Introduction
Osteoarthritis (OA) is the most common degenerative
joint disease and leading cause of pain and disability among
middle-aged and elderly individuals, which is a worldwide health
concern (1). OA is mainly
characterised by the degeneration of articular cartilage and
inflammatory response (2). The
degeneration of articular cartilage is influenced by multiple
factors, such as ageing, obesity, joint strain, trauma and
inflammatory disease (3). As this
disease increases in prevalence and remains difficult to treat,
further clinical and experimental studies are needed to explore the
molecular mechanisms of OA.
During the past several decades, the downregulation
and dysfunction of messenger RNAs, long non-coding RNAs (lncRNAs)
and microRNAs (miRNAs/miRs) (4,5) have
become evident in OA. It has been reported that lncRNAs, with
transcripts >200 nucleotides in length, are involved in OA
progression by regulating cartilage degradation (6). Furthermore, studies have demonstrated
the therapeutic potential of non-coding RNAs including lncRNAs in
the treatment of OA (7). Long
intergenic non-coding RNAs (lincRNAs), a subclass of lncRNAs, have
emerged as key regulators of mammalian gene expression. Several
thousand lincRNAs have been identified in the mouse genome
(8,9). Moreover, lincRNAs are reported to be
associated with human inflammatory diseases and tumorigenesis
(10,11). lincRNA-Cox2 is one of the best
characterised lincRNAs, which has been reported to regulate the
transcription of distinct classes of immune-related genes in the
inflammatory response, thus being regarded as an immune-inducible
lincRNA (12). In a previous study,
Elling et al (13) found
that lincRNA-Cox2 regulates critical innate immune-related genes,
dependently or independently of prostaglandin G/H synthase 2.
Moreover, Tong et al (14)
demonstrated a novel mechanism of epigenetic modulation by
lincRNA-Cox2 on Il12b transcription, suggesting an important
role for lincRNAs in the regulation of intestinal epithelial
inflammatory responses. However, the role of lincRNA-Cox2 in
cartilage degradation and the development of OA remains
unclear.
miRNAs are a class of non-coding RNAs 18-25
nucleotides in length, which are small, evolutionarily conserved
and regarded as novel biomarkers for the diagnosis, prognosis and
treatment of OA (7). miR-150 is
aberrantly expressed in cancer and infectious diseases, and plays a
crucial role in the progression of these diseases (15,16).
Moreover, peripheral blood mononuclear cells and synovial fluid
from patients with rheumatoid arthritis exhibit increased
expression of miR-150(17).
However, whether miR-150 is involved in OA progression remains
unclear.
Chondrocytes are important cells in cartilage, and
their proliferation, differentiation and apoptosis are regulated to
maintain a dynamic equilibrium. The dysfunction of chondrocytes is
responsible for OA development (18); thus, chondrocytes are commonly used
to induce an ex vivo OA model through the stimulation of
IL-1β. The apoptosis and proliferation of chondrocytes has also
been found to be modulated by lncRNAs, such as lncRNA XIST, PVT1
and PART-1 (19-21).
The present study investigated the effect of lincRNA-Cox2 on the
proliferation and apoptosis of chondrocytes, demonstrating an
important role for lincRNA-Cox2 in the development of OA.
Materials and methods
Isolation and culture of primary mouse
chondrocytes
Primary murine chondrocytes were isolated from
new-born mice as described previously (22). Briefly, 5-day-old C57BL/6 mice were
sacrificed by an intraperitoneal injection of pentobarbital (100
mg/kg) and considered dead when respiratory and cardiac arrest were
observed and muscles relaxed. Subsequently, the articular cartilage
was isolated and then digested with 3 mg/ml of collagenase D for 90
min at 37˚C under 5% CO2 and 0.5 mg/ml of collagenase D
overnight at 37˚C. After centrifugation at 400 x g for 10 min, the
supernatant was discarded, and the cell precipitate was resuspended
in Dulbecco's modified Eagle's medium supplemented with 10% foetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 100 IU/ml of
penicillin and 0.1 mg/ml of streptomycin. The chondrocytes were
then seeded on a culture dish at a density of 8x103
cells/cm2. The culture medium was changed after 2 days
of culture, and the isolated chondrocytes achieved confluence 6-7
day later. Only passages one to three were used for further
experiments. For IL-1β treatment assay, mouse chondrocytes were
stimulated using various concentrations (0, 1, 5, 10 and 20 ng/ml)
of IL-1β (PeproTech, Inc.) for 24 h at 37˚C. All protocols were
approved by The Ethics Committee of Qingdao No.6 People's Hospital
[Qingdao, China; approval no. (2018)11].
Induction of an OA mouse model
An experimental OA model was induced by surgical
destabilisation of the medial meniscus as described previously
(23). A total of 20 C57BL/6J male
mice (10-weeks-old; weight, 20-22 g) were purchased from the Animal
Centre of the Chinese Academy of Sciences, housed in plastic cages
with free access to drinking water and a pellet-based diet. The 20
mice were then randomly divided into sham and OA groups (n=10 per
group). In the OA group, under general anaesthesia of pentobarbital
(50 mg/kg) by intraperitoneal injection, the medial collateral
ligament and medial meniscus of the right knee were resected under
a microscope. As a control, the mice in the sham group underwent
skin incision and closure without meniscectomy. After 8 weeks, the
mice were sacrificed by intraperitoneal injection of pentobarbital
(100 mg/kg) and considered dead when respiratory arrest and cardiac
arrest were observed, their nerve reflexes disappeared and muscles
relaxed, followed by the detection of lincRNA-Cox2 expression. All
protocols were approved by The Ethics Committee of Qingdao No. 6
People's Hospital. All mice were sacrificed for experiments.
Mouse chondrocytes transfection
The miR-150 mimic (5'-UCUCCCAACCCUUGUACCAGUG-3'; 30
nM), miR-150 inhibitor (5'-CACUGGUACAAGGGUUGGGAGA-3'; 30 nM) and
their respective scrambled negative controls (NCs;
5'-CTCCCAACCCTTGTCCAGTG-3' and 5'-CCGAAACCUCGGUUGAUUGCGG-3'; 30 nM)
were synthesised by Shanghai GenePharma Co., Ltd. The full-length
wide-type lincRNA-Cox2 sequence was inserted in the pEX-2 plasmid
(Shanghai GenePharma Co., Ltd.). An empty pEX-2 plasmid was
transfected as an NC. siRNA specific for lincRNA-Cox2 was inserted
in a U6/Neo plasmid (Shanghai GenePharma Co., Ltd.). An empty
U6/Neo plasmid with non-targeting sequences was transfected as an
NC. The mass of all plasmids used for transfection was 1 µg.
Transfections of primary mouse chondrocytes were conducted using
Lipofectamine® 3000 reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) per the manufacturer's instructions. After 48 h
at 37˚C, the transfection efficiency was detected using reverse
transcription-quantitative (RT-q)PCR.
