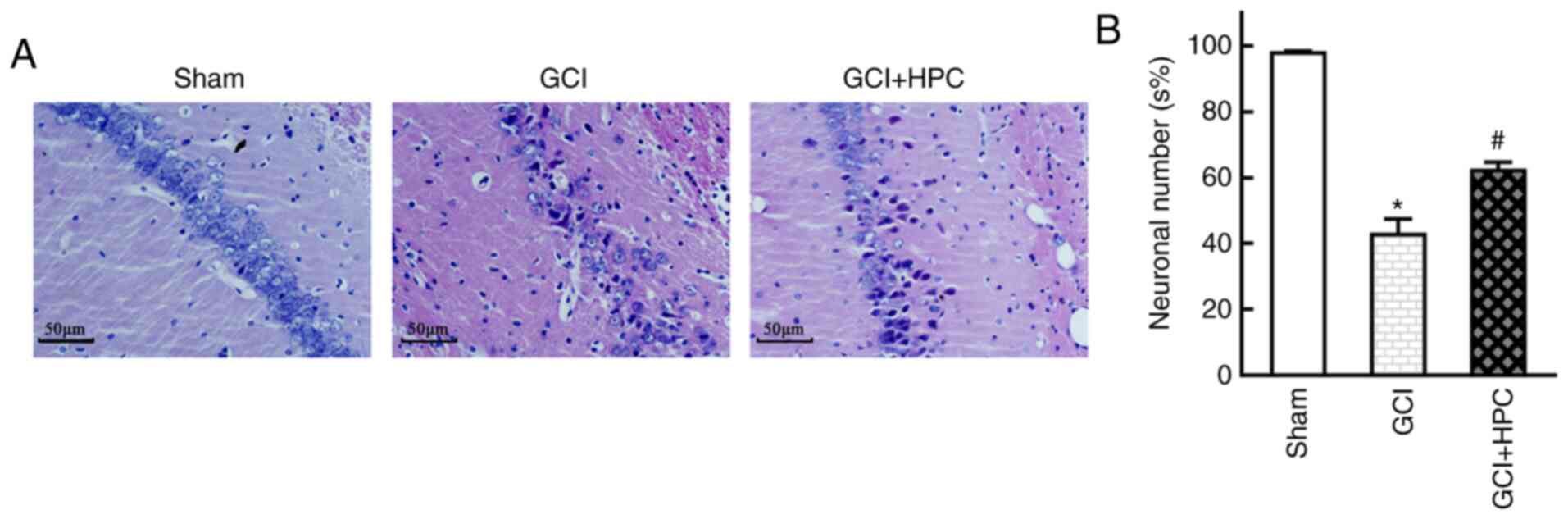
HPC promotes neuronal survival in rats
with GCI
Histopathological abnormalities were observed in the
hippocampus of the rats, and GCI group rats exhibited marked
neuronal loss and degeneration of neuronal structure in the CA1
region. HPC attenuated this neuronal loss and reversed the
structural injury, as reflected by the number and morphology of
neurons on HE staining examination (P<0.05). These results
suggested that HPC inhibited hippocampal neuron death in rats with
GCI (Fig. 3).
