According to the most recent global cancer
statistics, colorectal cancer (CRC) is the second most common cause
of cancer-associated death in males and females combined (1,2).
In 2020, ~1,1 million new CRC cases were reported along with
576,858 deaths, accounting for nearly 10.0% of new cases and 5.8%
of all cancer-associated deaths globally (1). While up to 10-15% of CRC patients
carry one or more inherited pathogenic mutations associated with
inherited syndromes (such as familial adenomatous polyposis or
hereditary non-polyposis CRC) (3), 85% of CRC cases are sporadic and
attributable to risk factors including age, race and sex, as well
as modifiable risk factors including diet, tobacco use, diabetes
and obesity (4). In the US, it is
estimated that ~20% of newly diagnosed CRC patients have metastatic
disease (mCRC) at the time of presentation (2), of which 3-5% harbor high
microsatellite instability (MSI-H) and deficiency in mismatch
repair mechanism (dMMR) of their genome (5,6).
MSI is the result of somatic or germline mutations
in the DNA mismatch repair (MMR) genes MutL homolog 1, MutS homolog
2 (MSH2), MSH6, post-meiotic segregation homolog 2 and epithelial
cell adhesion molecule. This defect leads to frame-shift mutations
due to the accumulation of DNA replication errors in the
microsatellites of DNA coding regions. As a result, tumors with
MSI-H/dMMR molecular profile tend to accumulate multiple
insertion/deletion mutations that translate into frame-shift
peptides (FSPs) expressed on tumor cell surfaces as neoantigens and
recognized by the immune system. This has made MSI-H/dMMR mCRC
attractive targets for immunotherapies that enhance self-immunity
against cancer via exploitation of these neoantigens (7). Prior to the development of immune
checkpoint inhibitors (ICIs), the mainstay of first-line therapy
for mCRC was combination chemotherapy plus an anti-vascular
endothelial growth factor (VEGF) or anti-epidermal growth factor
antibody, depending on tumor characteristics without accounting for
MSI-H/dMMR molecular status (8);
yet, most patients progressed within 1 year of treatment with these
systemic regimens (9). The
introduction of ICIs, on the other hand, has since revolutionized
cancer therapy and demonstrated impressive activity in patients
with mCRC as well as other types of solid tumor that are MSI-H/dMMR
(7,10,11). In the present review, the
molecular rationale behind the use of immunotherapy in patients
with MSI-H/dMMR mCRC is described, available clinical data
supporting its use are discussed and possible approaches and future
directions to overcome current therapeutic challenges immunotherapy
is facing in this select patient population are highlighted.
ICIs rely on the ability of tumor cells to suppress
the innate immune system by exploiting the interaction between
major histocompatibility complex (MHC)-T-cell receptor (TCR) and
key ligands on the surface of tumor cells, known as immune
checkpoints. These up-regulated immune checkpoints include
programmed death 1 (PD-1), PD-1 ligand (PD-L1) and cytotoxic
T-lymphocytes-associated protein 4 (CTLA-4), all of which act by
inducing anergy or 'dampening' of the immune system (12-14).
PD-1 is expressed on the surface of T-cells,
B-cells, dendritic cells and natural killer (NK) cells and becomes
overexpressed in inflammatory microenvironments such as the tumor
microenvironment (TME) (12).
When PD-1 binds to PD-L1 on tumor cells, an inhibitory signaling
cascade is initiated and results in i) direct inhibition of tumor
cell apoptosis, ii) conversion of effector T cells into regulatory
T-cells and iii) kinase-dependent down-regulation of cytokine
production required to stimulate proliferation and function of
effector T cells (12,13). This is the main mechanism by which
tumor cells escape immune surveillance (15). Similarly, CTLA-4 is another
co-inhibitory molecule expressed on tumor cells that functions as
an immune checkpoint by binding to B7-1 (CD80) and B7-2 (CD86) on
antigen-presenting cells, resulting in down-regulation of
tumor-reactive T-cell activation, expansion and anti-tumor effects
(14).
ICIs specifically target these checkpoints by
disrupting the interaction between tumor-expressed inhibitory
signals and cells of the immune system. ICIs were initially
indicated to improve survival of patients with metastatic melanoma
and non-small cell lung cancer (NSCLC) leading to the approval of
ipilimumab (anti-CTLA-4), pembrolizumab and nivolumab (anti-PD-1)
(16-20). These initial results were further
consolidated with long-term follow-up studies revealing prolonged
survival (for ≥10 years) after treatment with ipilimumab (21). These robust responses have been
explained by the high mutation prevalence commonly observed in both
melanoma and NSCLC, suggesting that tumor cells with a high tumor
mutation burden (TMB) generate more new peptides expressed as
neoantigens on their MHC surface molecules; these neoantigens are
recognized as non-self and result in priming of T-cell activation
and cytotoxic killing (22,23). More recently, the US Food and Drug
Administration (FDA) approved the use of pembrolizumab for all
solid tumors with high TMB, further consolidating the concept of
neoantigenicity promoting response to ICIs (24).
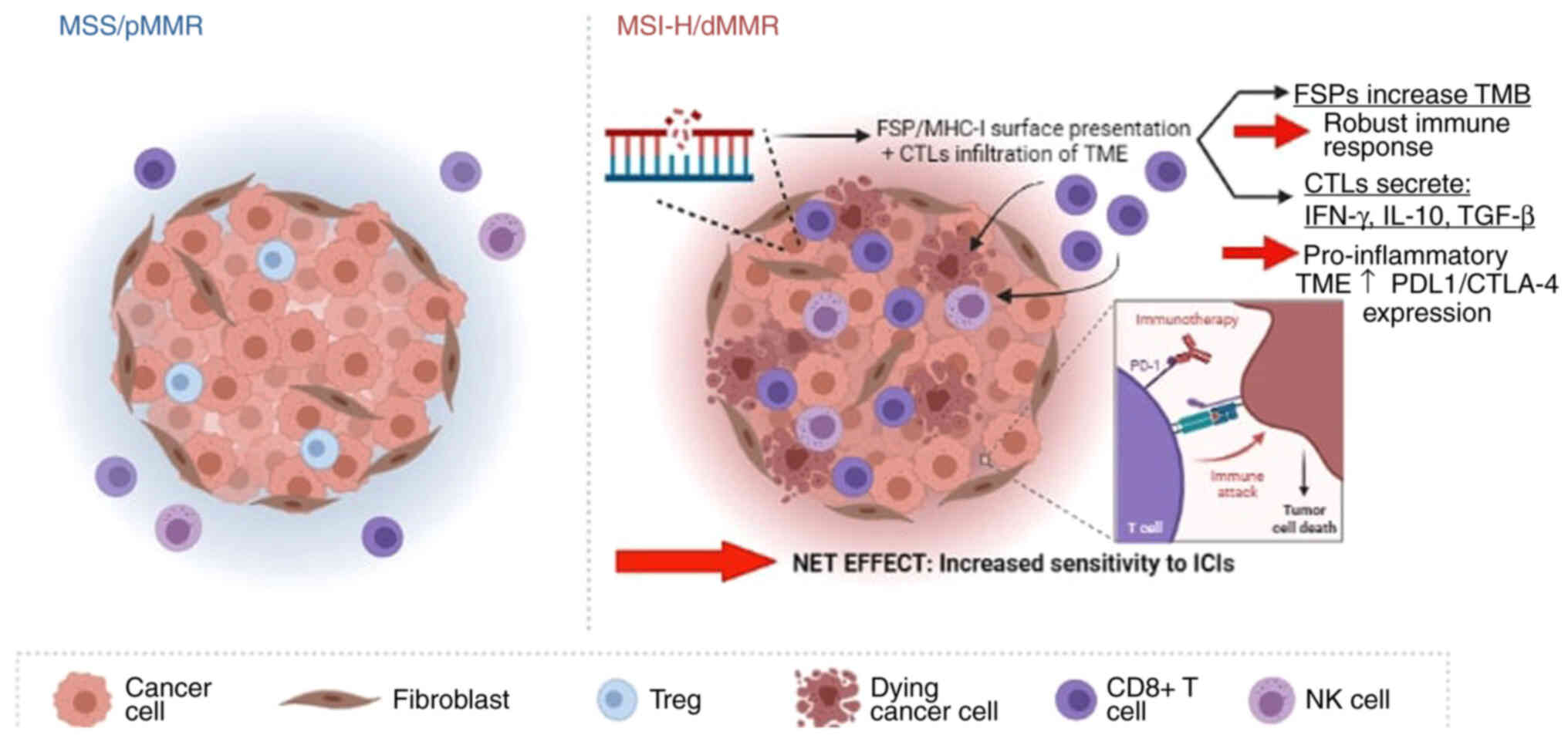
The increased sensitivity of MSI-H/dMMR mCRC tumors
and their susceptibility to ICIs has been attributed to multiple
immunologic, molecular and genetic factors. First, MSI-H/dMMR
tumors have an abundance of TILs, specifically cytotoxic T
lymphocytes, when compared with MMR proficient (pMMR) tumors
(29). This observation has been
linked to MSI-H tumors having a more favorable prognosis and
disease course (30). While the
mechanism behind this effect has remained to be fully elucidated,
it has been hypothesized that the abundance of TILs creates an
inflammatory TME that paradoxically does not eradicate the cancer
but rather, triggers the up-regulation of several immune checkpoint
molecules, including PD-1, PDL-1 and CTLA-4, in what appears an
adaptive immune phenomenon that launches the immune escape
mechanism discussed earlier (31). Part of this checkpoint-mediated
immune escape adopted by cancer cells involves the increased
conversion from effector T-cells to regulatory T-cells that secrete
immunosuppressive molecules such as TGF-β, IFN-γ and IL-10,
ultimately resulting in immune anergy (31). IFN-γ specifically has been
indicated to up-regulate the expression of PD-L1, which is
significantly higher in MSI-H tumors in comparison to
microsatellite-stable (MSS) tumors (31). ICIs, by virtue of their mechanism
of action, exploit these elevated levels of checkpoint inhibitors
on cancer cells to disrupt the interaction between cancer cells and
immune cells, ultimately 'blocking' the immune escape and
reinvigorating the host's immune system. It is specifically the
higher levels of checkpoint molecule expression in MSI-H tumors,
originally adaptive to escape the host's immune system, that render
this molecular subtype more sensitive and responsive to ICIs. Yet,
high levels of PD-L1 would not solely explain this robust response
to ICIs, as multiple studies have indicated that levels of PD-L1
expression alone are insufficient to predict response to ICIs,
pointing towards the requirement for other molecular and genomic
biomarkers [reviewed in (32)].