RT-qPCR
RT-qPCR assays were performed to detect the
expression of mouse lincRNA-Cox2 and miR-150 in chondrocytes after
transfection and IL-1β treatment. Total RNA of the chondrocytes was
extracted using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). RNA was reverse transcribed into cDNA
using a reverse transcription kit (Vazyme Biotech Co., Ltd.), with
the following temperature protocol: 37˚C for 15 min, 85˚C for 5 sec
and 4˚C for the end. RT-qPCR was performed using a SYBR Green
Master Mix (Applied Biosystems; Thermo Fisher Scientific, Inc.),
and the setting parameters were as follows: 95˚C For 10 min,
followed by 95˚C for 15 sec and 60˚C for 30 sec, lasting 40 cycles.
The cycle threshold (Cq) values were obtained, normalised to the
level of GAPDH and compared with the control. Data were quantified
using the 2-∆∆Cq method (24). The primer sequences were as follows:
Mouse lincRNA-Cox2 forward, 5'-AAGGAAGCTTGGCGTTGTGA-3' and reverse:
5'-GAGAGGTGAGGAGTCTTATG-3'; and GAPDH forward,
5'-CTGCCCAGAACATCATCCCT-3' and reverse,
5'-TGGTCCTCAGTGTAGCCCAAG-3'.
Proliferation assay
The proliferative capability of the chondrocytes was
assessed using an MTT assay. First, 5x103 cells were
seeded on 96-well plates for 24 h; cells were then transfected with
small interfering (si)-negative control (NC), si-Cox2, NC inhibitor
and miR-150 inhibitor as aforementioned. After 48 h of
transfection, 20 µl of MTT (5 mg/ml) (Sigma-Aldrich; Merck KGaA)
was added, and the cells were incubated for 4 h at 37˚C under 5%
CO2. Subsequently, the supernatant was discarded, and
200 ml of dimethyl sulfoxide was added. Finally, the OD 490 nm
value was measured using a microplate reader to evaluate the
proliferative capability of chondrocytes.
Apoptosis assay
The apoptosis of chondrocytes was determined using
the Annexin V fluorescein isothiocyanate (FITC) Apoptosis Detection
kit (BD Biosciences). After 10 ng/ml of IL-1β treatment and/or
relevant transfection, the cells were collected, washed with
phosphate-buffered saline (PBS), and resuspended in 100 µl Binding
Buffer (BD Biosciences) at a concentration of 1x106
cells/ml. Subsequently, 5 µl of Annexin V FITC and propidium iodide
were added. After incubation for 15 min at room temperature in the
dark, the apoptotic cells were quantitatively analysed by
FACSCalibur flow cytometry (BD Biosciences) using CellQuest Pro
software (BD Biosciences).
Western blotting
After the indicated treatment, chondrocytes were
collected and washed with PBS, and then lysed on ice with
radioimmunoprecipitation assay lysis buffer supplemented with 10 mM
of phenylmethylsulfonyl fluoride (Beyotime Institute of
Biotechnology) for 15 min. Total protein was quantified using the
BCA Protein Assay kit (Beijing Solarbio Science & Technology
Co., Ltd.). Proteins in equal amounts (20 µg/lane) were loaded per
lane then subjected to electrophoresis using 10% SDS-PAGE gels and
transferred onto polyvinylidene difluoride membranes (Bio-Rad
Laboratories, Inc.). After blocking with 5% skimmed milk for 2 h at
room temperature, the membranes were incubated with specific
primary antibodies against Ki67 (cat. no. sc-23900), proliferating
cell nuclear antigen (PCNA; cat. no. sc-25280), Bax (cat. no.
sc-70408), Caspase-3 (cat. no. sc-56053), Caspase-9 (cat. no.
sc-56076), glycogen synthase kinase (GSK)-3β (cat. no. sc-81462),
phosphorylated-GSK (p-GSK)-3β (ser9) (cat. no. sc-81494), β-catenin
(cat. no. sc-7963), p-β-catenin (cat. no. sc-57535), cyclin D1
(cat. no. sc-8396), c-Myc (cat. no. sc-40) and GAPDH (cat. no.
sc-32233) (all 1:1,000; Santa Cruz Biotechnology, Inc.) overnight
at 4˚C. The membranes were then incubated with IgG-horseradish
peroxidase-conjugated secondary antibody (cat. no. ab6728; Abcam;
1:5,000) for 1 h at room temperature, and protein bands were
visualised using electrochemiluminescence plus (Cytiva) according
to the manufacturer's instructions. Densitometry analysis of the
bands was performed using ImageJ software (v1.53a; National
Institutes of Health). GAPDH was used as an endogenous protein for
normalisation.
Dual luciferase activity assay
The 3'UTR target site was generated using PCR, and
the luciferase reporter constructs with the lincRNA-Cox2 sequences
carrying a putative miR-150-binding site in a pMiR-report vector
were amplified using PCR. Mouse chondrocytes were co-transfected
with the reporter construct, control vector and miR-150 or scramble
NC using Lipofectamine as aforementioned. After 48 h of
transfection, reporter assays were conducted using the
dual-luciferase assay system (Promega Corporation) following the
manufacturer's instructions. Renilla luciferase activity was
used for normalization, and the binding site of lincRNA-Cox2 and
miR-150 was predicted using DIANA tools (http://carolina.imis.athena-innovation.gr/diana_tools/web/).
Statistical analyses
All results were observed in at least three
independent experiments. Statistical analysis was carried out using
SPSS 19.0 (IBM Corp.), and data are presented as the mean ±
standard deviation (unless otherwise shown). Statistical
differences between two groups were determined using the unpaired
two-tailed Student's t-test. Differences among more than two groups
were estimated using one-way ANOVA and adjusted using Bonferroni's
correction or Dunnett's post hoc test. The linear relationships
among levels of lincRNA-Cox2 and miR-150 in OA mice were analysed
using Spearman's correlation coefficient. P<0.05 was considered
to indicate a statistically significant difference.