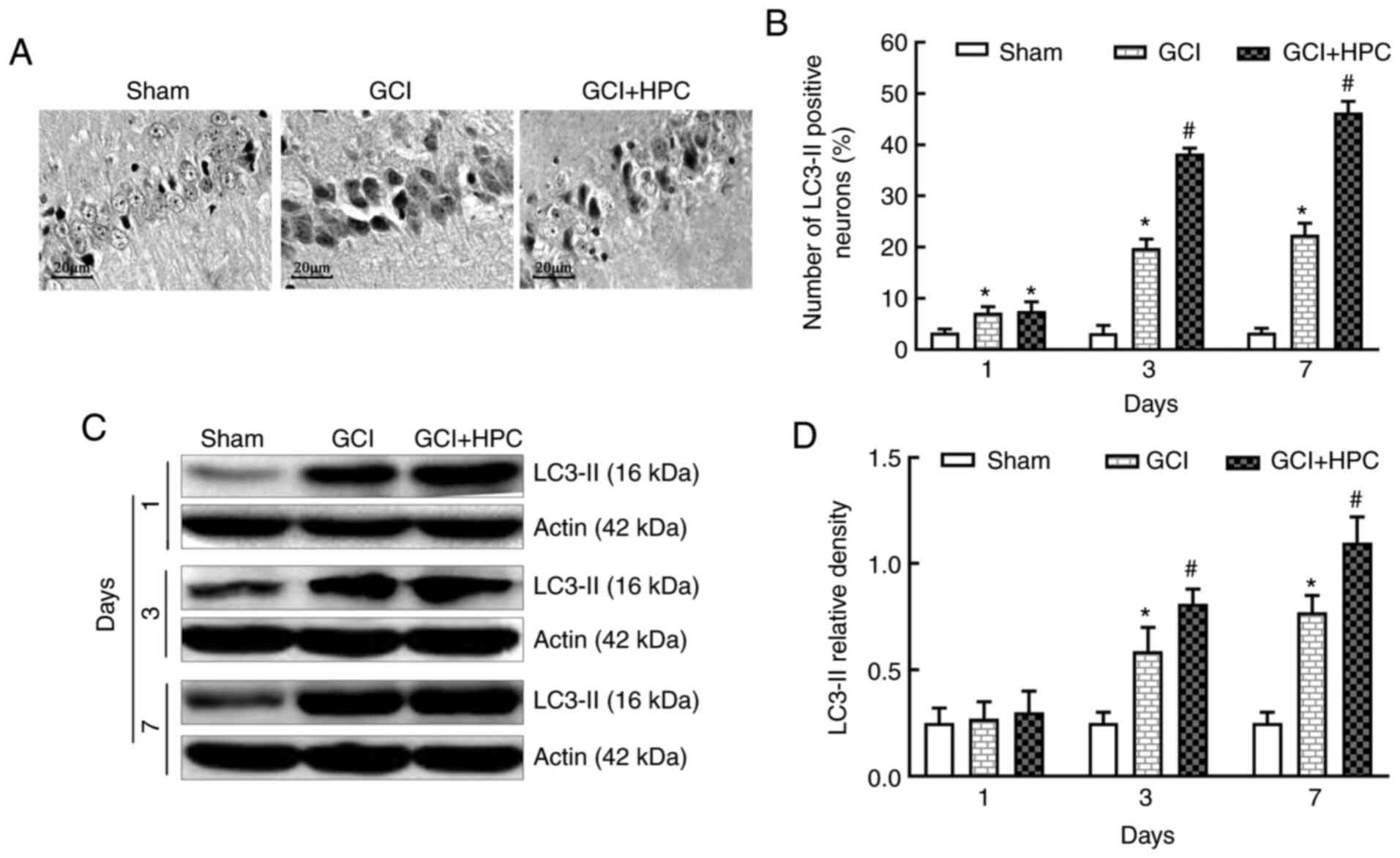
HPC increases autophagy in the
hippocampal CA1 area in rats with GCI
To identify autophagy, the expression of the
autophagy marker protein, LC3-II, was detected in the hippocampus
using immunohistochemistry and western blotting at 1, 3 and 7 days
after GCI surgery. Fewer positive cells were visible in the
hippocampal CA1 region of the rats in the sham group under an
optical microscope (Fig. 4). By
contrast, there were abundant positive cells in the GCI group.
Furthermore, the number of LC3-II positive cells was significantly
increased in the GCI + HPC group compared with that in the GCI
group (P<0.05), which indicated that autophagy was activated
further. Low levels of LC3-II were present in the hippocampal CA1
region of the rats in the sham group. However, the expression
levels of LC3-II were significantly increased in the GCI group
(P<0.05) and increased further in the GCI + HPC group
(P<0.05). These results suggested that HPC upregulated autophagy
in rats with GCI (Fig. 4).