The TMB, which quantifies the total number of mutations present in
a tumor specimen, has emerged as a promising quantitative genomic
biomarker for response to ICIs, independent of the PD-L1 expression
status [reviewed in (33)]. This
becomes of unique relevance with MSI-H/dMMR mCRC that harbor a high
level of somatic frame-shift mutations as a result of their dMMR
mechanism. These molecular defects that translate into short
stretches of DNA (micro-satellites) serve as neoantigens (as FSPs),
ultimately exposing cancer cells to the host's immune system,
namely in the T-cell-infiltrated TME of MSI-H/dMMR tumors (7). The accumulation of neoantigens
elicits a robust host immune response when recognized by the TILs
(34), but also by involvement of
macrophages and dendritic cells that serve as biologic immune
intermediates for neoantigen presentation and delivery on the one
hand, and as a pro-inflammatory vehicles that release inflammatory
cytokines on the other hand, further enhancing the immunologic
effect described earlier (35).
In fact, MSI-H/dMMR tumors produce a significantly higher TMB in
comparison to MSS tumors, in the realm of 10-fold or higher.
Numerous types of these mutations are sequences of tri-nucleotide
repeats within the introns of protein-coding sequences (36). Of these mutations, the majority
result in altered amino acid sequences and neoantigen peptides.
Thus, patients with MSI-H/dMMR tumors represent a unique population
of patients with mCRC that have been indicated to benefit the most
from immune-based therapies (37). In a recent study by Valero et
al (38), patients with
MSI-H/dMMR with high TMB levels were reported to have better
overall survival (OS) compared to MSS and this survival benefit was
further prolonged in patients treated with ICIs compared to non-ICI
therapy.
Finally, the impact of the MSI-H/dMMR status on
triggering an immune response and predicting a durable response to
ICIs reaches beyond the mere quantitative load of neoantigens to
the actual identity and genomic function of the neo-peptides. An
early study of the mutations causing amino acid alterations
suggested that, within the selected CRC samples, there were roughly
7 unique epitopes that were involved in tumorigenesis (39). Early identified FSPs included
transforming growth factor-β receptor 2 (TGFBR2), phosphatase and
tensin homolog, asteroid homolog 1, AIM2 and caspase 5 (36,39-42) and these FSPs, along with others
such as HT001, AIM2 and TAF1B, were detected at significantly
higher frequencies in MSI-H CRC compared to MSS (41).
More recent studies using serologic and
bio-informatics approaches have further identified a wider range of
FSPs and certain FSPs have potential functional genetic
implications (43-46). In general, mutations in coding
exons that are generated as a result of deficient MMR in MSI-H CRC
result in complete functional inactivation (47). While functional validation is
still pending for numerous FSPs, these observations open ways for
novel mechanisms that explain the exquisite sensitivity of
MSI-H/dMMR mCRC to ICIs: Beyond their immunogenicity as FSPs,
certain mutated genes may further exert a functional anti-tumor
effect that amplifies the immune efficiency of ICI in MSI-H/dMMR
mCRC. For instance, loss of TGFBR2 activity promotes an
inflammatory response within the TME by inhibiting
anti-inflammatory cytokines, increasing tumor-associated macrophage
infiltration and NK T-cell activation (48). AIM2 on the other hand has been
indicated to have tumor suppressive properties via promotion of an
inflammatory response and inhibition of CRC cell proliferation and
migration, similar to myristoylated alanine-rich protein kinase C
substrate, another identified FSP that inhibits the proliferation
of CRC (49-51). Inactivating mutation of these
genes in MSI-H/dMMR mCRC may thus not result in promoting
anti-tumor activity but may at least offer potential targets for
precision therapy along with ICIs. The roles of other FSPs, such as
TAF1B and ZNF294, remain to be identified (47,52) while others such as HT001 are known
to be non-coding with a strict immunogenic function (53). The extent to which such mutations
and their resulting FSPs have genomic implications in ICI therapy
warrants further study.
Yet, and irrespective of their potential with regard
to genomic function, the identification of these FSPs that are
present at higher frequencies in MSI-H/dMMR mCRC carries a
significant potential for future vaccine development and other
humoral FSP-based therapeutic strategies (42). With the mechanisms of response of
MSI-H/dMMR to ICIs in mind, clinical evidence for the effectiveness
of ICI for MSI has been provided in multiple studies, which is
discussed in the following section.
Evidence indicates that in patients with mCRC,
response to ICIs is limited to cases with MSI-H/dMMR molecular
status (54,55). In fact, the most recent practice
guidelines across major National and International organizations
recommend molecular testing for all newly diagnosed or recurrent
cases of advanced and mCRC for MMR status and MSI markers (56-59). While those with pMMR are treated
with systemic chemotherapy regimens, ICIs have been indicated for
patients with MSI-H/dMMR who have failed systemic therapies and
more recently, as an upfront therapy following an accelerated
approval by the FDA as the 1st line therapy (60,61). In this section, evidence from key
phase II and III trials supporting the use of ICIs in MSI-H/dMMR
mCRC, as well as ongoing trials, are reviewed (Table I).
A systematic review was conducted according to the
PRISMA guidelines with the last update of the search performed on
March 31, 2021. The search was conducted in PubMed as well as major
conference proceedings (American Society of Clinical Oncology;
European Society of Medical Oncology) using the following query
terms: (colon cancer OR rectal cancer OR colorectal cancer OR
colorectal neoplasm cancer) AND (MSI-H OR dMMR OR MSI-H/dMMR) OR
(immunotherapy OR ICI OR immune therapy OR anti-PD-1 OR anti-PD-L1
OR anti-CTLA-4). In addition, the clinical trials registry
(clinicaltrials.gov) was searched to
identify ongoing trials that have with so far unpublished reports.
Studies were included if they evaluated checkpoint inhibitors as a
monotherapy or in combination with any other agent in a clinical
trial setting in patients with MSI-H/dMMR mCRC. Studies were
excluded if they evaluated checkpoint inhibitor therapy or systemic
therapies for localized CRC or patients with MSS/pMMR, if a study
was a protocol-only publication without data or if it reported
overlapping data. In the latter case, the study with the most
recent and/or most comprehensive data was included. The initial
search identified a total of 29,980 studies. After review by title,
abstract and full-text review, 9 studies were included in the final
review (Table I). Furthermore, 28
additional ongoing and unpublished studies were identified via
clinicaltrials. gov (Table
II).
Pembrolizumab is currently approved for
chemo-refractory MSI-H/dMMR mCRC and as a 1st-line agent for this
population (61). It was first
evaluated in a phase II study (NCT01876511) involving patients with
mCRC who had at least two or more previous chemotherapy regimens
and consisting of three cohorts: dMMR (n=11), pMMR (n=21) and nine
patients with non-CRC dMMR gastrointestinal cancers (7). The primary endpoint was objective
response rate (ORR) evaluated by 'Response evaluation criteria in
solid tumors' (RECIST) v1.1. In the dMMR mCRC cohort, ORR was 40%
compared to 0% in the pMMR cohort. No complete response (CR) was
observed but patients with dMMR status had a high disease control
rate (DCR) of 90%, consisting of 40% partial responses (PR) and 50%
of patients with stable disease (SD) when evaluated at 12 weeks.
The median follow-up time was 36 weeks in the dMMR mCRC cohort and
20 weeks in the pMMR cohort, with a median progression-free
survival (PFS) that was not reached in the dMMR group [vs. 2.2
months for the pMMR group; hazard ratio (HR)=0.10; P<0.001]. At
week 20, PFS rates were 78 and 11%, respectively, and median OS was
not reached in the dMMR group (vs. 5.0 months for pMMR mCRCs). In a
follow-up study (KEYNOTE-016; NCT01876511) comprising 86 patients
with different refractory dMMR cancers, 40 patients had dMMR mCRC
(10). Compared to the previous
study, the ORR was 52% but CR was achieved in 12% of patients with
an average time to CR of 42 weeks. Neither median PFS nor OS were
reached (median follow-up of 12.5 months) but later follow-up
revealed a 2-year PFS of 59% and 2-year OS of 72% (10,62). In a subsequent phase II study
(KEYNOTE-164; NCT02460198) involving cases of MSI-H/dMMR
unresectable advanced or mCRC, pembrolizumab was administered to
patients who had received at least two prior therapies including
fluoropyrimidine, oxaliplatin and irinotecan (cohort A; n=61) or at
least one prior therapy (cohort B; n=63) (62). The primary endpoint (ORR) was
similar in both cohorts (33%) but patients with lesser prior
treatment (cohort B) had higher CR rates at 7.9% (vs. 3.3%). It is
worth noting that the ORR in this study (33%) was smaller than that
of KEYNOTE-016 (52%) and this may be attributed to a smaller cohort
size and the use of the immune-related RECIST (ir-RECIST) rather
than RECIST in the latter study. Similarly, the median PFS was
higher in cohort B (4.1 vs. 2.3 months) with an estimated 12-month
PFS rate of 41% (vs. 34% in cohort A). As far as the median OS was
concerned, it was not reached in the less pretreated cohort
compared to 31.4 months in the group with more previous lines of
treatment, with an estimated 1-year OS rate of 76 and 72%,
respectively. Besides confirming prior findings of a durable
clinical benefit in pretreated MSI-H/dMMR mCRC, the study indicated
a potential benefit from using anti-PD-1 in earlier stages of the
disease. Based on pooled data from both the KEYNOTE-016 and -0164
trials (n=90; pooled ORR: 36%) the FDA approved pembrolizumab for
patients with pretreated MSI-H/dMMR mCRC (63).
As a first-line treatment, pembrolizumab was
evaluated in a phase III trial, in which investigators evaluated
the efficacy and safety of pembrolizumab (n=153) vs. investigator's
choice of standard of care (SOC) chemotherapy (n=154) in dMMR mCRC
(KEYNOTE-177; NCT02563002) (64).
The primary endpoint of the study was median PFS with significantly
longer intervals in the pembrolizumab cohort (16.5 vs. 8.2 months;
HR=0.6 and P=0.0002) at the median follow-up (28.4 months). This
was also clinically meaningful with close to half of the patients
(48.3%) in the pembrolizumab arm without disease progression at 2
years, while patients experienced less drug-related adverse events
compared to the SOC group (grade 2-5 toxicities; 22 vs. 66%
respectively). Based on these results, the FDA approved
pembrolizumab as a first-line treatment option for patients with
MSI-H/dMMR mCRC (61). An updated
analysis of the results with the final PFS, PFS-2 (time from
randomization to progression on the next line of therapy or any
cause of death), as well as a health-related quality of life
(HR-QoL), was recently published (65). At the median follow-up of 32.4
months (range, 24.0-48.3 months), pembrolizumab continued to be
superior to SOC for PFS with a median PFS of 16.5 months (vs. 8.2
months; HR 0.60; P=0.0002), as well as 12- and 24-month PFS rates
of 55.3 and 48.3% (vs. 37.3 and 18.6% with chemotherapy),
respectively. The confirmed ORR was 43.8% (vs. 33.1%) for
pembrolizumab with longer durations of response (DOR; median DOR
not reached vs. 10.6 months in SOC). Similarly, PFS-2 was longer
with pembrolizumab (median not reached vs. 23.5 months; HR=0.63)
along with improved HR-QoL scores.
