Introduction
Breast cancer is currently one of the leading causes
of mortality due to cancer in females worldwide. Approximately
230,480 new cases of invasive breast cancer and 39,520 deaths due
to breast cancer were expected to occur among women in the USA in
2011 (1). Similar to other tumor
types, breast carcinoma is thought to arise following the
activation of oncogenes and inactivation of tumor suppressor genes.
p16, one of the most commonly inactivated tumor suppressor genes in
human cancer, is a cyclin-dependent kinase inhibitor that regulates
tumor progression through the G1 phase of the cell cycle (2). The downregulation of p16 expression
due to promoter hypermethylation frequently occurs in breast
cancer. A number of studies have shown that p16 hypermethylation is
an early and likely critical step in breast cancer development
(3,4). Aberrant DNA hypermethylation has been
increasingly recognized as a frequent molecular alteration in
cancer (5,6). This epigenetic modification occurs at
the cytosines of CpG dinucleotide-rich regions (CpG islands), which
are mostly unmethylated in normal tissues. The hypermethylation of
CpG islands in the gene promoter regions of numerous tumor
suppressor and DNA repair genes is associated with chromatin
condensation, replication delay, inhibition of the initiation of
transcription and gene silencing (7). Similar to a number of other genes,
p16 is commonly inactivated by the hypermethylation of its CpG-rich
promoter region (8). The
association between p16 promoter methylation and breast cancer
susceptibility has been extensively studied; however, the results
are inconsistent (8–16). Thus, a comprehensive meta-analysis
on the most recent and relevant articles was performed to identify
statistical evidence of the association between p16 gene
hypermethylation and breast cancer risk. In addition, the
correlation of p16 methylation with estrogen receptor (ER) and
progesterone receptor (PR) status in breast cancer differed from
those in previous studies. Therefore, a systematic review and
meta-analysis was conducted to evaluate this correlation
quantitatively.
Materials and methods
Meta-analysis
The meta-analysis was performed according to the
Preferred Reporting Items for Systematic Reviews and Meta-Analysis
(17) and the recommendations of
the Cochrane Collaboration (18).
The study was approved by the Research Ethics Committee of Jinling
Hospital.
Identification and eligibility of
relevant studies
To gather eligible articles, the terms ‘p16,’
‘methylation,’ ‘breast cancer,’ ‘breast tumor,’ ‘breast carcinoma,’
‘estrogen receptor,’ ‘ER,’ ‘progesterone receptor’ and ‘PR’ were
searched for in PubMed, Web of Science and EBSCO with an English
language restriction. The search results were updated until
February 1, 2012. Only published full-text articles were included
in the analysis. The following criteria were used in selecting
eligible articles: (a) articles dealing with p16 methylation and
breast cancer risk; (b) independent case-control, malignant and
benign breast cancer tissue studies with reliable methods; and (c)
sufficient data for estimating odds ratios (ORs) with 95%
confidence intervals (CIs). Articles concerning p16 methylation and
the ER and PR status in breast cancer were also searched.
Data extraction
Wang and Nie independently conducted reviews by
extracting data via a standardized approach. The publication
information (year of publication and name of first author), study
characteristics (sample size and distribution of ER and PR) and p16
hypermethylation rates were collected using standard data
extraction forms. The discrepancies were resolved through
discussions. Subgroup analyses were stratified using the ER and PR
status in breast cancer cases.