HPC upregulates autophagy via a
SIRT1/FoxO1-dependent pathway in rats with GCI
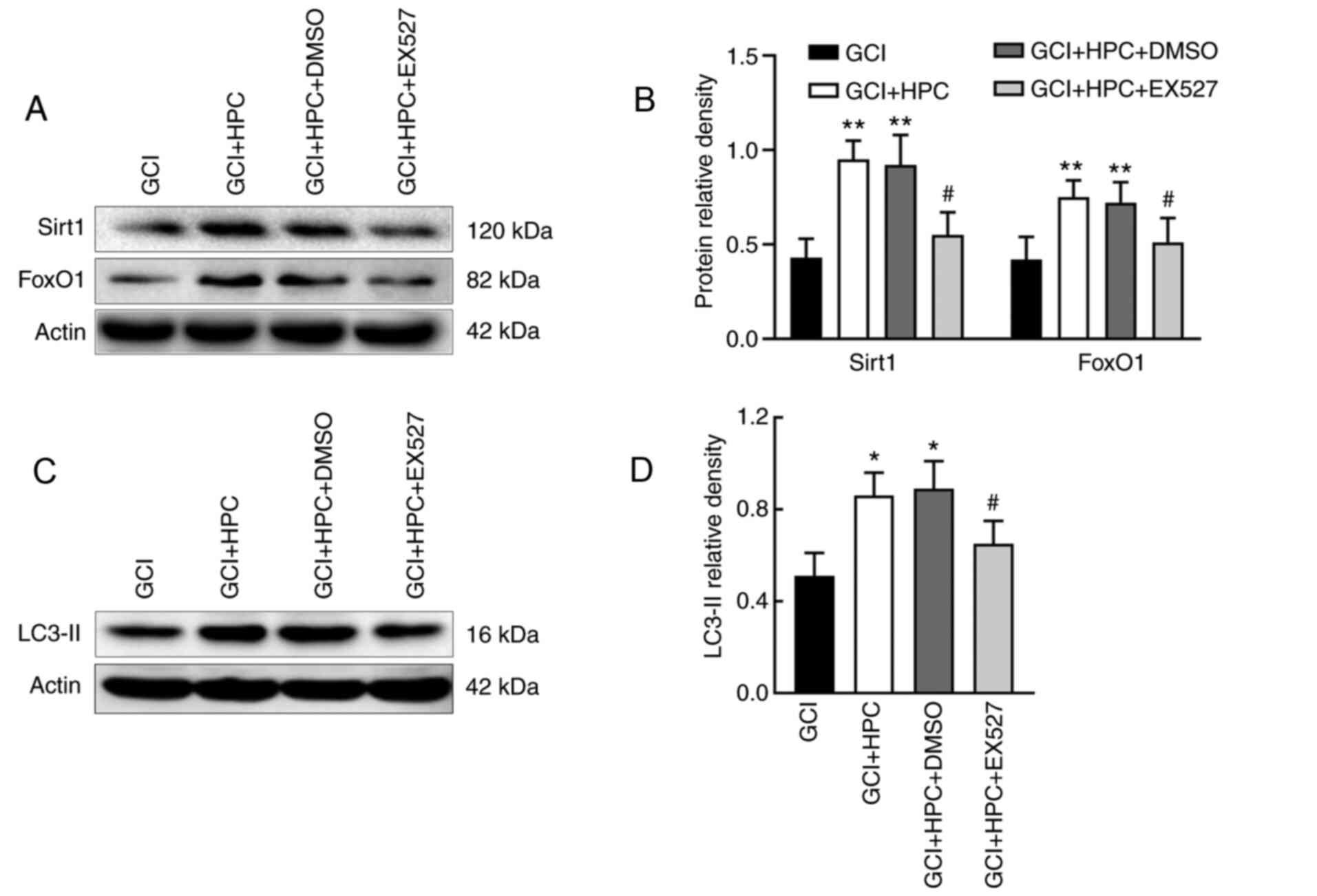
The role of the SIRT1/FoxO1 axis in GCI-induced
autophagy was assessed by measuring the relative protein levels of
SIRT1 and FoxO1 in the four groups. EX527, a SIRT1 inhibitor, was
able to reduce the expression levels of SIRT1 and FoxO1
(P<0.05); it simultaneously decreased the expression levels of
LC3-II in GCI rats compared with those in GCI + HPC + DMSO group
rats (P<0.05; Fig. 5). These
results indicated that HPC may upregulate autophagy through a
SIRT1/FoxO1-dependent pathway in rats with GCI.