Results
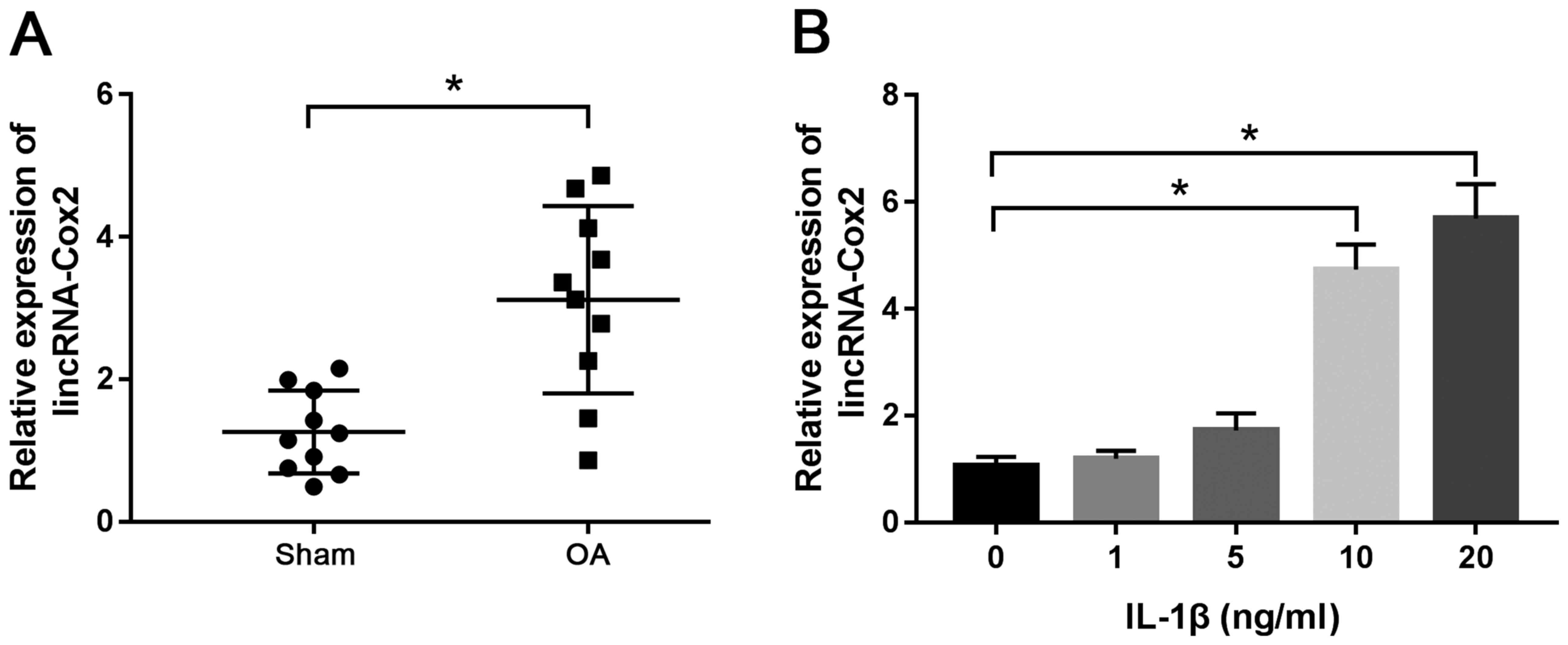
lincRNA-Cox2 expression is
up-regulated in cartilage tissues of OA and IL-1β-treated
chondrocytes
To detect the expression of lincRNA-Cox2 in
cartilage tissues of OA, RT-qPCR was performed using cartilage
specimens from 10 OA mice and 10 sham mice. The results
demonstrated that the expression of lincRNA-Cox2 was markedly
higher in OA cartilage tissues compared with that in tissues of the
sham group (3.12±1.32 vs. 1.26±0.58; P<0.05; Fig. 1A). Moreover, the expression of
lincRNA-Cox2 was significantly up-regulated in chondrocytes
stimulated by IL-1β at 10 ng/ml (4.73±0.47) and 20 ng/ml
(5.69±0.64) compared with that observed in non-stimulated
chondrocytes (1.06±0.17) (both P<0.05; Fig. 1B). The results indicated that
lincRNA-Cox2 played a crucial role in OA.
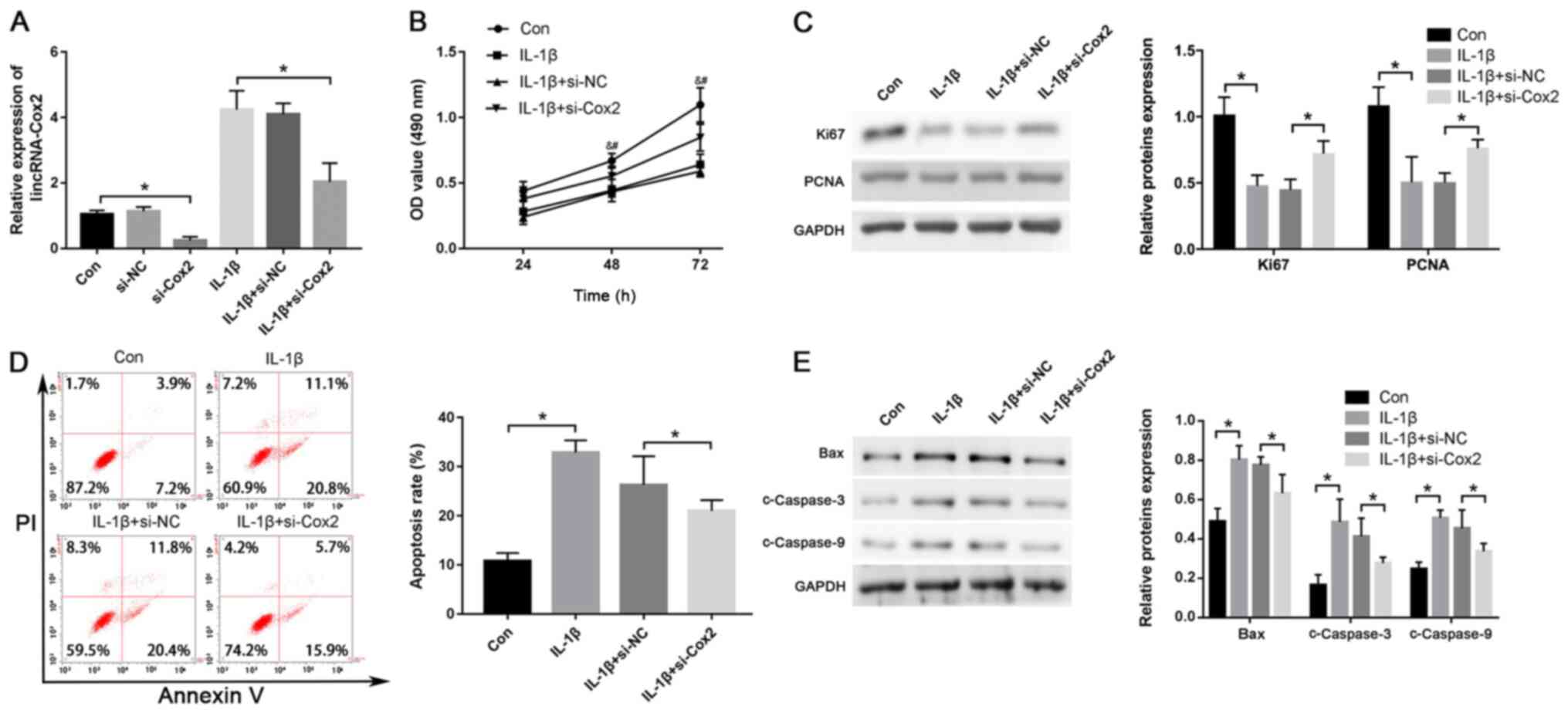
Knockdown of lincRNA-Cox2 promotes
proliferation and inhibits apoptosis in IL-1β-treated
chondrocytes
To explore the effect of lincRNA-Cox2 on the
proliferation and apoptosis of IL-1β-treated chondrocytes, si-Cox2
was used to knock down the expression of lincRNA-Cox2 and the
efficiency of transfection is shown in Fig. S1A (0.32±0.09 vs. 0.95±0.09;
P<0.05). Its abundance was knocked down using si-Cox2 in
IL-1β-treated (10 ng/ml) chondrocytes (2.03±0.58 vs. 4.24±0.58;
P<0.05; Fig. 2A). Moreover, the
MTT assay showed that knockdown of lincRNA-Cox2 markedly restored
the cell viability decreased by treatment of IL-1β in chondrocytes
(P<0.05; Fig. 2B). The protein
levels of major proliferation-related genes, including those
encoding for Ki67 and PCNA, were significantly increased after
silencing lincRNA-Cox2 expression in IL-1β-treated chondrocytes
(Ki67, 0.73±0.09 vs. 0.45±0.07; PCNA, 0.77±0.06 vs. 0.51 0.07; both
P<0.05; Fig. 2C). In addition,
after transfection with si-Cox2, a lower proportion of apoptotic
cells was shown in chondrocytes treated with IL-1β (21.44±1.44% vs.