The clinical benefit of targeting PD-1 was also
demonstrated with nivolumab. CheckMate-142 (NCT02060188) is a
non-randomized phase II trial that evaluated nivolumab with or
without ipilimumab (anti-CTLA-4) in MSI-H/dMMR mCRC patients that
had been heavily pretreated (two or 3 previous lines of therapy)
(11). In the initial part of the
study, nivolumab was administered as monotherapy to 74 patients
with ORR as a primary endpoint evaluated by RECIST v1.1. The
results revealed an ORR of 32.4% with 2.7% CR and 29.7% PR (central
review). The median DOR was not reached, 1-year PFS was 50.4% and
1-year OS was 73.4%. Disease control (defined as CR, PR or stable
disease of 12 weeks or longer) was achieved by 63.5% of patients.
This study was the first to demonstrate durable response and
disease control with nivolumab in this patient population. Of note,
the benefits of anti-PD-1 monotherapy were regardless of tumor
PD-L1 expression, TILs status, mutational status (BRAF, KRAS) or
germline dMMR status. Based on data from CheckMate-142, the FDA
extended the approval of nivolumab for patients with MSI-H/dMMR
mCRC who are refractory to 5-fluorouracil, oxaliplatin and
irinotecan-based therapy (66).
In a second step of CheckMate-142, the efficacy of
combining nivolumab with ipilimumab (anti-CTLA-4) was analyzed in
119 patients with dMMR mCRC who had received two or three lines of
previous therapy (11). The
rationale behind this combination is that both ICIs operate at
different points of the immune response and when combined are able
to act synergistically to promote the anti-tumor response (67). While the ORR as a primary endpoint
reached 49% (4% CR; 45% PR), the median PFS and OS were not
reached, with one-year estimates of 71 and 85%, respectively. The
DCR for 12 weeks or longer and durable response for 12 months or
longer was 79 and 83%, respectively. The analysis of this cohort
revealed better efficacy in combining nivolumab and ipilimumab
compared to nivolumab monotherapy. Finally, the third cohort in
CheckMate-142 consisted of untreated patients with MSI-H/dMMR who
received nivolumab plus low-dose ipilimumab and the results
revealed an ORR of 60%, DCR of 84% and 12-month PFS of 77%
(68). Those results further
granted ipilimumab extended approval for use in patients with
MSI-H/dMMR mCRC (69).
Unlike monotherapy with anti-PD1 agents, which has
demonstrated clinical efficacy in the treatment of MSI-H/dMMR mCRC
cases, treatment with single-agent anti-CTLA-4 monotherapy
(ipilimumab or tremelimumab) failed to provide a clinical benefit
in this population. In a single-arm phase II study of anti-CTLA-4
monotherapy with tremelimumab that involved 47 heavily pre-treated
mCRC patients, 45 were considered response-evaluable, of which 44
did not reach the second dose of therapy (43 had progressive
disease and 1 discontinued due to treatment-related adverse events)
(70). Only one patient had
stable disease for 6 months and the study did not demonstrate any
clinically meaningful activity of single-agent tremilimumab. Thus,
for now, anti-CTLA-4 therapy in MSI-H/dMMR mCRC is reserved for
combined use with anti-PD-1 therapy, as discussed further
above.
Of note, the nivolumab/ipilimumab combination has
also been tested as a neo-adjuvant treatment for early CRC. In a
recent phase Ib/II trial, nivolumab was administered in combination
with ipilimumab to both dMMR (n=20) and pMMR (n=15) early-stage (I,
II or III) CRC prior to surgery (71). In the dMMR cohort, 100% (20/20)
achieved pathological response, with 60% CR and 95% major responses
(defined as <10% residual tumor). Surprisingly, pMMR tumors also
responded to the ICIs combination, albeit to a lesser extent (27%
pathological response, 13.3% CR, 6.7% PR and 20% major responses).
Regarding molecular markers, increased
CD8+/PD-1+ T-cell infiltration was predictive
of response in these tumors.
Atezolizumab targets the PD-L1 immune checkpoint. To
date, there has not been any completed large clinical study in
MSI-H/dMMR mCRC; however, early-phase studies involving anti-PD-L1
agents have been reported: In a phase I dose-escalation study of
atezolizumab, one of the patients with unselected (unknown MSI or
MMR status) mCRC achieved a durable PR (72). Another anti-PD-L1 monocloncal
antibody (BMS-936559) was evaluated in 207 patients with advanced
solid tumors, including 18 with unselected mCRC but no clinical
response was observed (73).
Atezolizumab has also been evaluated in combination
with SOC therapies. In a phase III trial (IMblaze370; NCT 02788279)
performed by Eng et al (74), 363 patients with unresectable,
locally advanced CRC or mCRC were treated with a combination of
atezolizumab and cobimetinib/MEK 1/2 inhibitor (cohort 1; n=183) or
atezolizumab monotherapy (cohort 2; n=80) or SOC regoraginib
(cohort 3; n=183). Patients with dMMR tumors constituted 5% of all
CRC cases. The results revealed no CR in any cohort along with low
rates of PR with no difference between the 3 different cohorts.
Similarly, no difference in PFS (1.91 vs. 1.94 vs. 2.0 months,
respectively) or OS (8.9 vs. 7.1 vs. 8.5 months, respectively) was
observed.
Atezolizumab has also been evaluated in a phase II
study as a first-line treatment in unresectable wild-type BRAF mCRC
in combination with fluoropyrimidine-bevacizumab treatment (MODUL;
NCT02291289) (75). Compared to
chemotherapy only, there was no improvement in PFS (HR=0.96) or OS
(HR=0.86).
Two large trials involving atezolizumab are
currently underway: In the COMMIT trial (NCT02997228), a phase III
study, patients newly diagnosed with mCRC (n=373, expected sample
size) are randomly assigned to either receive FOLFOX with
bevacizumab (control arm) with or without atezolizumab or
atezolizumab monotherapy (76).
The primary trial end-point was PFS, and secondary end-points
included OS and ORR. This awaited study may potentially provide
evidence for incorporating ICIs with SOC first-line agents in
MSI-H/dMMR mCRC. Another ongoing phase III randomized trial,
(ATOMIC ALLIANCE A021502; NCT02912559) is allocating stage III
MSI-H/dMMR CRC to either SOC adjuvant chemotherapy monotherapy
(FOLFOX for 6 months) or combined with atezolizumab, with an
additional 6-months maintenance treatment with anti-PD-L1 (77). This study will investigate the
ability of ICIs to eradicate any minimal residual disease in this
patient population.
Beyond the marked efficacy in patients with
MSI-H/dMMR mCRC, the use of ICIs in this patient population
revealed certain unique patterns of response previously uncommonly
observed with mCRC conventional therapy. Pseudo-progression is one
unique striking pattern encountered in mCRC, also reported in
melanoma, whereby an initial radiographic tumor enlargement is
observed after ICI therapy is initiated, followed by measurable
tumor regression months to years after therapy initiation (78,79). This phenomenon may be explained by
a transient increase in immune-cell tumor infiltration and
pro-inflammatory cytokine release that generate edema (79). As a consequence, RECIST was
modified to account for pseudo-progression through the development
of immune-related response criteria (ir-RECIST) to assess responses
to ICIs (80). In the trials
reviewed above, RECIST was used for response assessment except in
KEYNOTE-016, where ir-RECIST was used for primary endpoint
assessment (10). This may
explain why higher response rates were observed in KEYNOTE-016
compared to KEYNOTE-164 (where a pseudo-progression would be
considered a progression as per the RECIST criteria).
Another aspect particular to ICIs in MSI-H/dMMR mCRC
is the rate of CRs achieved compared to chemotherapy. In this
population, CR rates with combination chemotherapy have been
reported to be 1-2% (81), in
contrast to 3% with pembrolizumab [median follow-up, 12.6 months;
KEYNOTE-164; (62)], 3% for
nivolumab plus ipilimumab (median follow-up, 13.4 months;
CheckMate-142) and 3% for nivolumab monotherapy (median follow-up,
13 months) with an increase to 9% at a median follow-up of 21
months, thus achieving a deepening response over time
[CheckMate-142; (11,82)]. These radiologic responses further
translated to a clinical benefit as patients in CheckMate-142 who
had CR or PR to nivolumab achieved a 100% 2-year OS compared to 50%
among those with stable disease (83).
In addition to improved radiologic and clinical
responses, the use of ICIs in mCRC has been unique in terms of DOR;
for instance, the median DOR was not reached in CheckMate-142 after
a median follow-up of 21 months (82). This has translated into prolonged
median efficacy outcomes in MSI-H/dMMR mCRC in terms of median OS
and 1-year OS as discussed earlier.
In terms of safety, ICIs have demonstrated a
distinct safety profile compared to chemotherapy with toxicities
relating to the skin (rash, pruritis), the gastrointestinal system
(colitis) and endocrinopathies (thyroiditis, hypophysitis and
adrenal insufficiency) (84).
These toxicities are most likely secondary to autoimmune-like
T-cell-mediated toxicities induced by disruption of the
checkpoint's function. In the trials reviewed above, the most
common immune-related toxicities (seen in at least 5% of patients
receiving ICIs) included the following: Rash, pruritis, dry skin,
diarrhea, colitis, nausea and vomiting, pancreatitis, gastritis,
hepatic transaminitis, hypo/hyperthyroidism, hypophysitis, adrenal
insufficiency, acute kidney injury, pneumonitis and myocarditis
(10,11,84). Most of these adverse events
developed within 12 weeks of treatment initiation and resolved
within 12 weeks of onset or were easily reversed by appropriate
therapy. Compared to toxicities experienced with systemic therapy,
ICIs offer not only a unique profile but also significantly better
safety in terms of side effects.
At present, PD1 and CTLA-4 blockade are the only
FDA-approved ICIs for the treatment of MSI-H/dMMR mCRC. However,
several other immune checkpoint interactions regulate T-cell
activation in the TME. These include the following: T-cell
immunoglobulin mucin receptor 3 (TIM3), lymphocyte activation gene
3 and T-cell Ig and ITIM domains; in pre-clinical studies, they
have all been indicated to contribute to T-cell exhaustion and
promotion of CRC progression (85,86). These immune checkpoints are
currently being evaluated in phase I clinical trials to assess
their safety profile either as single agents or in combination with
PD-L1 blockade (Table II)
(87). Other agents with a
different mechanism of action are also being evaluated; they act as
antibody agonists of co-stimulatory immune receptors to enhance the
host's immune system response against tumor cells. These include
CD27, OX40 (CD134), 4-1BB (CD137) and glucocorticoid-induced TNF
receptor-related gene (GITR; CD357 and CD40) (88).