HPC ameliorates cognitive function via
a SIRT1/FoxO1-dependent pathway in rats with GCI
To explore the mechanism through which HPC mitigates
GCI-induced cognitive function injury, the SIRT1 inhibitor, EX527,
was introduced, and cognitive function was determined using the MWM
test. As shown in Fig. 6, EX527
reversed the decrease in escape latency and the increase in the
number of platform crossings induced by HPC. Repeated measures
ANOVA revealed that the total effect was not statistically
significant (F=3.483, P=0.1438); however, the groups effect was
significant (F=325.3, P=0.001). These results indicated that HPC
ameliorated cognitive function via a SIRT1/FoxO1-dependent pathway
in rats with GCI.
Discussion
Similar to humans, experimental GCI induced in
animal models has been shown to result in selective and delayed
neuronal death of pyramidal neurons in the hippocampal CA1 region,
as well as cognitive impairment; therefore, experimental GCI in
animals has become a valuable model for the study of GCI (39,40).
The present study demonstrated that the cognitive function of rats
was impaired and weight loss was obvious after GCI. GCI group rats
exhibited marked neuronal loss and degeneration of neuronal
structure in the CA1 region, whereas HPC was able to alleviate
these changes. These results are similar to those of earlier
studies reporting that HPC treatment may confer neuroprotection
against brain ischemia or injury (35,41-43).
In addition, by measuring the expression of the autophagy marker
protein LC3-II in the hippocampus, it was observed that there were
more positive cells in the hippocampal CA1 region of the GCI group
compared with that of the sham group. Interestingly, HPC can
upregulate the expression of LC3-II. Further studies have found
that HPC can upregulate autophagy by activating the SIRT1/FoxO1
signaling pathway, thereby reducing neuronal death and ameliorating
cognitive function impairment. Taken together, all the
aforementioned findings indicate that HPC treatment may represent a
new therapeutic modality for multiple neurological disorders.
Autophagy is a highly regulated process that
involves the degradation of cytoplasmic macromolecules and
organelles. In mammalian cells, this catabolic mechanism utilizes
the lysosomal system and has a homeostatic function in normal cell
growth and development, helping to maintain a balance between the
synthesis, degradation and subsequent recycling of cellular
products (44). Autophagy is a
programmed and physiologically conserved self-degradation process
(45) and disruption of autophagy
may have detrimental consequences. It was previously demonstrated
that autophagy prevents neuronal death in response to GCI (46). In the present study, a specific
marker for autophagosomes, LC3-II, was used to test whether
autophagic activity can be induced by HPC treatment. As shown in
Fig. 4, the expression of LC3-II
was markedly elevated following GCI, and this phenomenon was
enhanced by HPC treatment, which suggests that HPC treatment
effectively suppresses GCI-induced neuronal death in the
hippocampal CA1 region by further activating autophagy. Recently,
it was also demonstrated that pre-treatment with inhibitors of
autophagy markedly increased the infarct area in neonatal and adult
rats with MCAO (38). Therefore, it
may be hypothesized that GCI activates neuronal autophagy, but this
is not sufficient to cause the self-digestion of damaged organelles
and to induce neuronal death, which is particularly crucial during
neurodegeneration. Furthermore, the neuroprotective effect of HPC
may be associated with further upregulation of GCI-induced neuronal
autophagy in the hippocampal CA1 region.
As previous studies have demonstrated that various
signaling pathways contribute to neuronal autophagy, it was
inferred that neuronal autophagy may be the downstream target of
SIRT1/FoxO1 in GCI rats (47,48).
SIRT1 overexpression may promote autophagy of neurons by regulating
FoxO1-mediated target genes, which eventually results in cell
autophagy, indicating that the SIRT1/FoxO1 axis may be one of the
pathways involved in neuroprotection (49). In the present study, western blot
analysis demonstrated that SIRT1 expression was upregulated by HPC
treatment, leading to an increasing expression level of FoxO1. The
increased expression of SIRT1, FoxO1 and LC3-II was markedly
rescued by treatment with a selective SIRT1 inhibitor.
Surprisingly, the ability of HPC to upregulate autophagy was
suppressed, and its ability to improve cognitive function was also
inhibited. These results suggest that SIRT1/FoxO1 may be a key
pathway involved in HPC-induced neuron autophagy. This is in
accordance with the findings of Jiang et al (50), who also demonstrated that FoxO1 can
upregulate autophagy to reduce neuron death in vascular dementia
rats. In addition, Wu et al (51) also reported that SIRT1 upregulated
the deacetylation of FoxO1 and promoted its nuclear translocation,
thus increasing cell autophagy to resist ischemia-induced damage.