Statistical analysis
The ORs and 95% CIs were calculated to estimate the
strength of the association between p16 hypermethylation and breast
cancer risk. A Chi-square-based Q-test was performed to assess
between-study heterogeneity. If P>0.10, the studies were
considered to lack heterogeneity and the pooled ORs were calculated
using the fixed-effects model according to the Mantel-Haenszel
method. Otherwise, the random-effect model was used according to
the DerSimonian-Laird method. A sensitivity analysis was performed
to assess the stability of the meta-analysis results by repeating
the meta-analysis and omitting one study at each iteration
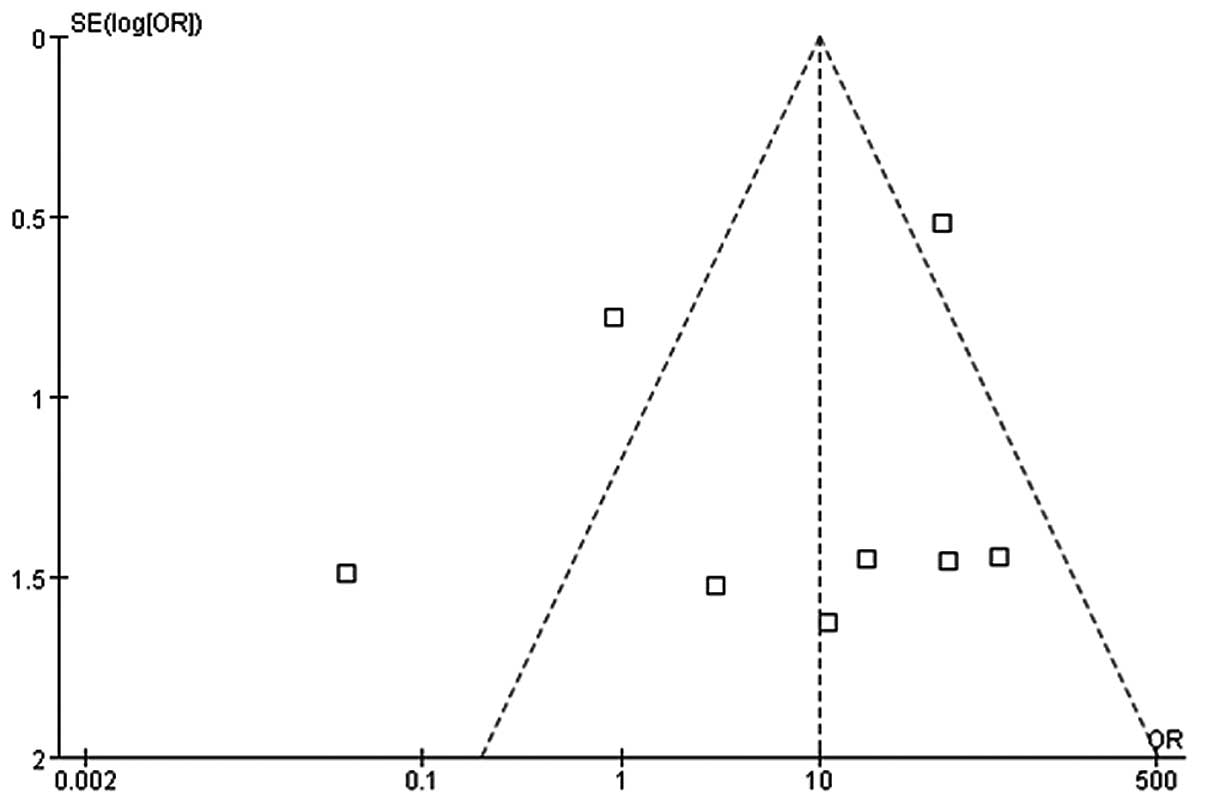
(19). Begg’s funnel plot and
Egger’s linear regression test were used to estimate potential
publication bias. An asymmetric funnel plot indicated possible
publication. In Egger’s test, P<0.05 indicated the existence of
statistically significant publication bias. The shapes of the
funnel plots did not reveal any evidence of asymmetry (Fig. 1) and the Egger’s test results did
not suggest any evidence of publication bias (P>1.00). All
statistical analyses in the current study were performed using
Review Manager 5 and Stata 11.0 software.
Results
Study characteristics
A total of eight articles met the inclusion criteria
and were used in the current meta-analysis. The studies involved
691 breast cancer cases and 525 controls. When the same
investigators reported results obtained from the same cohort of
patients in several publications, only the largest series was
included in the analysis. A cohort of patients was excluded due to
duplicate reports (20). Only four
articles were included in the analysis for the quantitative
evaluation of the correlation between p16 hypermethylation and the
ER and PR status in breast cancer (2,11,13,21).
The main characteristics of these studies are listed in Tables I–III.
 | Table IData of p16 methylation included in
this study. |
Table I
Data of p16 methylation included in
this study.