The use of ICIs in the treatment of MSI-H/dMMR mCRC
has also been explored in combination with other conventional CRC
therapies, including chemotherapy, targeted monoclonal antibodies
(mAbs), radiation therapy (RT) and small-molecule tyrosine kinase
inhibitors (TKIs). The rationale of combining ICIs with cytotoxic
chemotherapy relies on evidence suggesting that cytotoxic cell
killing results in cellular fragmentation that is taken up and
presented to T-cells by antigen-presenting cells (89). Furthermore, chemotherapy-induced
bone marrow suppression was indicated to decrease
immune-suppressive T-regulatory cells as well as induce
proliferation of other T-cells, resulting in stimulation of the
immune system (90). Trials
evaluating these treatments are summarized in Table II. Similar combinations are also
being explored in patients with MSS/unknown MSS status mCRC
[reviewed in (91)].
Another strategy to enhance the host immune response
includes combining ICIs with mAbs that block growth factor
receptors. In addition to this blockage, mAbs are thought to induce
antibody-dependent cell-mediated cytotoxicity, hence justifying the
above combination. However, current trials evaluating this
combination are not specifically targeted to MSI-H/dMMR mCRC but
rather serve as a potential strategy to sensitize the
immune-insensitive MSS/pMMR CRC tumors. Small-molecule TKIs such as
bevacizumab, an anti-VEGF molecule, have been evaluated in patients
with MSI-H/dMMR mCRC. In preclinical studies, targeting VEGF has
been demonstrated to offer potential therapeutic avenues for mCRC
via suppression of tumor-associated macrophages, increase of
interactions between dendritic cells and antigen-presenting cells,
as well as augmenting endothelial vasculature to increase
lymphocyte chemotaxis and T-cell tumor infiltration (92). This translated into a phase Ib
clinical study that evaluated atezolizumab and bevacizumab
combination in 10 pretreated patients with MSI-H/dMMR mCRC
(93). At 11 months, the median
OS had not been reached, the ORR was 30% and the median DOR was 7.8
months with 90% DCR. The ongoing COMMIT trial discussed earlier
(NCT02997228) is investigating the combination of bevacizumab with
chemo-immunotherapy (mFOLFOX6 and atezolizumab) as a first-line
therapy in patients with MSI-H/dMMR mCRC (76).
Finally, the combination of ICIs with RT has also
been considered in the treatment of mCRC based on the abscopal
effect theory, whereby radiation of cancer cells is thought to
induce immunogenic cell death (with increased neo-antigen
exposure), resulting in immune activation against tumor cells at
more distant sites (94). This
theory remains anecdotal with pre-clinical evidence [reviewed in
(95)], as well as clinical
evidence of ipilimumab/RT combination in melanoma achieving marked
tumor regression at non-irradiated metastatic sites (96). This synergistic treatment modality
has not been previously studied or explored in patients with
MSI-H/dMMR mCRC but has been evaluated as a way of sensitizing
MSS/pMMR mCRC to immunotherapy. In a phase II study (NCT02437071),
pembrolizumab was evaluated in combination with RT demonstrating
good tolerance, but modest effects in terms of response to ICI
monotherapy with only one patient achieving PR (ORR, 4.5%)
(97). A subsequent study
evaluated the effect of combining RT with dual ICIs: NSABP FC-9
(NCT02701400), a phase II single-arm study, evaluated the use of
durvalumab (anti-PD-L1) plus tremelimumab (anti-CTLA-4) following
hypofractionated RT in 20 patients with refractory MSS/pMMR mCRC
(98). The initial results
suggested a good overall safety profile and tolerance. Only two PRs
were observed and lasted >44 weeks, along with 2 patients with
stable disease for 12 and 16 weeks.
Despite the marked responses to ICI compared to
traditional therapies in patients with MSI-H/dMMR mCRC, up to 50%
of patients ultimately acquire resistance to immunotherapy with
subsequent progression and disease recurrence. While the MSI-H/dMMR
status in itself, along with the TMB status, are powerful
biomarkers of response to ICIs in mCRC, a requirement remains to
understand the mechanisms by which tumor cells develop resistance
to ICIs and eventually manage to re-escape the host's immune
surveillance mechanism.
Resistance to ICIs has been described as either
innate, termed as 'immunological ignorance' pertaining to natural
lack of immune response to developing tumors, or acquired following
treatment with ICIs (99). Innate
resistance is frequently observed in patients with systemic
immune-suppression (e.g. HIV patients) or in those that express few
molecular targets that are recognized by the immune system (e.g.
non-virally-induced tumors with low TMB expression, referred to as
'cold'). This is not the case in patients with MSI-H/dMMR mCRC with
high TMB and a TME rich in immune infiltrate. Those patients
respond well to ICIs but eventually develop acquired resistance to
immunotherapy. Acquired resistance has been looked at as a
by-product of two elements: i) The tumor's transcriptional profile
or gene signature, but more extensively ii) the molecular type of
the TME as classified by O'Donnell et al (99).
As far as the genomic signature is concerned,
gene-set enrichment analysis studies revealed that transcriptional
profiles enriched with IFN-γ response genes are associated with
better prognosis and response to anti-cancer immune therapies
(100). Conversely, tumors with
the innate anti-PD-1 resistance transcriptional profile lack any
response to anti-PD-1 ICIs (101). In MSI-H/dMMR mCRC, IFN-γ
response genes are enriched (31), accounting for the significant
response to ICIs. Other potential genomic markers of
response/resistance among this patient population have been
investigated and preliminary results did not indicate any
predictive impact of the RAS/RAF mutational status, PD-L1
expression or the origin of the MMR deficiency status (inherited
Lynch syndrome vs. acquired/sporadic origin) (7,11,82,102). Numerous potential mechanisms of
resistance are under investigation, including deleterious mutations
in JAK, loss of MHC molecules or beta-2-microglobulin (B2M)
loss-of-function mutations (103,104). Similarly, the amount of
extracellular mucin (assessed by histopathology) has been suggested
to be associated with resistance to ICI (105).
In terms of types of TME, MSI-H/dMMR mCRC tumors
belong to type 1 TME which includes tumors with high TMB and an
inflammatory gene signature. The three other types include
different combinations of TMB levels along with presence/absence of
inflammatory gene signature [reviewed in (99)]. The type of TME dictates the
interaction between tumor cells and the immune system, and in type
1, high TMB levels and the presence of an inflammation gene
signature are indicative of the existence of an ongoing but
functionally suppressed immune response (106). Despite having the best
probability of responding to the re-invigorative effect of ICIs,
MSI-H/dMMR tumors exploit immune-suppressive/evasive strategies
within the TME signaling pathway, which includes the following:
Adaptive immune resistance pathways, loss of tumor antigen
expression, insensitivity to interferons and cytokine/metabolite
dysregulation (99).
Understanding these mechanisms of resistance may provide additional
potential targets to treat MSI-H/dMMR tumors. Table III summarizes the mechanisms of
resistance to ICIs.
As mentioned in prior sections, elevated levels of
type I and II interferons as well as immunosuppressive cytokines
(IL-6, IL-12, TGF-β) lead to up-regulation of PD-L1 on immune cells
and potentiate immune escape. While this interaction between PD-1
and PD-L1 is successfully targeted and disrupted by ICIs, the
actual expression of PD-L1 is regulated in a complex manner beyond
cytokine release and this includes influences by genomic
alterations, transcriptional control mechanisms, mRNA stability as
well as oncogenic signaling and protein stability (107). These alternative regulators of
PD-L1 expression potentially represent novel targets to augment the
efficacy of ICIs and extend the DOR. Similarly, it was indicated
that tumors are able to release exosomes that express PD-L1,
further contributing to immune suppression (108). Other adaptive immune resistance
pathways have also been characterized, including overexpression of
CD155 in both tumor cells and tumor-infiltrating myeloid cells
(109). Binding of CD155 to CD96
and TGIT on tumor cells results in inhibition of the TILs and
conversely, binding to CD266 on T-cells and NK cells competes with
the prior inhibitory interaction (110). Thus, therapies targeting CD96
and TGIT may present a possible way to overcome resistance with
evidence from preclinical tumor models demonstrating efficacy and
synergy when PD-1 inhibition is combined with TGIT targeting
(109-111).
Finally, the adaptive immune resistance to PD-1
inhibition has been indicated to be mediated through T-cell induced
production of macrophage colony-stimulating factor 1 (MCSF-1),
suggesting co-treatment of patients with anti-PD1 and MCSF-1
inhibitors as a possible strategy (112).
Another mechanism for acquired resistance is the
loss of the ability to present neoantigens, mostly via mutations
affecting the antigen-presenting machinery such as proteasome
subunits or transporters associated with antigen processing, or
proteins involved in folding and sub-cellular translocation of MHC
molecules (113). For instance,
loss-of-function mutations of B2M, a chaperone protein essential
for the folding and transport of MHC molecules to the cell surface,
limits the recognition of tumor antigens by T-cells (114). This was clinically associated
with resistance to ICIs (101,104,115). Epigenetic events associated with
tumor development and progression constitute another mechanism that
alter human leukocyte antigen expression in tumor cells; unlike
genetic alterations, epigenetic changes may potentially be reversed
pharmacologically (113).
As indicated earlier, IFN-γ is a major regulator of
anti-tumor immunity that may also promote tumor resistance.
Exposure of tumors to IFN-γ in pre-clinical studies resulted in
genetic instability in these tumors that translated into higher
copy number variation associated with the DNA damage response and
repair genes (116). However,
IFN-γ was also reported to induce neoantigen loss/down-regulation,
thus favoring tumor escape (116,117). Similarly, in tumor analysis from
melanoma patients who responded poorly or acquired resistance to
anti-CTLA-4 therapy, a higher frequency of mutations within genes
involved in the IFN-γ signaling pathway was noted and this included
IFN-γ receptor 1 (IFNGR1), IFNGR2, JAK2 and IFN regulatory factor 1
(104,118). Similar observations of immune
escape have also been made through loss of sensitivity to TNF
(119). While the precise
mechanisms behind this loss of sensitivity in both pathways have
not been clearly defined, these observations suggest a potential
benefit from genetic screening to identify new immunotherapy
targets that operate in previously unknown pathways.
Hypoxia in TME promotes the accumulation of
extra-cellular ATP that is metabolized to adenosine (120). One of the downstream effects of
adenosine accumulation is the induction of strong immunosuppression
within the TME: Adenosine signaling may impair effector T-cells and
NK cells (120-122), and ADP-mediated
immunosuppression through production of adenosine has been reported
as a mechanism of tumor cell escape from PD-1/PD-L1 inhibition
(123). Based on these
observations, co-targeting enzymes involved in ATP degradation or
blocking adenosine receptors may improve the efficacy of PD-1/PD-L1
inhibition (120). Tryptophan is
another metabolite that mediates immunosupression when catabolised
in the TME, via upregulation of indoleamine 2,3-dioxygenase 1
(IDO1), an IFN-γ-inducible enzyme (124). Preclinical work in cancer mouse
models indicated that IDO promotes immunosuppression and IDO1
inhibitors had synergistic effects with ICIs (125). Despite promising early-phase
trials of IDO1 inhibitors, the first phase III trial of an IDO1
inhibitor in combination with an anti-PD-1 antibody in melanoma
patients did not achieve its primary endpoint (NCT02752074).