These findings suggest that deacetylation of FoxO1 by activated
SIRT1 is one of the mechanisms through which HPC exerts its
effects. Although it has been demonstrated that SIRT1 activation is
a promising candidate for treating or preventing several diseases
and disorders, some studies suggest that SIRT1 inhibition may have
beneficial effects on some pathological conditions. For example,
Nikseresht et al (24)
demonstrated that the mortality and cerebral infarct volume of rats
can be reduced in a focal model of cerebral ischemia-reperfusion by
using EX527 to inhibit SIRT1. In addition, Jiang et al also
demonstrated that inhibiting SIRT1 may conserve cellular
NAD+ levels and protect neurons against ischemic damage.
Additionally, SIRT1 may play a dual role, protective as well as
destructive, in Huntington's disease (52,53).
However, the results of the present study suggest that HPC exert
neuroprotective effects by activating the SIRT1/FoxO1 pathway.
It has been demonstrated that upregulating the level
of autophagy in neurons can reduce cell death and improve recovery.
HPC has emerged as a viable new therapy for GCI, offering a
non-invasive approach to reducing neurological injury. However,
there are several pathways that mediate autophagy, and the
SIRT1/FoxO1 pathway is only one of them, so it cannot be
definitively concluded that the neuroprotective effects were
entirely due to HPC. Additional studies are needed to confirm the
current results and elucidate the mechanisms underlying the
neuroprotective properties of HPC.
In conclusion, taken together, the data of the
present study indicated that HPC treatment exerts beneficial
effects on neuronal survival and preservation of cognitive function
following GCI, which may be mediated via activation of the
SIRT1/FoxO1 pathway that increases neuronal autophagy. Based on
these beneficial effects, and the fact that HPC is a non-invasive
and cost-effective therapy, HPC may hold promise as a potential
treatment in humans for ameliorating the poor cognitive outcomes
following GCI.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Hebei Medical
Science Research Project, China (grant no. 20190106); the Tangshan
City Science and Technology Project, China (grant no. 18130232a);
and the North China University of Science and Technology Research
and Research Project (grant no. Z201736).
Availability of materials and data
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JL and YZ designed the study; JL, JW, XW and JW
performed the experiments; JX analyzed the data; SL complementary
experiments; JL and ZG wrote the manuscript. All the authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
All procedures used in the present study were
approved by the Institutional Animal Care and Use Committee of
North China University of Science and Technology (no. 2015-99) and
were conducted in accordance with the guidelines of the National
Institutes of Health of the United State of America for animal
research.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jalife J: The tornadoes of sudden cardiac
arrest. Nature. 555:597–598. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yeung J and Moulaert V: Focus on the brain
of the cardiac arrest survivor. Resuscitation. 88:A5–A6.
2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Weihs W, Warenits AM, Ettl F, Magnet IA,
Teubenbacher U, Hilpold A, Schober A, Testori C, Tiboldi A, Mag KT,
et al: Reduced long-term memory in a rat model of 8 minutes
ventricular fibrillation cardiac arrest: A pilot trial. BMC Vet
Res. 12(103)2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Woods D and Chantavarin S: Serial
neuropsychological assessment of an adolescent girl after suffering
a sudden out-of-hospital-cardiac-arrest following recreational
inhalant use. Appl Neuropsychol Child. 6:378–387. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wolman RL, Nussmeier NA, Aggarwal A,
Kanchuger MS, Roach GW, Newman MF, Mangano CM, Marschall KE, Ley C,
Boisvert DM, et al: Cerebral injury after cardiac surgery:
Identification of a group at extraordinary risk. Multicenter Study
of Perioperative Ischemia Research Group (McSPI) and the ischemia
research education foundation (IREF) investigators. Stroke.