26.76±5.18%; P<0.05; Fig. 2D),
and the apoptosis-related protein levels of Bax (0.64±0.08 vs.
0.79±0.02), cleaved Caspase-3 (c-Caspase-3) (0.29±0.01 vs.
0.42±0.07) and cleaved Caspase-9 (c-Caspase-9) (0.35±0.02 vs.
0.47±0.07) were also down-regulated compared with those in the
control group (all P<0.05; Fig.
2E). The current results suggested that lincRNA-Cox2 inhibited
proliferation and enhanced apoptosis of chondrocytes.
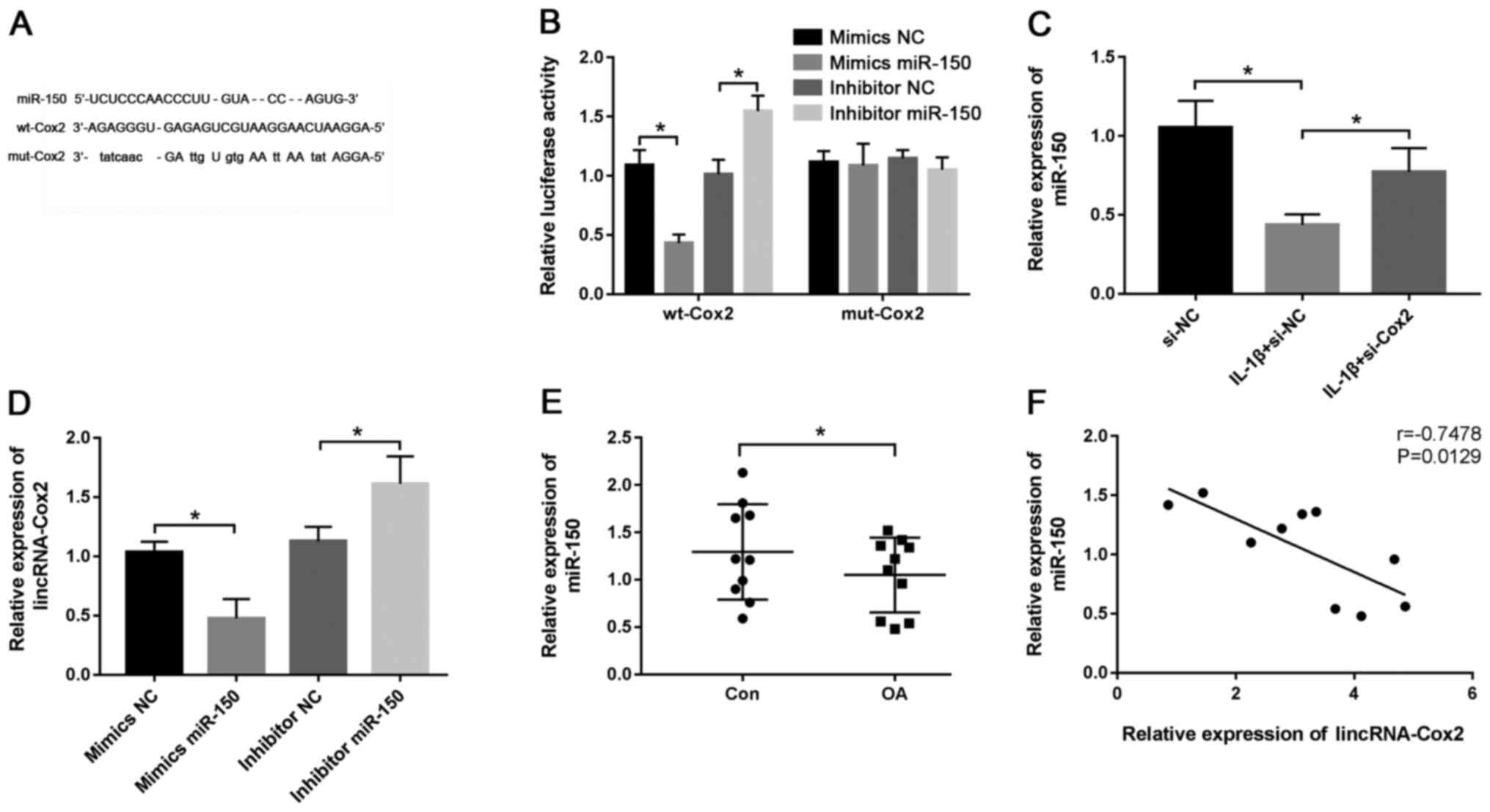
lincRNA-Cox2 directly targets
miR-150
It was predicted that there were putative
complementary sequences of lincRNA-Cox2 and miR-150 using DIANA
tools (Fig. 3A). To confirm the
potential relationship between lincRNA-Cox2 and miR-150, the
luciferase reporter vectors wild-type (wt)-Cox2 and mutant
(mut)-Cox2 were constructed and transfected into chondrocytes. The
results revealed that the mimics miR-150 induced a notable
reduction in luciferase activity (0.43±0.07 vs. 1.09±0.13;
P<0.05; Fig. 3B) and inhibitor
miR-150 led to an increase in luciferase activity in the wt-Cox2
group (1.55±0.13 vs. 1.01±0.12; P<0.05; Fig. 3B), while little effect was observed
on the activity in the mut-Cox2 group (Fig. 3B). The efficiency of mimics miR-150
(1.77±0.13 vs. 1.08±0.06; P<0.05) and inhibitor miR-150
(0.39±0.07 vs. 0.97±0.07; P<0.05) are shown in Fig. S1B and C, respectively. Additionally, the effect
of lincRNA-Cox2 on miR-150 in chondrocytes was evaluated, and the
results showed that the expression of miR-150 was significantly
increased by the interference of lincRNA-Cox2 (0.77±0.15 vs.
0.44±0.07; P<0.05; Fig. 3C).
Moreover, the expression of lincRNA-Cox2 in chondrocytes was
reduced by the overexpression of miR-150 (1.03±0.07 vs. 0.48±0.13;
P<0.05; Fig. 3D), while it was
increased by knockdown of miR-150 (1.61±0.19 vs. 1.13±0.10;
P<0.05; Fig. 3D), compared with
their respective NCs. In cartilage tissues of the OA mouse model,
miR-150 expression was significantly decreased (1.05±0.39 vs.