However, the study still demonstrated the importance of concurrent
biomarker and target development along with ICI development. In
addition to upregulation of immunosuppressive metabolites, tumor
cells frequently overexpress immunosuppressive cytokines, including
VEGF and TGF-β. VEGF promotes upregulation of PD-1, CTLA-4 and TIM3
on TILs, thus promoting T-cell dysfunction (126,127). Similarly, TGF-β was indicated to
upregulate PD-L1 expression and to promote metastasis of lung
tumors, thus suggesting the potential of co-targeting PD-L1 and
TGF-β receptor (128).
Immunotherapy with checkpoint inhibitors has
revolutionized the treatment of MSI-H/dMMR mCRC. This success is
primarily based on the discovery of the immune escape phenomenon on
the one hand and the advent of mAbs that block immune checkpoints
and re-invigorate the immune system on the other hand. This
strategy has proven successful in the MSI-H/dMMR population
specifically based on this subtype's high mutation burden and its
ability to present the FSP as neoantigens on MHC class I molecules
to prime T-cells to recognize them as foreign. This has culminated
in the FDA approval of anti-PD-L1 and anti-CTLA-4 ICIs for the
treatment of MSI-H/dMMR mCRC. Despite this success, challenges have
been encountered in primary non-responders and in those who develop
acquired resistance to ICIs. As the current knowledge of the immune
system and its intricacies continues to grow, a better
understanding of the different mechanisms behind immune resistance
is gradually being achieved, allowing for the development of novel
therapies, therapeutic combinations and strategies to overcome
resistance.
At present, new-generation ICIs are being evaluated
alone or in combination, with the hope of augmenting initial host
anti-tumor immune response and enhance therapeutic efficacy. Other
strategies include combination of ICIs with conventional therapy
(chemotherapy, radiation, TKIs), as well as with new immune
modulators. Furthermore, and based on the high frequency of
generated FSPs in MSI-H/dMMR mCRC populations, efforts have been
directed towards exploiting these mechanisms to develop
individualized therapies, either by developing epitope-based
vaccines or through harnessing adoptive cellular therapies via
ex-vivo T-cell manipulation and chimeric antigen receptor
T-cell generation. Although still in their early phases of clinical
research, these novel approaches in conjunction with the ongoing
trials outlined in the present article offer promising avenues to
further advance immunotherapy in patients with MSI-H/dMMR mCRC.
Data sharing is not applicable.
AS and AB conceived and designed the structure of
this manuscript. KC and MR reviewed the literature and collated
data from major studies and clinical trials. KC, MR and RP provided
the initial draft of the manuscript, including the tables. AS, AB,
KC and RP revised the manuscript for important intellectual
content. All authors read and approved the final manuscript. Data
authentication is not applicable.
Not applicable.
Not applicable.
AS reports research grants (to institution) for
unrelated research work from Astrazeneca, Bristol Myers Squibb,
Merck, Exelixis and Clovis and Advisory board/consultant fees from
Astrazeneca, Bristol Myers Squibb, Merck and Pfizer. The remaining
authors declare that they have no competing interests.
Not applicable.
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249. 2021.
View Article : Google Scholar
|
|
2
|
Siegel RL, Miller KD, Goding Sauer A,
Fedewa SA, Butterly LF, Anderson JC, Cercek A, Smith RA and Jemal
A: Colorectal cancer statistics, 2020. CA Cancer J Clin.
70:145–164. 2020. View Article : Google Scholar
|
|
3
|
Pearlman R, Frankel WL, Swanson B, Zhao W,
Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ, et
al: Prevalence and spectrum of germline cancer susceptibility gene
mutations among patients with early-onset colorectal cancer. JAMA
Oncol. 3:464–471. 2017. View Article : Google Scholar
|
|
4
|
Hull R, Francies FZ, Oyomno M and Dlamini
Z: Colorectal cancer genetics, incidence and risk factors: In
search for targeted therapies. Cancer Manag Res. 12:9869–9882.
2020. View Article : Google Scholar
|
|
5
|
Koopman M, Kortman GAM, Mekenkamp L,
Ligtenberg MJ, Hoogerbrugge N, Antonini NF, Punt CJ and van Krieken
JH: Deficient mismatch repair system in patients with sporadic
advanced colorectal cancer. Br J Cancer. 100:266–273. 2009.
View Article : Google Scholar
|
|
6
|
Venderbosch S, Nagtegaal ID, Maughan TS,
Smith CG, Cheadle JP, Fisher D, Kaplan R, Quirke P, Seymour MT,
Richman SD, et al: Mismatch repair status and BRAF mutation status
in metastatic colorectal cancer patients: A pooled analysis of the
CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res.
20:5322–5330. 2014. View Article : Google Scholar
|
|
7
|
Le DT, Uram JN, Wang H, Bartlett BR,
Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, et
al: PD-1 blockade in tumors with mismatch-repair deficiency. N Engl
J Med. 372:2509–2520. 2015. View Article : Google Scholar
|
|
8
|
Morse MA, Hochster H and Benson A:
Perspectives on treatment of metastatic colorectal cancer with
immune checkpoint inhibitor therapy. Oncologist. 25:33–45. 2020.
View Article : Google Scholar
|
|
9
|
Chau I and Cunningham D: Treatment in
advanced colorectal cancer: What, when and how? Br J Cancer.
100:1704–1719. 2009. View Article : Google Scholar
|
|
10
|
Le DT, Durham JN, Smith KN, Wang H,
Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et
al: Mismatch repair deficiency predicts response of solid tumors to
PD-1 blockade. Science. 357:409–413. 2017. View Article : Google Scholar
|
|
11
|
Overman MJ, McDermott R, Leach JL, Lonardi
S, Lenz HJ, Morse MA, Desai J, Hill A, Axelson M, Moss RA, et al:
Nivolumab in patients with metastatic DNA mismatch repair-deficient
or microsatellite instability-high colorectal cancer (CheckMate
142): An open-label, multicentre, phase 2 study. Lancet Oncol.
18:1182–1191. 2017. View Article : Google Scholar
|
|
12
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar
|
|
13
|
Saunders PA, Hendrycks VR, Lidinsky WA and
Woods ML: PD-L2:PD-1 involvement in T cell proliferation, cytokine
production, and integrin-mediated adhesion. Eur J Immunol.
35:3561–3569. 2005. View Article : Google Scholar
|
|
14
|
Sharma P and Allison JP: Immune checkpoint
targeting in cancer therapy: Toward combination strategies with
curative potential. Cell. 161:205–214. 2015. View Article : Google Scholar
|
|
15
|
Ijsselsteijn ME, Petitprez F, Lacroix L,
Ruano D, van der Breggen R, Julie C, Morreau H, Sautès-Fridman C,
Fridman WH and de Miranda NFDCC: Revisiting immune escape in
colorectal cancer in the era of immunotherapy. Br J Cancer.
120:815–818. 2019. View Article : Google Scholar
|
|
16
|
Hodi FS, O'Day SJ, McDermott DF, Weber RW,
Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel
JC, et al: Improved survival with ipilimumab in patients with
metastatic melanoma. N Engl J Med. 363:711–723. 2010. View Article : Google Scholar
|
|
17
|
Robert C, Long GV, Brady B, Dutriaux C,
Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C,
Kalinka-Warzocha E, et al: Nivolumab in previously untreated
melanoma without BRAF mutation. N Engl J Med. 372:320–330. 2015.
View Article : Google Scholar
|
|
18
|
Garon EB, Rizvi NA, Hui R, Leighl N,
Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L,
et al: Pembrolizumab for the treatment of non-small-cell lung
cancer. N Engl J Med. 372:2018–2028. 2015. View Article : Google Scholar
|
|
19
|
Brahmer J, Reckamp KL, Baas P, Crinò L,
Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE,
Holgado E, et al: Nivolumab versus docetaxel in advanced
squamous-cell non-small-cell lung cancer. N Engl J Med.
373:123–135. 2015. View Article : Google Scholar
|
|
20
|
Borghaei H, Paz-Ares L, Horn L, Spigel DR,
Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al:
Nivolumab versus docetaxel in advanced nonsquamous non-small-cell
lung cancer. N Engl J Med. 373:1627–1639. 2015. View Article : Google Scholar
|
|
21
|
Schadendorf D, Hodi FS, Robert C, Weber
JS, Margolin K, Hamid O, Patt D, Chen TT, Berman DM and Wolchok JD:
Pooled analysis of long-term survival data from phase II and phase
III trials of ipilimumab in unresectable or metastatic melanoma. J
Clin Oncol. 33:1889–1894. 2015. View Article : Google Scholar
|
|
22
|
Rizvi NA, Hellmann MD, Snyder A, Kvistborg
P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al:
Mutational landscape determines sensitivity to PD-1 blockade in
non-small cell lung cancer. Science. 348:124–128. 2015. View Article : Google Scholar
|
|
23
|
Schumacher TN and Schreiber RD:
Neoantigens in cancer immunotherapy. Science. 348:69–74. 2015.
View Article : Google Scholar
|
|
24
|
Subbiah V, Solit DB, Chan TA and Kurzrock
R: The FDA approval of pembrolizumab for adult and pediatric
patients with tumor mutational burden (TMB) ≥10: A decision
centered on empowering patients and their physicians. Ann Oncol.
31:1115–1118. 2020. View Article : Google Scholar
|
|
25
|
Mlecnik B, Tosolini M, Kirilovsky A,
Berger A, Bindea G, Meatchi T, Bruneval P, Trajanoski Z, Fridman
WH, Pagès F and Galon J: Histopathologic-based prognostic factors
of colorectal cancers are associated with the state of the local
immune reaction. J Clin Oncol. 29:610–618. 2011. View Article : Google Scholar
|
|
26
|
Galon J, Costes A, Sanchez-Cabo F,
Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M,
Berger A, Wind P, et al: Type, density, and location of immune
cells within human colorectal tumors predict clinical outcome.
Science. 313:1960–1964. 2006. View Article : Google Scholar
|
|
27
|
Galon J, Fridman WH and Pagès F: The
adaptive immunologic microenvironment in colorectal cancer: A novel
perspective. Cancer Res. 67:1883–1886. 2007. View Article : Google Scholar
|
|
28
|
Franke AJ, Skelton WP, Starr JS, Parekh H,
Lee JJ, Overman MJ, Allegra C and George TJ: Immunotherapy for
colorectal cancer: A review of current and novel therapeutic
approaches. J Natl Cancer Inst. 111:1131–1141. 2019. View Article : Google Scholar
|
|
29
|
Shia J, Holck S, DePetris G, Greenson JK
and Klimstra DS: Lynch syndrome-associated neoplasms: A discussion
on histopathology and immunohistochemistry. Fam Cancer. 12:241–260.