30:514–522. 1999.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tsunekawa T, Sawada M, Kato T, Motoji Y,
Kinoshita T, Hirakawa A, Okawa Y and Tomita S: The prevalence and
distribution of occlusive lesions of the cerebral arteries in
patients undergoing coronary artery bypass graft surgery. Semin
Thorac Cardiovasc Surg. 30:413–420. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Ji NN, Wu L, Shao BM, Meng QX, Xu JN, Zhu
HW and Zhang YM: CTL-Derived Granzyme B participates in hippocampal
neuronal apoptosis induced by cardiac arrest and resuscitation in
rats. Front Neurol. 10(1306)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lee D, Pearson T, Proctor JL, Rosenthal RE
and Fiskum G: Oximetry-Guided normoxic resuscitation following
canine cardiac arrest reduces cerebellar Purkinje neuronal damage.
Resuscitation. 140:23–28. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Harukuni I and Bhardwaj A: Mechanisms of
brain injury after global cerebral ischemia. Neurol Clin. 24:1–21.
2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kim YM, Yim HW, Jeong SH, Klem ML and
Callaway CW: Does therapeutic hypothermia benefit adult cardiac
arrest patients presenting with non-shockable initial rhythms? A
systematic review and meta-analysis of randomized and
non-randomized studies. Resuscitation. 83:188–196. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kim KA, Kim D, Kim JH, Shin YJ, Kim ES,
Akram M, Kim EH, Majid A, Baek SH and Bae ON: Autophagy-mediated
occludin degradation contributes to blood-brain barrier disruption
during ischemia in bEnd.3 brain endothelial cells and rat ischemic
stroke models. Fluids Barriers CNS. 17(21)2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chen R, Zhang YY, Lan JN, Liu HM, Li W, Wu
Y, Leng Y, Tang LH, Hou JB, Sun Q, et al: Ischemic postconditioning
alleviates intestinal ischemia-reperfusion injury by enhancing
autophagy and suppressing oxidative stress through the
Akt/GSK-3β/Nrf2 pathway in mice. Oxid Med Cell Longev.
2020(6954764)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Liu Z, Zhang J, Zhang F and Chang Y:
Propofol post-conditioning lessens renal
ischemia/reperfusion-induced acute lung injury associated with
autophagy and apoptosis through MAPK signals in rats. Gene.
741(144562)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Wu B, Luo H, Zhou X, Cheng CY, Lin L, Liu
BL, Liu K, Li P and Yang H: Succinate-induced neuronal
mitochondrial fission and hexokinase II malfunction in ischemic
stroke: Therapeutical effects of kaempferol. Biochim Biophys Acta
Mol Basis Dis. 1863:2307–2318. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wu X, He L, Cai Y, Zhang G, He Y, Zhang Z,
He X, He Y, Zhang G and Luo J: Induction of autophagy contributes
to the myocardial protection of valsartan against ischemia
reperfusion injury. Mol Med Rep. 8:1824–1830. 2013.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lv B, Li F, Han J, Xu L, Sun C, Hua T,
Zhang Z, Feng Z and Jiang X: Hif-1α overexpression improves
transplanted bone mesenchymal stem cells survival in rat MCAO
stroke model. Front Mol Neurosci. 10(80)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu
H, Su DF, Pei G and Miao CY: ARRB1/β-arrestin-1 mediates
neuroprotection through coordination of BECN1-dependent autophagy
in cerebral ischemia. Autophagy. 10:1535–1548. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Deng Z, Li Y, Liu H, Xiao S, Li L, Tian J,
Cheng C, Zhang G and Zhang F: The role of sirtuin 1 and its
activator, resveratrol in osteoarthritis. Biosci Rep.
39(BSR20190189)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cao W, Dou Y and Li A: Resveratrol boosts
cognitive function by targeting SIRT1. Neurochem Res. 43:1705–1713.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Gonfloni S, Iannizzotto V, Maiani E,
Bellusci G, Ciccone S and Diederich M: P53 and Sirt1: Routes of
metabolism and genome stability. Biochem Pharmacol. 92:149–156.
2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hwang JW, Yao H, Caito S, Sundar IK and
Rahman I: Redox regulation of SIRT1 in inflammation and cellular
senescence. Free Radic Biol Med. 61:95–110. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Khan M, Ullah R, Rehman SU, Shah SA, Saeed
K, Muhammad T, Park HY, Jo MH, Choe K, Rutten BPF and Kim MO:
17β-estradiol modulates SIRT1 and halts oxidative stress-mediated
cognitive impairment in a male aging mouse model. Cells.