1.29±0.50; P<0.05; Fig. 3E) and
negatively correlated with lincRNA-Cox2 expression (r=-0.7478;
P=0.0129; Fig. 3F). These results
indicated that lincRNA-Cox2 directly targeted miR-150 to promote
the development of OA.
Deficiency of miR-150 reverses the
effect of lincRNA-Cox2-knockdown on IL-1β-induced injury in
chondrocytes
To explore whether lincRNA-Cox2 exerts its function
by miR-150 in chondrocytes, si-Cox2 and miR-150 inhibitors were
co-transfected into chondrocytes and the efficiency of both si-Cox2
and miR-150 inhibitors was shown in Fig. S1D. As shown in Fig. 4A, knockdown of lincRNA-Cox2 enhanced
the proliferation of IL-1β-treated chondrocytes, while inhibition
of miR-150 suppressed the proliferation of IL-1β-treated
chondrocytes compared with their respective NCs (both P<0.05).
The expression of both Ki67 and PCNA were also promoted by the
knockdown of lincRNA-Cox2 compared with that in the si-NC group
(Ki67, 0.63±0.01 vs. 0.56±0.00; PCNA, 0.52±0.00 vs. 0.33±0.02; both
P<0.05; Fig. 4B). However, these
increases were inhibited by miR-150 inhibitors (Ki67, 0.47±0.06 vs.
0.63±0.01; PCNA, 0.28±0.05 vs. 0.52±0.00; both P<0.05; Fig. 4B). Moreover, the anti-apoptotic
effect of lincRNA-Cox2-knockdown (10.22±0.69% vs. 22.13±2.27%) and
pro-apoptotic effect of miR-150 inhibitors (33.06±2.79% vs.
22.13±2.27%) were also evidently reversed by co-transfection with
si-Cox2 and miR-150 inhibitors (lincRNA-Cox2 knockdown, 19.40±3.32%
vs. 10.22±0.69%; miR-150 inhibitor, 19.40±3.32% vs. 33.06±2.79%;
all P<0.05; Fig. 4C). The
decreased expression of Bax, c-Caspase-3 and c-Caspase-9 in the
si-Cox2 group were reversed by co-transfection with si-Cox2 and
miR-150 inhibitors (Bax, 0.56±0.01 vs. 0.40±0.01; c-Caspase-3,
0.90±0.01 vs. 0.79±0.01; c-Caspase-9, 0.65±0.06 vs. 0.48±0.01; all
P<0.05; Fig. 4D). The above
results further confirmed that lincRNA-Cox2 promote the development
of OA by targeting miR-150.
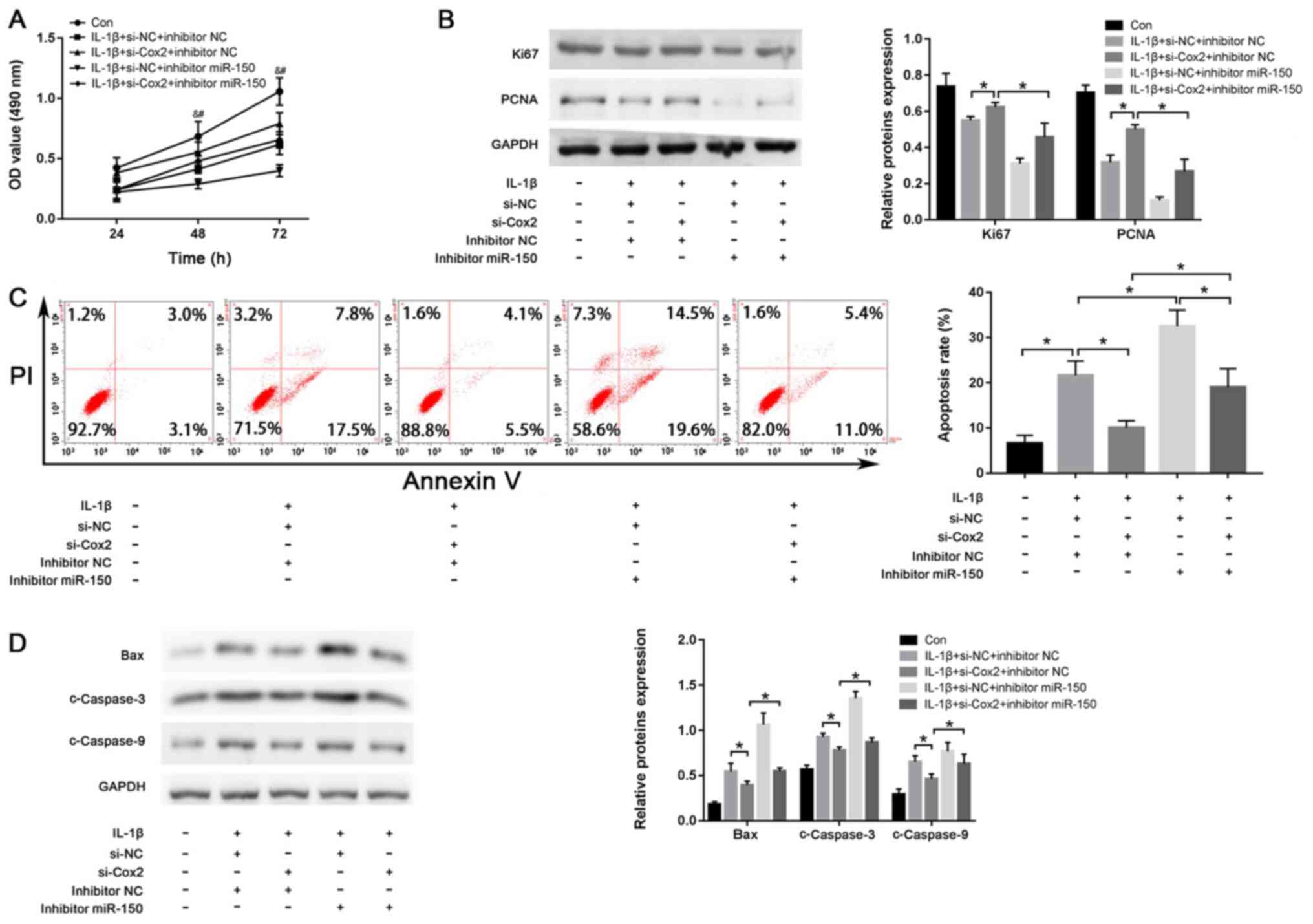 | Figure 4miR-150 inhibitor reverses the effect
of lincRNA-Cox2-knockdown on IL-1β-induced injury in chondrocytes.