2013. View Article : Google Scholar
|
|
30
|
Choi YY, Bae JM, An JY, Kwon IG, Cho I,
Shin HB, Eiji T, Aburahmah M, Kim HI, Cheong JH, et al: Is
microsatellite instability a prognostic marker in gastric cancer? A
systematic review with meta-analysis. J Surg Oncol. 110:129–135.
2014. View Article : Google Scholar
|
|
31
|
Llosa NJ, Cruise M, Tam A, Wicks EC,
Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS,
et al: The vigorous immune microenvironment of microsatellite
instable colon cancer is balanced by multiple counter-inhibitory
checkpoints. Cancer Discov. 5:43–51. 2015. View Article : Google Scholar
|
|
32
|
Lagos GG, Izar B and Rizvi NA: Beyond
tumor PD-L1: Emerging genomic biomarkers for checkpoint inhibitor
immunotherapy. Am Soc Clin Oncol Educ Book. 40:1–11. 2020.
|
|
33
|
Choucair K, Morand S, Stanbery L, Edelman
G, Dworkin L and Nemunaitis J: TMB: A promising immune-response
biomarker, and potential spearhead in advancing targeted therapy
trials. Cancer Gene Ther. 27:841–853. 2020. View Article : Google Scholar
|
|
34
|
Bever KM and Le DT: An expanding role for
immunotherapy in colorectal cancer. J Natl Compr Canc Netw.
15:401–410. 2017. View Article : Google Scholar
|
|
35
|
Vignali DA and Kuchroo VK: IL-12 family
cytokines: Immunological playmakers. Nat Immunol. 13:722–728. 2012.
View Article : Google Scholar
|
|
36
|
Kim TM, Laird PW and Park PJ: The
landscape of microsatellite instability in colorectal and
endometrial cancer genomes. Cell. 155:858–868. 2013. View Article : Google Scholar
|
|
37
|
Price TJ, Thavaneswaran S, Burge M,
Segelov E, Haller DG, Punt CJ, Arnold D, Karapetis CS, Tebbutt NC,
Pavlakis N, et al: Update on optimal treatment for metastatic
colorectal cancer from the ACTG/AGITG expert meeting: ECCO.
2015.Expert Rev Anticancer Ther. 16:557–571. 2016. View Article : Google Scholar
|
|
38
|
Valero C, Lee M, Hoen D, Zehir A, Berger
MF, Seshan VE, Chan TA and Morris LGT: Response rates to anti-PD-1
immunotherapy in microsatellite-stable solid tumors with 10 or more
mutations per megabase. JAMA Oncol. 7:739–743. 2021. View Article : Google Scholar
|
|
39
|
Segal NH, Parsons DW, Peggs KS, Velculescu
V, Kinzler KW, Vogelstein B and Allison JP: Epitope landscape in
breast and colorectal cancer. Cancer Res. 68:889–892. 2008.
View Article : Google Scholar
|
|
40
|
Kloor M and von Knebel Doeberitz M: The
immune biology of microsatellite-unstable cancer. Trends Cancer.
2:121–133. 2016. View Article : Google Scholar
|
|
41
|
Bauer K, Nelius N, Reuschenbach M, Koch M,
Weitz J, Steinert G, Kopitz J, Beckhove P, Tariverdian M, von
Knebel Doeberitz M and Kloor M: T cell responses against
microsatellite instability-induced frameshift peptides and
influence of regulatory T cells in colorectal cancer. Cancer
Immunol Immunother. 62:27–37. 2013. View Article : Google Scholar
|
|
42
|
Kloor M, Reuschenbach M, Pauligk C,
Karbach J, Rafiyan MR, Al-Batran SE, Tariverdian M, Jäger E and von
Knebel Doeberitz M: A frameshift peptide neoantigen-based vaccine
for mismatch repair-deficient cancers: A phase I/IIa clinical
trial. Clin Cancer Res. 26:4503–4510. 2020. View Article : Google Scholar
|
|
43
|
Reuschenbach M, Kloor M, Morak M,
Wentzensen N, Germann A, Garbe Y, Tariverdian M, Findeisen P,
Neumaier M, Holinski-Feder E and von Knebel Doeberitz M: Serum
antibodies against frameshift peptides in microsatellite unstable
colorectal cancer patients with Lynch syndrome. Fam Cancer.
9:173–179. 2010. View Article : Google Scholar
|
|
44
|
Ballhausen A, Przybilla MJ, Jendrusch M,
Haupt S, Pfaffendorf E, Draxlbauer M, Seidler F, Krausert S,
Ahadova A, Kalteis MS, et al: The shared neoantigen landscape of
MSI cancers reflects immunoediting during tumor evolution. bioRxiv.
Jan 1–2019.Epub ahead of print.
|
|
45
|
Rus Bakarurraini NAA, Ab Mutalib NS and
Jamal R: Abu N. The landscape of tumor-specific antigens in
colorectal cancer. Vaccines (Basel). 8:3712020. View Article : Google Scholar
|
|
46
|
Yang G, Zheng R and Jin Z: Correlations
between microsatellite instability and the biological behaviour of
tumours. J Cancer Res Clin Oncol. 145:2891–2899. 2019. View Article : Google Scholar
|
|
47
|
Kim NG, Rhee H, Li LS and Kim H, Lee JS,
Kim JH, Kim NK and Kim H: Identification of MARCKS, FLJ11383 and
TAF1B as putative novel target genes in colorectal carcinomas with
microsatellite instability. Oncogene. 21:5081–5087. 2002.
View Article : Google Scholar
|
|
48
|
Itatani Y, Kawada K and Sakai Y:
Transforming growth factor-β signaling pathway in colorectal cancer
and its tumor microenvironment. Int J Mol Sci. 20:58222019.
View Article : Google Scholar
|
|
49
|
Bickeböller M, Tagscherer KE, Kloor M,
Jansen L, Chang-Claude J, Brenner H, Hoffmeister M, Toth C,
Schirmacher P, Roth W and Bläker H: Functional characterization of
the tumor-suppressor MARCKS in colorectal cancer and its
association with survival. Oncogene. 34:1150–1159. 2015. View Article : Google Scholar
|
|
50
|
Hornung V, Ablasser A, Charrel-Dennis M,
Bauernfeind F, Horvath G, Caffrey DR, Latz E and Fitzgerald KA:
AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating
inflammasome with ASC. Nature. 458:514–518. 2009. View Article : Google Scholar
|
|
51
|
Xu M, Wang J, Li H, Zhang Z and Cheng Z:
AIM2 inhibits colorectal cancer cell proliferation and migration
through suppression of Gli1. Aging (Albany NY). 13:1017–1031. 2020.
View Article : Google Scholar
|
|
52
|
Ivanov I, Lo KC, Hawthorn L, Cowell JK and
Ionov Y: Identifying candidate colon cancer tumor suppressor genes
using inhibition of nonsense-mediated mRNA decay in colon cancer
cells. Oncogene. 26:2873–2884. 2007. View Article : Google Scholar
|
|
53
|
Tougeron D, Fauquembergue E, Rouquette A,
Le Pessot F, Sesboüé R, Laurent M, Berthet P, Mauillon J, Di Fiore
F, Sabourin JC, et al: Tumor-infiltrating lymphocytes in colorectal
cancers with microsatellite instability are correlated with the
number and spectrum of frameshift mutations. Mod Pathol.
22:1186–1195. 2009. View Article : Google Scholar
|
|
54
|
Topalian SL, Hodi FS, Brahmer JR,
Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD,
Sosman JA, Atkins MB, et al: Safety, activity, and immune
correlates of anti-PD-1 antibody in cancer. N Engl J Med.
366:2443–2454. 2012. View Article : Google Scholar
|
|
55
|
Brahmer JR, Drake CG, Wollner I, Powderly
JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller
TL, et al: Phase I study of single-agent anti-programmed death-1
(MDX-1106) in refractory solid tumors: Safety, clinical activity,
pharmacodynamics, and immunologic correlates. J Clin Oncol.
28:3167–3175. 2010. View Article : Google Scholar
|
|
56
|
Giardiello FM, Allen JI, Axilbund JE,
Boland CR, Burke CA, Burt RW, Church JM, Dominitz JA, Johnson DA,
Kaltenbach T, et al: Guidelines on genetic evaluation and
management of Lynch syndrome: A consensus statement by the US
multi-society task force on colorectal cancer. Gastroenterology.
147:502–526. 2014. View Article : Google Scholar
|
|
57
|
Stoffel EM, Mangu PB, Gruber SB, Hamilton
SR, Kalady MF, Lau MW, Lu KH, Roach N and Limburg PJ; American
Society of Clinical Oncology; European Society of Clinical
Oncology: Hereditary colorectal cancer syndromes: American society
of clinical oncology clinical practice guideline endorsement of the
familial risk-colorectal cancer: European society for medical
oncology clinical practice guidelines. J Clin Oncol. 33:209–217.
2015. View Article : Google Scholar
|
|
58
|
Luchini C, Bibeau F, Ligtenberg MJL, Singh
N, Nottegar A, Bosse T, Miller R, Riaz N, Douillard JY, Andre F and
Scarpa A: ESMO recommendations on microsatellite instability
testing for immunotherapy in cancer, and its relationship with
PD-1/PD-L1 expression and tumour mutational burden: A systematic
review-based approach. Ann Oncol. 30:1232–1243. 2019. View Article : Google Scholar
|
|
59
|
Monahan KJ, Bradshaw N, Dolwani S, Desouza
B, Dunlop MG, East JE, Ilyas M, Kaur A, Lalloo F, Latchford A, et
al: Guidelines for the management of hereditary colorectal cancer
from the British society of gastroenterology (BSG)/association of
coloproctology of Great Britain and Ireland (ACPGBI)/United Kingdom
cancer genetics group (UKCGG). Gut. 69:411–444. 2020. View Article : Google Scholar
|
|
60
|
Stein A, Moehler M, Trojan J, Goekkurt E
and Vogel A: Immuno-oncology in GI tumours: Clinical evidence and
emerging trials of PD-1/PD-L1 antagonists. Crit Rev Oncol Hematol.
130:13–26. 2018. View Article : Google Scholar
|
|
61
|
FDA approves pembrolizumab for first-line
treatment of MSI-H/dMMR colorectal cancer. FDA; 2020
|
|
62
|
Le DT, Kim TW, Van Cutsem E, Geva R, Jäger
D, Hara H, Burge M, O'Neil B, Kavan P, Yoshino T, et al: Phase II
open-label study of pembrolizumab in treatment-refractory,
microsatellite instability-high/mismatch repair-deficient
metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 38:11–19.