| | Case (n) | Control (n) |
|---|
| |
|
|
|---|
| First author
(ref.) | Year | Methylated | Unmethylated | Methylated | Unmethylated |
|---|
| Di Vinci A (8) | 2005 | 9 | 5 | 10 | 5 |
| Cavusoglu CA
(10) | 2010 | 15 | 47 | 0 | 4 |
| Jing F (11) | 2008 | 30 | 72 | 0 | 20 |
| Jing F (12) | 2010 | 15 | 35 | 0 | 50 |
| Raish M (13) | 2009 | 112 | 239 | 4 | 347 |
| Vallian S (14) | 2009 | 25 | 45 | 0 | 70 |
| Van Zee KJ (15) | 1998 | 16 | 17 | 11 | 0 |
| Woodcock DM (16) | 1999 | 5 | 4 | 0 | 4 |
| Table IIIThe data of methylation of p16 in
different PR statuses included in this study |
Table III
The data of methylation of p16 in
different PR statuses included in this study
| | Methylated (n) | Unmethylated
(n) |
|---|
| |
|
|
|---|
| First author
(ref.) | Year | PR(+) | PR(−) | PR(+) | PR(−) |
|---|
| Jing F (11) | 2008 | 21 | 9 | 28 | 44 |
| Li S (21) | 2006 | 18 | 8 | 102 | 60 |
| Tao MH (2) | 2009 | 78 | 56 | 247 | 146 |
| Raish M (13) | 2009 | 93 | 19 | 178 | 61 |
Main results
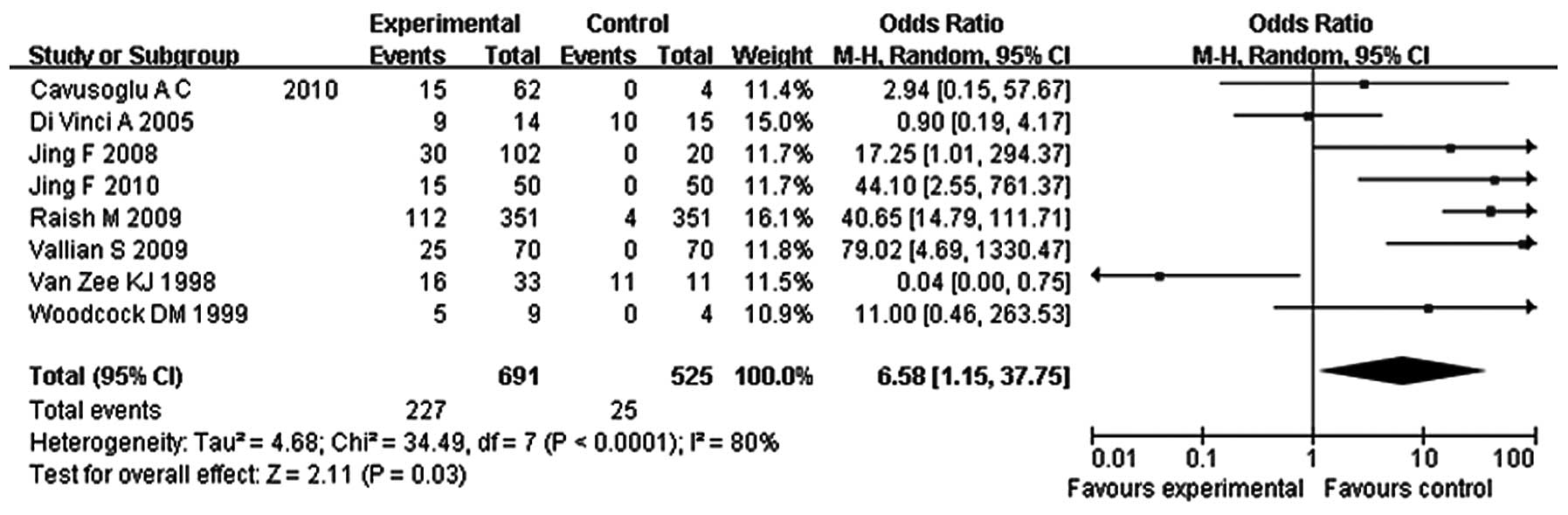
p16 hypermethylation in breast cancer
and control groups
Data for the p16 hypermethylation comparison among
the breast cancer and control groups were taken from eight studies.
The breast cancer and control groups included 227 (32.9%) and 25
(4.8%) p16 hypermethylated cases, respectively. The pooled analysis
shows that the ORs of the breast cancer group during p16
hypermethylation significantly increased compared with those of the
controls (OR, 6.58; 95% CI, 1.15–37.75; P=0.03). However, the
studies were heterogeneous (P<0.0001, I2=80%,
Fig. 2) and this heterogeneity was
incorporated into the random-effects model. The funnel plots did
not show any evidence of publication bias.