(A) Chondrocytes proliferation with si-Cox2 or miR-150 inhibitor
after treatment of IL-1β. #P<0.05, si-Cox2+inhibitor
miR-150 vs. si-Cox2+inhibitor NC; &P<0.05,
si-Cox2+inhibitor NC vs. si-NC+inhibitor NC (B) Ki67 and PCNA
levels in chondrocytes. (C) Apoptotic rate of chondrocytes after
transfection and IL-1β stimulation. (D) Protein levels of Bax,
c-Caspase-3 and c-Caspase-9 in chondrocytes. *P<0.05.
linc, long intergenic non-coding; PCNA, proliferating cell nuclear
antigen; c-, cleaved; si, small interfering; NC, negative control;
miR, microRNA; OD, optical density. |
lincRNA-Cox2 aggravates OA progression
through the Wnt/β-catenin pathway
To determine the function of the molecular
mechanisms induced by the lincRNA-Cox2/miR-150 axis, Wnt/β-catenin
pathway-related proteins including GSK-3β, p-GSK-3β, β-catenin,
p-β-catenin, cyclin D1 and c-Myc were detected using western
blotting (Fig. 5A). The results
showed that knockdown of lincRNA-Cox2 notably inhibited the
expression of p-GSK-3β/GSK-3β (0.75±0.03 vs. 0.90±0.06; P<0.05;
Fig. 5B) while promoting the
expression of p-β-catenin/β-catenin (1.06±0.06 vs. 0.88 ± 0.01;
P<0.05; Fig. 5C), cyclin D1
(0.98±0.03 vs. 0.72±0.07; P<0.05; Fig. 5D) and c-Myc (1.09±0.03 vs.
0.96±0.04; P<0.05; Fig. 5E).
However, the suppressive or promotive effect was reversed by
miR-150 inhibitors (p-GSK-3β/GSK-3β, 0.88±0.00 vs. 0.75±0.03;
p-β-catenin/β-catenin, 0.82±0.04 vs. 1.06±0.06; cyclin D1,
0.75±0.08 vs. 0.98±0.03; c-Myc, 0.91±0.04 vs. 1.09±0.03; all
P<0.05; Fig. 5). This results
revealed lincRNA-Cox2 aggravated OA progression through the
Wnt/β-catenin pathway.
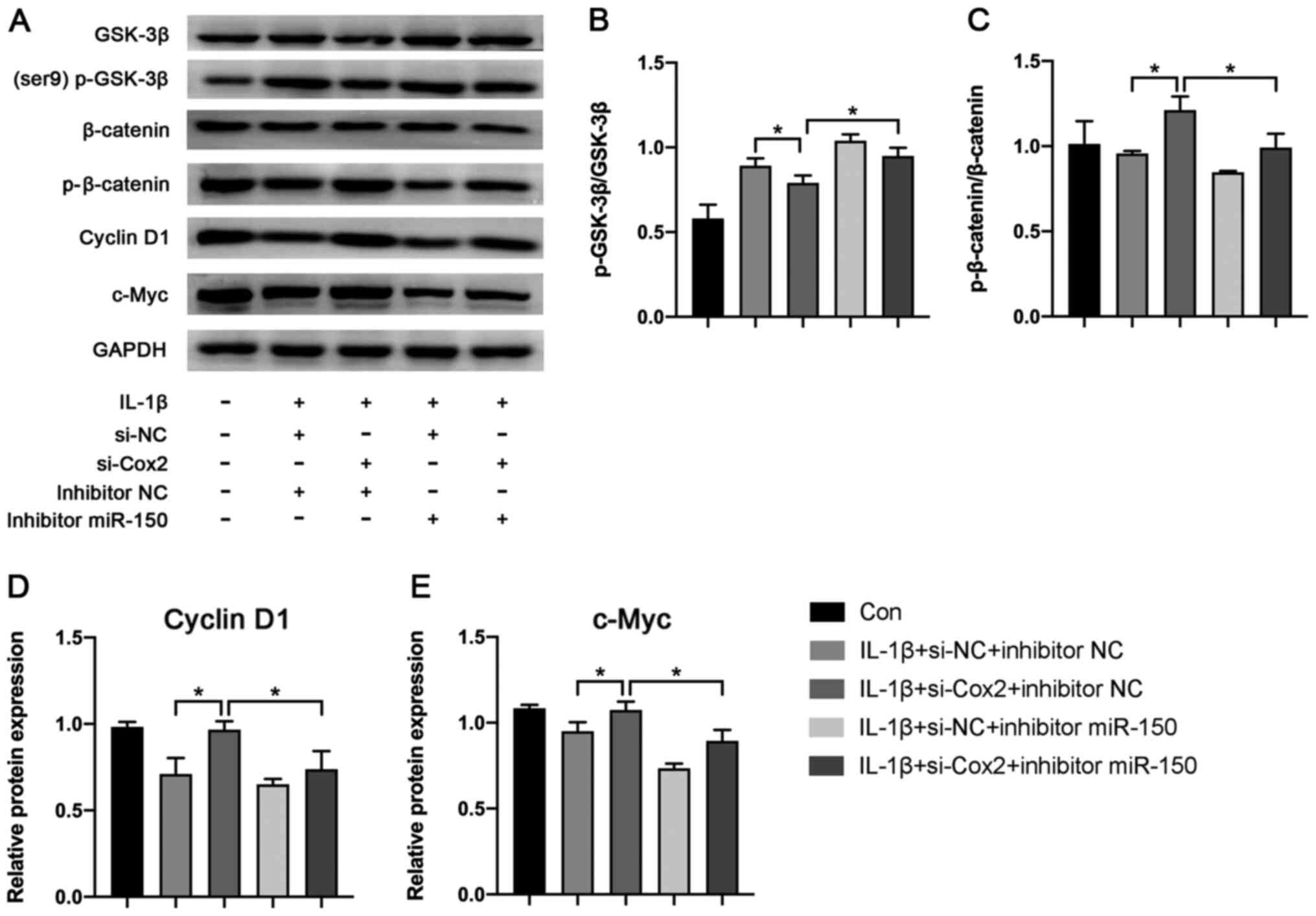 | Figure 5Effect of lincRNA-Cox2/miR-150 axis
on Wnt/β-catenin pathway. (A) Protein expression of p-GSK-3β,
GSK-3β, p-β-catenin, β-catenin, c-Myc and cyclin D1 in chondrocytes
after transfection with si-Cox2 or miR-150 inhibitor. (B) The ratio
of p-GSK-3β/GSK-3β. (C) The ratio of p-β-catenin/β-catenin. (D) The
relative expression of Cyclin D1. (E) The relative expression of
c-Myc. *P<0.05. linc, long intergenic non-coding;
Con, control; NC, negative control; p-, phosphorylated; si, small
interfering; miR, microRNA. |
Discussion
OA is a chronic, progressive and degenerative
disease affecting multiple joint tissues and resulting in
significant reductions in patient quality of life including pain,
stiffness, movement difficulties and progressive disability
(25). The main characteristics of
OA are degeneration of articular cartilage and chronic inflammation
(25). However, despite the diverse
aetiologies and pathogenesis of OA, a detailed pathogenic mechanism
has not yet been elucidated. Recent focus on the
epigenetic-regulating mechanisms of OA has revealed that numerous
lncRNAs serve important functions in the development of
inflammatory diseases including OA, such as lncRNAs XIST and PVT1
(19,20). lincRNA-Cox2, a class of lncRNAs
localised to both the cytosolic and nuclear compartments, affects
the expression of hundreds of inflammatory genes (such as Tlr1,
Il6 and Il23a) and regulates the inflammatory response
(12). Moreover, lincRNA-Cox2
mediates neuroinflammation by regulating the NACHT, LRR and PYD
domains-containing protein 3 inflammasome and autophagy (26). However, whether lincRNA-Cox2 is
involved in the pathogenesis of OA remains unclear. The present
study reported that the expression of lincRNA-Cox2 was markedly
up-regulated in an OA model both in vivo and in
vitro, indicating that lincRNA-Cox2 may play a role in OA
development.