2020. View Article : Google Scholar
|
|
63
|
US. Food and drug administration: KEYTRUDA
(pembrolizumab) {package insert}. 2017.
|
|
64
|
Andre T, Shiu KK, Kim TW, Jensen BV,
Jensen LH, Punt CJA, Smith DM, Garcia-Carbonero R, Benavides M,
Gibbs P, et al: Pembrolizumab versus chemotherapy for
microsatellite instability-high/mismatch repair deficient
metastatic colorectal cancer: The phase 3 KEYNOTE-177 study. J Clin
Oncol. 38(18 Suppl): LBA42020. View Article : Google Scholar
|
|
65
|
Shiu KK, Andre T, Kim TW, Jensen BV,
Jensen LH, Punt cJA, Smith DM, Garcia-Carbonero R, Benavides M,
Gibbs P, et al: KEYNOTE-177: Phase III randomized study of
pembrolizumab versus chemotherapy for microsatellite
instability-high advanced colorectal cancer. J Clin Oncol. 39(3
Suppl): S62021. View Article : Google Scholar
|
|
66
|
FDA grants nivolumab accelerated approval
for MSI-H or dMMR colorectal cancer. FDA; 2017
|
|
67
|
Das R, Verma R, Sznol M, Boddupalli CS,
Gettinger SN, Kluger H, Callahan M, Wolchok JD, Halaban R,
Dhodapkar MV and Dhodapkar KM: Combination therapy with anti-CTLA-4
and anti-PD-1 leads to distinct immunologic changes in vivo. J
Immunol. 194:950–959. 2015. View Article : Google Scholar
|
|
68
|
Lenz HJJ, Van Cutsem E, Limon ML, Wong KY,
Hendlisz A, Aglietta M, Garcia-Alfonso P, Neyns B, Luppi G, Cardin
D, et al: Durable clinical benefit with nivolumab (NIVO) plus
low-dose ipilimumab (IPI) as first-line therapy in microsatellite
instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic
colorectal cancer (mCRC). Ann Oncol. 29(Suppl 8): viii7142018.
View Article : Google Scholar
|
|
69
|
FDA grants accelerated approval to
ipilimumab for MSI-H or dMMR metastatic colorectal cancer. FDA;
2018
|
|
70
|
Chung KY, Gore I, Fong L, Venook A, Beck
SB, Dorazio P, Criscitiello PJ, Healey DI, Huang B, Gomez-Navarro J
and Saltz LB: Phase II study of the anti-cytotoxic
T-lymphocyte-associated antigen 4 monoclonal antibody,
tremelimumab, in patients with refractory metastatic colorectal
cancer. J Clin Oncol. 28:3485–3490. 2010. View Article : Google Scholar
|
|
71
|
Chalabi M, Fanchi LF, Dijkstra KK, Van den
Berg JG, Aalbers AG, Sikorska K, Lopez-Yurda M, Grootscholten C,
Beets GL, Snaebjornsson P, et al: Neoadjuvant immunotherapy leads
to pathological responses in MMR-proficient and MMR-deficient
early-stage colon cancers. Nat Med. 26:566–576. 2020. View Article : Google Scholar
|
|
72
|
Herbst RS, Garon EB, Kim DW, Cho BC,
Perez-Gracia JL, Han JY, Arvis CD, Majem M, Forster MD, Monnet I,
et al: Long-term outcomes and retreatment among patients with
previously treated, programmed death-ligand 1-positive, advanced
non-small-cell lung cancer in the KEYNOTE-010 study. J Clin Oncol.
38:1580–1590. 2020. View Article : Google Scholar
|
|
73
|
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ,
Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, et al:
Safety and activity of anti-PD-L1 antibody in patients with
advanced cancer. N Engl J Med. 366:2455–2465. 2012. View Article : Google Scholar
|
|
74
|
Eng C, Kim TW, Bendell J, Argilés G,
Tebbutt NC, Di Bartolomeo M, Falcone A, Fakih M, Kozloff M, Segal
NH, et al: Atezolizumab with or without cobimetinib versus
regorafenib in previously treated metastatic colorectal cancer
(IMblaze370): A multicentre, open-label, phase 3, randomised,
controlled trial. Lancet Oncol. 20:849–861. 2019. View Article : Google Scholar
|
|
75
|
Grothey A, Tabernero J, Arnold D, De
Gramont A, Ducreux MP, O'Dwyer PJ, Van Cutsem E, Bosanac I, Srock
S, Mancao C, et al: Fluoropyrimidine (FP) + bevacizumab (BEV) +
atezolizumab vs FP/BEV in BRAFwt metastatic colorectal cancer
(mCRC): Findings from Cohort 2 of MODUL-a multicentre, randomized
trial of biomarker-driven maintenance treatment following
first-line induction therapy. Ann Oncol. 29(Suppl 8):
viii714–viii715. 2018. View Article : Google Scholar
|
|
76
|
Lee JJ, Yothers G, Jacobs SA, Sanoff HK,
Cohen DJ, Guthrie KA, Henry NL, Ganz PA, Kopetz S, Lucas PC, et al:
Colorectal cancer metastatic dMMR immuno-therapy (COMMIT) study
(NRG-GI004/SWOG-S1610): A randomized phase III study of
mFOLFOX6/bevacizumab combination chemotherapy with or without
atezolizumab or atezolizumab monotherapy in the first-line
treatment of patients with deficient DNA mismatch repair (dMMR)
metastatic colorectal cancer. J Clin Oncol. 36(Suppl 15):
TPS36152018. View Article : Google Scholar
|
|
77
|
Sinicrope FA, Ou FS, Shi Q, Nixon AB, Mody
K, Levasseur A, Dueck AC, Dhanarajan AR, Lieu CH, Cohen DJ, et al:
Randomized trial of FOLFOX alone or combined with atezolizumab as
adjuvant therapy for patients with stage III colon cancer and
deficient DNA mismatch repair or microsatellite instability
(ATOMIC, Alliance A021502). J Clin Oncol. 35(Suppl 15):
TPS36302017. View Article : Google Scholar
|
|
78
|
Chiou VL and Burotto M: Pseudoprogression
and Immune-Related Response in Solid Tumors. J Clin Oncol.
33:3541–3543. 2015. View Article : Google Scholar
|
|
79
|
Solinas C, Porcu M, Hlavata Z, De Silva P,
Puzzoni M, Willard-Gallo K, Scartozzi M and Saba L: Critical
features and challenges associated with imaging in patients
undergoing cancer immunotherapy. Crit Rev Oncol Hematol. 120:13–21.
2017. View Article : Google Scholar
|
|
80
|
Seymour L, Bogaerts J, Perrone A, Ford R,
Schwartz LH, Mandrekar S, Lin NU, Litière S, Dancey J, Chen A, et
al: iRECIST: Guidelines for response criteria for use in trials
testing immunotherapeutics. Lancet Oncol. 18:e143–e152. 2017.
View Article : Google Scholar
|
|
81
|
Ducreux M, Bennouna J, Hebbar M, Ychou M,
Lledo G, Conroy T, Adenis A, Faroux R, Rebischung C, Bergougnoux L,
et al: Capecitabine plus oxaliplatin (XELOX) versus
5-fluorouracil/leucovorin plus oxaliplatin (FOLFOX-6) as first-line
treatment for metastatic colorectal cancer. Int J Cancer.
128:682–690. 2011. View Article : Google Scholar
|
|
82
|
Overman MJ, Bergamo F, McDermott RS,
Aglietta M, Chen F, Gelsomino F, Wong M, Morse M, Van Cutsem E,
Hendlisz A, et al: Nivolumab in patients with DNA mismatch
repair-deficient/microsatellite instability-high (dMMR/MSI-H)
metastatic colorectal cancer (mCRC): Long-term survival according
to prior line of treatment from CheckMate-142. J Clin Oncol.
36(Suppl 4): S5542018. View Article : Google Scholar
|
|
83
|
Overman MJ, Lonardi S, Wong KYM, Lenz HJ,
Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill
A, et al: Durable clinical benefit with nivolumab plus ipilimumab
in DNA mismatch repair-deficient/microsatellite instability-high
metastatic colorectal cancer. J Clin Oncol. 36:773–779. 2018.
View Article : Google Scholar
|
|
84
|
Brahmer JR, Lacchetti C, Schneider BJ,
Atkins MB, Brassil KJ, Caterino JM, Chau I, Ernstoff MS, Gardner
JM, Ginex P, et al: Management of immune-related adverse events in
patients treated with immune checkpoint inhibitor therapy: American
society of clinical oncology clinical practice guideline. J Clin
Oncol. 36:1714–1768. 2018. View Article : Google Scholar
|
|
85
|
Woo SR, Turnis ME, Goldberg MV, Bankoti J,
Selby M, Nirschl CJ, Bettini ML, Gravano DM, Vogel P, Liu CL, et
al: Immune inhibitory molecules LAG-3 and PD-1 synergistically
regulate T-cell function to promote tumoral immune escape. Cancer
Res. 72:917–927. 2012. View Article : Google Scholar
|
|
86
|
Ngiow SF, von Scheidt B, Akiba H, Yagita
H, Teng MW and Smyth MJ: Anti-TIM3 antibody promotes T cell
IFN-γ-mediated antitumor immunity and suppresses established
tumors. Cancer Res. 71:3540–3551. 2011. View Article : Google Scholar
|
|
87
|
Anderson AC, Joller N and Kuchroo VK:
Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized
functions in immune regulation. Immunity. 44:989–1004. 2016.
View Article : Google Scholar
|
|
88
|
Ward-Kavanagh LK, Lin WW, Šedý JR and Ware
CF: The TNF receptor superfamily in co-stimulating and
co-inhibitory responses. Immunity. 44:1005–1019. 2016. View Article : Google Scholar
|
|
89
|
Kroemer G, Galluzzi L, Kepp O and Zitvogel
L: Immunogenic cell death in cancer therapy. Annu Rev Immunol.
31:51–72. 2013. View Article : Google Scholar
|
|
90
|
Sun X, Suo J and Yan J: Immunotherapy in
human colorectal cancer: Challenges and prospective. World J
Gastroenterol. 22:6362–6372. 2016. View Article : Google Scholar
|
|
91
|
Ganesh K, Stadler ZK, Cercek A, Mendelsohn
RB, Shia J, Segal NH and Diaz LA Jr: Immunotherapy in colorectal
cancer: Rationale, challenges and potential. Nat Rev Gastroenterol
Hepatol. 16:361–375. 2019. View Article : Google Scholar
|
|
92
|
Limagne E, Euvrard R, Thibaudin M, Rébé C,
Derangère V, Chevriaux A, Boidot R, Végran F, Bonnefoy N, Vincent
J, et al: Accumulation of MDSC and Th17 cells in patients with
metastatic colorectal cancer predicts the efficacy of a
FOLFOX-bevacizumab drug treatment regimen. Cancer Res.
76:5241–5252. 2016. View Article : Google Scholar
|
|
93
|
Hochster HS, Bendell JC, Cleary JM, Foster
P, Zhang W, He X, Hernandez G, Iizuka K and Eckhardt SG: Efficacy
and safety of atezolizumab (atezo) and bevacizumab (bev) in a phase
Ib study of microsatellite instability (MSI)-high metastatic
colorectal cancer (mCRC). J Clin Oncol. 35(Suppl 4): S6732017.