8(928)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tang X, Zhao Y, Zhou Z, Yan J, Zhou B, Chi
X, Luo A and Li S: Resveratrol mitigates sevoflurane-induced
neurotoxicity by the SIRT1-dependent regulation of BDNF expression
in developing mice. Oxid Med Cell Longev.
2020(9018624)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Nikseresht S, Khodagholi F and Ahmadiani
A: Protective effects of ex-527 on cerebral ischemia-reperfusion
injury through necroptosis signaling pathway attenuation. J Cell
Physiol. 234:1816–1826. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Huang J, Tian R, Yang Y, Jiang R, Dai J,
Tang L and Zhang L: The SIRT1 inhibitor EX-527 suppresses mTOR
activation and alleviates acute lung injury in mice with
endotoxiemia. Innate Immun. 23:678–686. 2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Calnan DR and Brunet A: The FoxO code.
Oncogene. 27:2276–2288. 2008.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yan X, Yu A, Zheng H, Wang S, He Y and
Wang L: Calycosin-7-O-β-D-glucoside attenuates OGD/R-induced damage
by preventing oxidative stress and neuronal apoptosis via the
SIRT1/FOXO1/PGC-1α pathway in HT22 cells. Neural Plast.
2019(8798069)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Susanti VY, Sasaki T, Yokota-Hashimoto H,
Matsui S, Lee YS, Kikuchi O, Shimpuku M, Kim HJ, Kobayashi M and
Kitamura T: Sirt1 rescues the obesity induced by insulin-resistant
constitutively-nuclear FoxO1 in POMC neurons of male mice. Obesity
(Silver Spring). 22:2115–2119. 2014.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Fan L, Chen D, Wang J, Wu Y, Li D and Yu
X: Sevoflurane ameliorates myocardial cell injury by inducing
autophagy via the deacetylation of LC3 by SIRT1. Anal Cell Pathol
(Amst). 2017(6281285)2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang Z, Li D, Xu L and Li HP: Sirt1
improves functional recovery by regulating autophagy of astrocyte
and neuron after brain injury. Brain Res Bull. 150:42–49.
2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang Y, Hao Y, Zhang H, Xu L, Ding N, Wang
R, Zhu G, Ma S, Yang A, Yang Y, et al: DNA hypomethylation of
miR-30a mediated the protection of hypoxia postconditioning against
aged cardiomyocytes hypoxia/reoxygenation injury through inhibiting
autophagy. CIRC J. 84:616–625. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Albrecht M, Zitta K, Groenendaal F, van
Bel F and Peeters-Scholte C: Neuroprotective strategies following
perinatal hypoxia-ischemia: Taking aim at NOS. Free Radic Biol Med.
142:123–131. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Nguyen HL, Ruhoff AM, Fath T and Jones NM:
Hypoxic postconditioning enhances functional recovery following
endothelin-1 induced middle cerebral artery occlusion in conscious
rats. Exp Neurol. 306:177–189. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhan L, Li D, Liang D, Wu B, Zhu P, Wang
Y, Sun W and Xu E: Activation of Akt/FoxO and inactivation of
MEK/ERK pathways contribute to induction of neuroprotection against
transient global cerebral ischemia by delayed hypoxic
postconditioning in adult rats. Neuropharmacology. 63:873–882.
2012.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhu P, Zhan L, Zhu T, Liang D, Hu J, Sun
W, Hou Q, Zhou H, Wu B, Wang Y and Xu E: The roles of p38 MAPK/MSK1
signaling pathway in the neuroprotection of hypoxic
postconditioning against transient global cerebral ischemia in
adult rats. Mol Neurobiol. 49:1338–1349. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tu J, Zhang X, Zhu Y, Dai Y, Li N, Yang F,
Zhang Q, Brann DW and Wang R: Cell-permeable peptide targeting the
Nrf2-Keap1 Interaction: A potential novel therapy for global
cerebral ischemia. J Neurosci. 35:14727–14739. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Rybnikova E, Vorobyev M, Pivina S and
Samoilov M: Postconditioning by mild hypoxic exposures reduces rat
brain injury caused by severe hypoxia. Neurosci Lett. 513:100–105.