Chondrocytes are the only cells found in the
cartilage and their dynamic equilibrium between proliferation,
differentiation and apoptosis is crucial to maintain the
appropriate cycles of biosynthesis and degradation of the
cartilaginous matrix (18). The
current study investigated the role of lincRNA-Cox2 in the
viability of chondrocytes using IL-1β-treated chondrocytes, and the
results demonstrated that lincRNA-Cox2 inhibited the viability of
chondrocytes. In addition, the protein levels of Ki67 and PCNA, two
main proliferation-related proteins (27), were also suppressed by lincRNA-Cox2.
Apoptosis is an important process associated with cell viability,
and the activation of Bax, c-Caspase 3 and c-Caspase 9 are
responsible for regulating apoptosis (28,29).
The present study demonstrated that both the rate of apoptosis and
expression of Bax, c-Caspase 3 and c-Caspase 9 were reduced after
knockdown of lincRNA-Cox2, suggesting an important pro-apoptotic
role of lincRNA-Cox2 in OA chondrocytes.
Previously, the hypothesis of competing endogenous
RNAs (ceRNAs) as an alternative function for lncRNAs has garnered
increasing attention (30). As a
novel regulatory mechanism, the crosstalk between lncRNAs and
miRNAs has been identified in various diseases including OA
(31). Zhang et al (32) demonstrated that lncRNA MALAT1
promoted OA by competing with miR-150-5p. Therefore, the present
study hypothesised that lincRNA-Cox2 may act as a ceRNA sponge for
miRNAs through which OA development is promoted. The current study
predicted direct binding between lincRNA-Cox2 and miR-150 and
confirmed this prediction using a luciferase activity assay.
Furthermore, it was reported that the expression of miR-150 was
decreased after silencing lincRNA-Cox2 expression, while
lincRNA-Cox2 was also found to negatively regulate miR-150
expression. Moreover, an inverse correlation between lincRNA-Cox2
and miR-150 was observed in OA cartilage tissues. These findings
illustrated that lincRNA-Cox2 exerted its functions on the
proliferation and apoptosis of chondrocytes by sponging miR-150.
However, this effect of lincRNA-Cox2 was reversed by miR-150
inhibitors. To the best of our knowledge, the present study is the
first to reveal that the lincRNA-Cox2/miR-150 axis mediates OA
progression.
Previous studies have indicated that the
Wnt/β-catenin pathway plays a pivotal role in the regulation of
inflammatory processes in various mammalian non-neuronal cells
(33) and is involved in OA
development (34). In addition, the
Wnt/β-catenin pathway is necessary to regulate the differentiation,
phenotypic maturation and function of chondrocytes (35). Inhibition of GSK-3β activates the
Wnt/β-catenin pathway and further leads to increased expression of
the Wnt/β-catenin target genes, such as those encoding for cyclin
D1 and c-Myc (36). Moreover,
silencing lncRNA HOTAIR regulates the proliferation and apoptosis
of synoviocytes, which are the main effector cells of knee OA
synovial fibrosis, in OA through inhibition of the Wnt/β-catenin
signalling pathway (37). The
present study demonstrated that knockdown of lincRNA-Cox2 inhibited
the expression of p-GSK-3β while promoting the expression of
p-β-catenin, cyclin D1 and c-Myc, but these trends were reversed by
miR-150 inhibitors. However, the underlying mechanism and whether
miR-150 directly binds to the downstream proteins need to be
further investigated in future studies. Our data demonstrated that
lincRNA-Cox2 triggered the activity of the Wnt/β-catenin pathway by
sponging miR-150 in chondrocytes, suggesting a novel pathogenic
mechanism for OA progression.
In conclusion, lincRNA-Cox2 is up-regulated in OA
cartilage tissues and IL-1β-treated chondrocytes. The
overexpression of lincRNA-Cox2 reduces the viability, enhances
apoptosis and aggravates the injury of chondrocytes, suggesting
that lincRNA-Cox2 may act as a useful marker and potential
therapeutic target in OA. It was further identified that
lincRNA-Cox2 exerts its effects partially through the
lincRNA-Cox2/miR-150/Wnt/β-catenin axis. This finding may improve
our understanding of the mechanisms involved in OA progression and
provide novel targets for the molecular treatment of OA.
Supplementary Material
Efficiency of transfection. Efficiency
of (A) si-Cox2, (B) mimics miR-150, (C) inhibitor miR-150, (D)
si-Cox2 and inhibitor miR-150 in chondrocytes when co-transfected
with si-Cox2 and miR-150 inhibitors. *P<0.05. linc,
long intergenic non-coding; si, small interfering; NC, negative
control; miR, microRNA.
Acknowledgements
Not applicable.
Funding
Funding: The study was supported by Qingdao No. 6 People's
Hospital (grant no. SPH2018011).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JC made substantial contributions to conception and
design. MJ and KX prepared the experimental materials and performed
the experiments. HR, MW and XH interpreted the data, performed the
statistical analysis and analyzed the results. JC revised and
approved the final version of the manuscript. MW and XH confirm the
authenticity of the data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The protocol of this research was approved by The
Ethics Committee of Qingdao No.6 People's Hospital [Qingdao, China;
approval no. (2018)11].
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Charlier E, Relic B, Deroyer C, Malaise O,
Neuville S, Collée J, Malaise MG and De Seny D: Insights on
molecular mechanisms of chondrocytes death in osteoarthritis. Int J
Mol Sci. 17(2146)2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Eveque-Mourroux MR, Rocha B, Barre FPY,
Heeren RMA and Cillero-Pastor B: Spatially resolved proteomics in
osteoarthritis: State of the art and new perspectives. J
Proteomics. 215(103637)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Goldring M and Goldring S: Osteoarthritis.
J Cell Physiol. 213:626–634. 2007.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Jiang S, Liu Y, Xu B, Zhang Y and Yang M:
Noncoding RNAs: New regulatory code in chondrocyte apoptosis and
autophagy. Wiley Interdiscip Rev RNA. 11(e1584)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhu J, Yu W, Wang Y, Xia K, Huang Y, Xu A,
Chen Q, Liu B, Tao H, Li F and Liang C: lncRNAs: Function and
mechanism in cartilage development, degeneration, and regeneration.
Stem Cell Res Ther. 10(344)2019.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Shui X, Xie Q, Chen S, Zhou C, Kong J and
Wang Y: Identification and functional analysis of long non-coding
RNAs in the synovial membrane of osteoarthritis patients. Cell
Biochem Funct. 38:460–471. 2020.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
Yuangang W, Xiaoxi L, Bin S and Yi Z: The
therapeutic potential and role of miRNA, lncRNA, and circRNA in
osteoarthritis. Curr Gene Ther. 19:255–263. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chen YG, Satpathy AT and Chang HY: Gene
regulation in the immune system by long noncoding RNAs. Nat
Immunol. 18:962–972. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Luo H, Sun S, Li P, Bu D, Cao H and Zhao
Y: Comprehensive characterization of 10,571 mouse large intergenic
noncoding RNAs from whole transcriptome sequencing. PLoS One.