View Article : Google Scholar
|
|
94
|
Kaminski JM, Shinohara E, Summers JB,
Niermann KJ, Morimoto A and Brousal J: The controversial abscopal
effect. Cancer Treat Rev. 31:159–172. 2005. View Article : Google Scholar
|
|
95
|
Gaipl US, Multhoff G, Scheithauer H,
Lauber K, Hehlgans S, Frey B and Rödel F: Kill and spread the word:
Stimulation of antitumor immune responses in the context of
radiotherapy. Immunotherapy. 6:597–610. 2014. View Article : Google Scholar
|
|
96
|
Postow MA, Callahan MK, Barker CA, Yamada
Y, Yuan J, Kitano S, Mu Z, Rasalan T, Adamow M, Ritter E, et al:
Immunologic correlates of the abscopal effect in a patient with
melanoma. N Engl J Med. 366:925–931. 2012. View Article : Google Scholar
|
|
97
|
Segal NH, Kemeny NE, Cercek A, Reidy DL,
Raasch PJ, War ren P, Hrabovsky AE, Campbell N, Shia J, Goodman KA,
et al: Non-randomized phase II study to assess the efficacy of
pembrolizumab (Pem) plus radiotherapy (RT) or ablation in mismatch
repair proficient (pMMR) metastatic colorectal cancer (mCRC)
patients. J Clin Oncol. 34(Suppl 15): S35392016. View Article : Google Scholar
|
|
98
|
Lee JJ, Yothers G, George TJ, Fakih MG,
Mallick AB, Mitchell EP, Wade JL, Krauss JC, Kayaleh OR, Heron DE,
et al: Abstract 2257: Phase II study of dual immune checkpoint
blockade (ICB) with durvalumab (Durva) plus tremelimumab (T)
following palliative hypofractionated radiotherapy (SBRT) in
patients (pts) with microsatellite-stable (MSS) metastatic
colorectal cancer (mCRC) progressing on chemotherapy: NSABP FC-9.
Cancer Res. 79(Suppl 13): S22572019.
|
|
99
|
O'Donnell JS, Long GV, Scolyer RA, Teng
MWL and Smyth MJ: Resistance to PD1/PDL1 checkpoint inhibition.
Cancer Treat Rev. 52:71–81. 2017. View Article : Google Scholar
|
|
100
|
Chen DS and Mellman I: Elements of cancer
immunity and the cancer-immune set point. Nature. 541:321–330.
2017. View Article : Google Scholar
|
|
101
|
Hugo W, Zaretsky JM, Sun L, Song C, Moreno
BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G,
et al: Genomic and transcriptomic features of response to anti-PD-1
therapy in metastatic melanoma. Cell. 165:35–44. 2016. View Article : Google Scholar
|
|
102
|
O'Neil BH, Wallmark JM, Lorente D, Elez E,
Raimbourg J, Gomez-Roca C, Ejadi S, Piha-Paul SA, Stein MN, Abdul
Razak AR, et al: Safety and antitumor activity of the anti-PD-1
antibody pembrolizumab in patients with advanced colorectal
carcinoma. PLoS One. 12:e01898482017. View Article : Google Scholar
|
|
103
|
Kopetz S, Andre T, Overman MJ, Zagonel V,
Lonardi S, Aglietta M, Gelsomino F, McDermott R, Wong KYM, Hendlisz
A, et al: Abstract 2603: Exploratory analysis of Janus kinase 1
(JAK1) loss-of-function (LoF) mutations in patients with DNA
mismatch repair-deficient/microsatellite instability-high
(dMMR/MSI-H) metastatic colorectal cancer (mCRC) treated with
nivolumab + ipilimumab in CheckMate-142. Cancer Res. 78(Suppl 13):
S26032018.
|
|
104
|
Zaretsky JM, Garcia-Diaz A, Shin DS,
Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY,
Abril-Rodriguez G, Sandoval S, Barthly L, et al: Mutations
associated with acquired resistance to PD-1 blockade in melanoma. N
Engl J Med. 375:819–829. 2016. View Article : Google Scholar
|
|
105
|
Llosa NJ, Luber B, Siegel N, Awan AH, Oke
T, Zhu Q, Bartlett BR, Aulakh LK, Thompson ED, Jaffee EM, et al:
Immunopathologic stratification of colorectal cancer for checkpoint
blockade immunotherapy. Cancer Immunol Res. 7:1574–1579. 2019.
View Article : Google Scholar
|
|
106
|
Chen YP, Zhang Y, Lv JW, Li YQ, Wang YQ,
He QM, Yang XJ, Sun Y, Mao YP, Yun JP, et al: Genomic analysis of
tumor microenvironment immune types across 14 solid cancer types:
Immunotherapeutic implications. Theranostics. 7:3585–3594. 2017.
View Article : Google Scholar
|
|
107
|
Sun C, Mezzadra R and Schumacher TN:
Regulation and function of the PD-L1 checkpoint. Immunity.
48:434–452. 2018. View Article : Google Scholar
|
|
108
|
Daassi D, Mahoney KM and Freeman GJ: The
importance of exosomal PDL1 in tumour immune evasion. Nat Rev
Immunol. 20:209–215. 2020. View Article : Google Scholar
|
|
109
|
Li XY, Das I, Lepletier A, Addala V, Bald
T, Stannard K, Barkauskas D, Liu J, Aguilera AR, Takeda K, et al:
CD155 loss enhances tumor suppression via combined host and
tumor-intrinsic mechanisms. J Clin Invest. 128:2613–2625. 2018.
View Article : Google Scholar
|
|
110
|
Blake SJ, Stannard K, Liu J, Allen S, Yong
MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, et
al: Suppression of metastases using a new lymphocyte checkpoint
target for cancer immunotherapy. Cancer Discov. 6:446–459. 2016.
View Article : Google Scholar
|
|
111
|
Harjunpää H, Blake SJ, Ahern E, Allen S,
Liu J, Yan J, Lutzky V, Takeda K, Aguilera AR, Guillerey C, et al:
Deficiency of host CD96 and PD-1 or TIGIT enhances tumor immunity
without significantly compromising immune homeostasis.
OncoImmunology. 7:e14459492018. View Article : Google Scholar
|
|
112
|
Neubert NJ, Schmittnaegel M, Bordry N,
Nassiri S, Wald N, Martignier C, Tillé L, Homicsko K, Damsky W,
Maby-El Hajjami H, et al: T cell-induced CSF1promotes melanoma
resistance to PD1 blockade. Sci Transl Med. 10:eaan33112018.
View Article : Google Scholar
|
|
113
|
Seliger B: The link between MHC class I
abnormalities of tumors, oncogenes, tumor suppressor genes, and
transcription factors. J Immunotoxicol. 11:308–310. 2014.
View Article : Google Scholar
|
|
114
|
Godfrey DI, Le Nours J, Andrews DM,
Uldrich AP and Rossjohn J: Unconventional T cell targets for cancer
immunotherapy. Immunity. 48:453–473. 2018. View Article : Google Scholar
|
|
115
|
Sade-Feldman M, Jiao YJ, Chen JH, Rooney
MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum
H, Blackmon SM, et al: Resistance to checkpoint blockade therapy
through inactivation of antigen presentation. Nat Commun.
8:11362017. View Article : Google Scholar
|
|
116
|
Takeda K, Nakayama M, Hayakawa Y, Kojima
Y, Ikeda H, Imai N, Ogasawara K, Okumura K, Thomas DM and Smyth MJ:
IFN-γ is required for cytotoxic T cell-dependent cancer genome
immunoediting. Nat Commun. 8:146072017. View Article : Google Scholar
|
|
117
|
Dunn GP, Ikeda H, Bruce AT, Koebel C,
Uppaluri R, Bui J, Chan R, Diamond M, White JM, Sheehan KC and
Schreiber RD: Interferon-gamma and cancer immunoediting. Immunol
Res. 32:231–245. 2005. View Article : Google Scholar
|
|
118
|
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He
Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al: Loss of IFN-γ
pathway genes in tumor cells as a mechanism of resistance to
anti-CTLA-4 therapy. Cell. 167:397–404.e9. 2016. View Article : Google Scholar
|
|
119
|
Kearney CJ, Vervoort SJ, Hogg SJ,
Ramsbottom KM, Freeman AJ, Lalaoui N, Pijpers L, Michie J, Brown
KK, Knight DA, et al: Tumor immune evasion arises through loss of
TNF sensitivity. Sci Immunol 3: eaar3451,. 2018.
|
|
120
|
Vijayan D, Young A, Teng MWL and Smyth MJ:
Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer.
17:709–724. 2017. View Article : Google Scholar
|
|
121
|
Ohta A, Gorelik E, Prasad SJ, Ronchese F,
Lukashev D, Wong MK, Huang X, Caldwell S, Liu K, Smith P, et al:
A2A adenosine receptor protects tumors from antitumor T cells. Proc
Natl Acad Sci USA. 103:13132–13137. 2006. View Article : Google Scholar
|
|
122
|
Beavis PA, Divisekera U, Paget C, Chow MT,
John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ and Darcy PK:
Blockade of A2A receptors potently suppresses the metastasis of
CD73+ tumors. Proc Natl Acad Sci USA. 110:14711–14716.
2013. View Article : Google Scholar
|
|
123
|
Chen L, Diao L, Yang Y, Yi X, Rodriguez
BL, Li Y, Villalobos PA, Cascone T, Liu X, Tan L, et al:
CD38-mediated immunosuppression as a mechanism of tumor cell escape
from PD-1/PD-L1 blockade. Cancer Discov. 8:1156–1175. 2018.
View Article : Google Scholar
|
|
124
|
Triplett TA, Garrison KC, Marshall N,
Donkor M, Blazeck J, Lamb C, Qerqez A, Dekker JD, Tanno Y, Lu WC,
et al: Reversal of indoleamine 2,3-dioxygenase-mediated cancer
immune suppression by systemic kynurenine depletion with a
therapeutic enzyme. Nat Biotechnol. 36:758–764. 2018. View Article : Google Scholar
|
|
125
|
Moon YW, Hajjar J, Hwu P and Naing A:
Targeting the indoleamine 2,3-dioxygenase pathway in cancer. J
Immunother Cancer. 3:512015. View Article : Google Scholar
|
|
126
|
Voron T, Colussi O, Marcheteau E, Pernot
S, Nizard M, Point AL, Latreche S, Bergaya S, Benhamouda N, Tanchot
C, et al: VEGF-A modulates expression of inhibitory checkpoints on
CD8+ T cells in tumors. J Exp Med. 212:139–148. 2015.
View Article : Google Scholar
|
|
127
|
Johnston CJ, Smyth DJ, Dresser DW and
Maizels RM: TGF-β in tolerance, development and regulation of
immunity. Cell Immunol. 299:14–22. 2016. View Article : Google Scholar
|
|
128
|
Strauss J, Heery CR, Schlom J, Madan RA,
Cao L, Kang Z, Lamping E, Marté JL, Donahue RN, Grenga I, et al:
Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein
targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer
Res. 24:1287–1295. 2018. View Article : Google Scholar
|