2012.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhang HL, Xu M, Wei C, Qin AP, Liu CF,
Hong LZ, Zhao XY, Liu J and Qin ZH: Neuroprotective effects of
pioglitazone in a rat model of permanent focal cerebral ischemia
are associated with peroxisome proliferator-activated receptor
gamma-mediated suppression of nuclear factor-κB signaling pathway.
Neuroscience. 176:381–395. 2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kirino T and Sano K: Selective
vulnerability in the gerbil hippocampus following transient
ischemia. ACTA Neuropathol. 62:201–208. 1984.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chen J, Zhu RL, Nakayama M, Kawaguchi K,
Jin K, Stetler RA, Simon RP and Graham SH: Expression of the
apoptosis-effector gene, Bax, is upregulated in vulnerable
hippocampal CA1 neurons following global ischemia. J Neurochem.
67:64–71. 1996.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhu T, Zhan L, Liang D, Hu J, Lu Z, Zhu X,
Sun W, Liu L and Xu E: Hypoxia-inducible factor 1α mediates
neuroprotection of hypoxic postconditioning against global cerebral
ischemia. J Neuropathol Exp Neurol. 73:975–986. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Solevag AL, Schmolzer GM and Cheung PY:
Novel interventions to reduce oxidative-stress related brain injury
in neonatal asphyxia. Free Radic Biol Med. 142:113–122.
2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Wolf MS, Bayir H, Kochanek PM and Clark R:
The role of autophagy in acute brain injury: A state of flux?
Neurobiol Dis. 122:9–15. 2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Mizushima N: Autophagy: Process and
function. Genes Dev. 21:2861–2873. 2007.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Baehrecke EH: Autophagy: Dual roles in
life and death? Nat Rev Mol Cell Biol. 6:505–510. 2005.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Ryan F, Khodagholi F, Dargahi L,
Minai-Tehrani D and Ahmadiani A: Temporal pattern and crosstalk of
necroptosis markers with autophagy and apoptosis associated
proteins in ischemic hippocampus. Neurotox Res. 34:79–92.
2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Marzetti E, Calvani R, Cesari M, Buford
TW, Lorenzi M, Behnke BJ and Leeuwenburgh C: Mitochondrial
dysfunction and sarcopenia of aging: From signaling pathways to
clinical trials. Int J Biochem Cell Biol. 45:2288–2301.
2013.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Martinez MA, Rodriguez JL, Lopez-Torres B,
Martínez M, Martínez-Larrañaga MR, Maximiliano JE, Anadón A and
Ares I: Use of human neuroblastoma SH-SY5Y cells to evaluate
glyphosate-induced effects on oxidative stress, neuronal
development and cell death signaling pathways. Environ Int.
135(105414)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lim CJ, Lee YM, Kang SG, Lim HW, Shin KO,
Jeong SK, Huh YH, Choi S, Kor M, Seo HS, et al: Aquatide activation
of SIRT1 reduces cellular senescence through a
SIRT1-FOXO1-autophagy axis. Biomol Ther (Seoul). 25:511–518.
2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jiang X, Niu XL, Guo Q, et al:
FoxO1-mediated autophagy plays an important role in the
neuroprotective effects of hydrogen in a rat model of vascular
dementia. Behav Brain Res. 356:98–106. 2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wu B, Feng JY, Yu LM, Wang YC, Chen YQ,
Wei Y, Han JS, Feng X, Zhang Y, Di SY, et al: Icariin protects
cardiomyocytes against ischaemia/reperfusion injury by attenuating
sirtuin 1-dependent mitochondrial oxidative damage. Br J Pharmacol.
175:4137–4153. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Jiang M, Wang J, Fu J, Du L, Jeong H, West
T, Xiang L, Peng Q, Hou Z, Cai H, et al: Neuroprotective role of
Sirt1 in mammalian models of Huntington's disease through
activation of multiple Sirt1 targets. Nat Med. 18:153–158.
2011.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Süssmuth SD, Haider S, Landwehrmeyer GB,
Farmer R, Frost C, Tripepi G, Andersen CA, Di Bacco M, Lamanna C,
Diodato E, et al: An exploratory double-blind, randomized clinical
trial with selisistat, a SirT1 inhibitor, in patients with
Huntington's disease. Brit J Clin Pharmaco. 79:465–476.
2015.PubMed/NCBI View Article : Google Scholar
|