8(e70835)2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Terracciano D, Terreri S, de Nigris F,
Costa V, Calin GA and Cimmino A: The role of a new class of long
noncoding RNAs transcribed from ultraconserved regions in cancer.
Biochim Biophys Acta Rev Cancer. 1868:449–455. 2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Dominguez-Andres J, Fanucchi S, Joosten
LAB, Mhlanga MM and Netea MG: Advances in understanding molecular
regulation of innate immune memory. Curr Opin Cell Biol. 63:68–75.
2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Carpenter S, Aiello D, Atianand MK, Ricci
EP, Gandhi P, Hall LL, Byron M, Monks B, Henry-Bezy M, Lawrence JB,
et al: A long noncoding RNA mediates both activation and repression
of immune response genes. Science. 341:789–792. 2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Elling R, Robinson EK, Shapleigh B, Liapis
SC, Covarrubias S, Katzman S, Groff AF, Jiang Z, Agarwal S, Motwani
M, et al: Genetic models reveal cis and Trans immune-regulatory
activities for lincRNA-Cox2. Cell Rep. 25:1511–1524.e6.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Tong Q, Gong AY, Zhang XT, Lin C, Ma S,
Chen J, Hu G and Chen XM: lincRNA-Cox2 modulates TNF-α-induced
transcription of Il12b gene in intestinal epithelial cells through
regulation of Mi-2/NuRD-mediated epigenetic histone modifications.
FASEB J. 30:1187–1197. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Brown AL, Al-Samadi A, Sperandio M, Soares
AB, Teixeira LN, Martinez EF, Demasi APD, Araújo VC, Leivo I, Salo
T and Passador-Santos F: miR-455-3p, miR-150 and miR-375 are
aberrantly expressed in salivary gland adenoid cystic carcinoma and
polymorphous adenocarcinoma. J Oral Pathol Med. 48:840–845.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Verma P, Pandey RK, Prajapati P and
Prajapati VK: Circulating microRNAs: Potential and emerging
biomarkers for diagnosis of human infectious diseases. Front
Microbiol. 7(1274)2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Niimoto T, Nakasa T, Ishikawa M, Okuhara
A, Izumi B, Deie M, Suzuki O, Adachi N and Ochi M: MicroRNA-146a
expresses in interleukin-17 producing T cells in rheumatoid
arthritis patients. BMC Musculoskelet Disord.
11(209)2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sandell LJ and Aigner T: Articular
cartilage and changes in arthritis. An introduction: Cell biology
of osteoarthritis. Arthritis Res. 3:107–113. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
19
|
Sun P, Wu Y, Li X and Jia Y: miR-142-5p
protects against osteoarthritis through competing with lncRNA XIST.
J Gene Med. 22(e3158)2020.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Lu X, Yu Y, Yin F, Yang C, Li B, Lin J and
Yu H: Knockdown of PVT1 inhibits IL-1β-induced injury in
chondrocytes by regulating miR-27b-3p/TRAF3 axis. Int
Immunopharmacol. 79(106052)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lu C, Li Z, Hu S, Cai Y and Peng K: LncRNA
PART-1 targets TGFBR2/Smad3 to regulate cell viability and
apoptosis of chondrocytes via acting as miR-590-3p sponge in
osteoarthritis. J Cell Mol Med. 23:8196–8205. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gosset M, Berenbaum F, Thirion S and
Jacques C: Primary culture and phenotyping of murine chondrocytes.
Nat Protoc. 3:1253–1260. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li Y, Wu Y, Jiang K, Han W, Zhang J, Xie
L, Liu Y, Xiao J and Wang X: Mangiferin prevents TBHP-induced
apoptosis and ECM degradation in mouse osteoarthritic chondrocytes
via restoring autophagy and ameliorates murine osteoarthritis. Oxid
Med Cell Longev. 2019(8783197)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hawker GA: Osteoarthritis is a serious
disease. Clin Exp Rheumatol. 120:3–6. 2019.PubMed/NCBI
|
|
26
|
Xue Z, Zhang Z, Liu H, Li W, Guo X, Zhang
Z, Liu Y, Jia L, Li Y, Ren Y, et al: lincRNA-Cox2 regulates NLRP3
inflammasome and autophagy mediated neuroinflammation. Cell Death
Differ. 26:130–145. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Jurikova M, Danihel L, Polak S and Varga
I: Ki67, PCNA, and MCM proteins: Markers of proliferation in the
diagnosis of breast cancer. Acta Histochem. 118:544–552.
2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yan X, Zhou R and Ma Z: Autophagy-cell
survival and death. In: Autophagy: Biology and Diseases: Basic
Science. Qin ZH (ed). Springer Singapore, Singapore, pp667-696,
2019.
|
|
29
|
Aghaei M, KhanAhmad H, Aghaei S, Ali
Nilforoushzadeh M, Mohaghegh MA and Hejazi SH: The role of Bax in
the apoptosis of Leishmania-infected macrophages. Microb Pathog.
139(103892)2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tay Y, Rinn J and Pandolfi PP: The
multilayered complexity of ceRNA crosstalk and competition. Nature.
505:344–352. 2014.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Jeffries MA: Osteoarthritis year in review
2018: Genetics and epigenetics. Osteoarthritis Cartilage.
27:371–377. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhang Y, Wang F, Chen G, He R and Yang L:
LncRNA MALAT1 promotes osteoarthritis by modulating miR-150-5p/AKT3
axis. Cell Biosci. 9(54)2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Takahashi T: Roles of nAChR and Wnt
signaling in intestinal stem cell function and inflammation. Int
Immunopharmacol. 81(106260)2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huang J, Chen C, Liang C, Luo P, Xia G,
Zhang L, Wang X, Wen Z, Cao X and Wu S: Dysregulation of the Wnt
signaling pathway and synovial stem cell dysfunction in
osteoarthritis development. Stem Cells Dev. 29:401–413.
2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Xiaoliang Y, Haiqing L, Hao H, Hai L,
Linfu L, Jianqiong Y, Weimei S, Weiyou L and Longhuo W: The key
role of canonical Wntβ-catenin signaling in cartilage chondrocytes.
Curr Drug Targets. 17:475–484. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Vallée A and Lecarpentier Y: Crosstalk
between peroxisome proliferator-activated receptor gamma and the
canonical WNT/β-catenin pathway in chronic inflammation and
oxidative stress during carcinogenesis. Front Immunol.
9(745)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mao T, He C, Wu H, Yang B and Li X:
Silencing lncRNA HOTAIR declines synovial inflammation and
synoviocyte proliferation and promotes synoviocyte apoptosis in
osteoarthritis rats by inhibiting Wnt/β-catenin signaling pathway.
Cell Cycle. 18:3189–3205. 2019.PubMed/NCBI View Article : Google Scholar
|