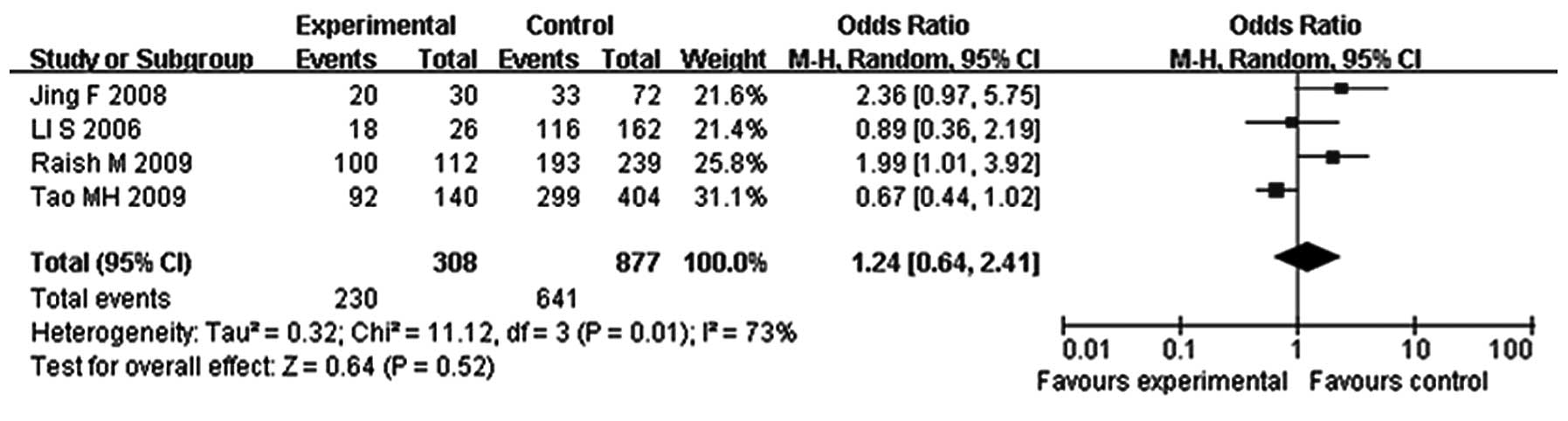
Correlation between p16 methylation
and the ER and PR status in breast cancer
A total of four studies calculated the ORs of p16
hypermethylation in ER-positive and -negative breast cancer
(Fig. 3). Of the 308
hypermethylated breast cancer cases, 230 were ER-positive (74.7%),
whereas among the 877 unmethylated breast cancer cases, 641 were
ER-positive (73.1%). The pooled analysis revealed no significantly
increased ORs in the correlation between the ER and methylation
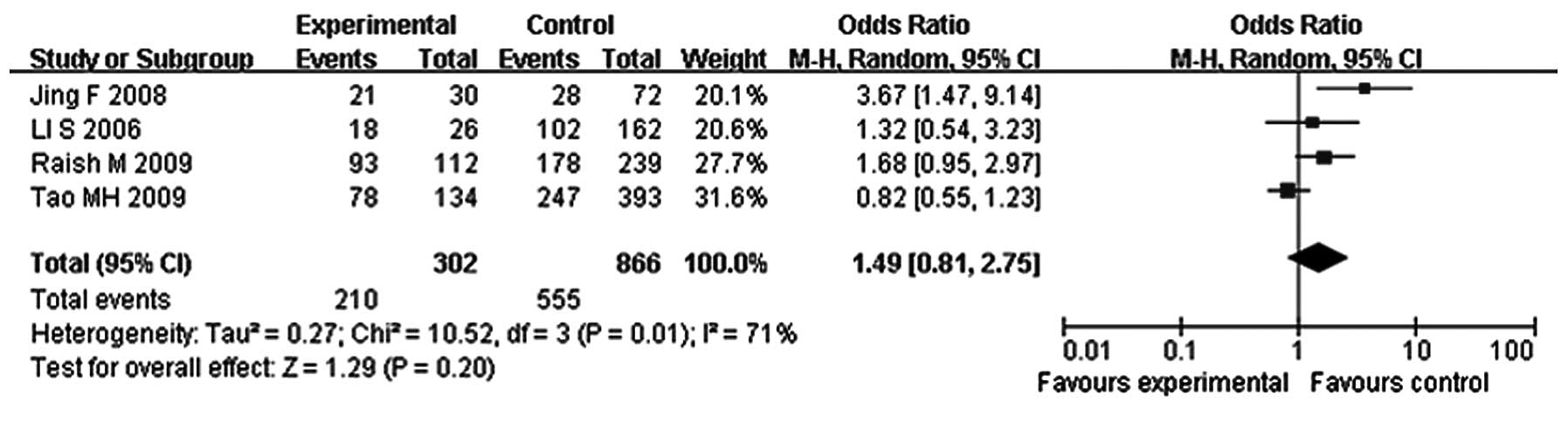
status in breast cancer (OR, 1.24; 95% CI, 0.64–2.41; P=0.52).
However, 210 out of the 302 hypermethylated breast cancer cases
were PR-positive (69.5%), whereas 555 out of the 866 unmethylated
breast cancer cases were PR-positive (64.1%). The pooled analysis
revealed no significantly increased ORs in the correlation between
the PR and methylation status in breast cancer (OR, 1.49; 95% CI,
0.81–2.75; P=0.20). However, the studies were heterogenous
(P<0.05, Figs. 3 and 4) and this heterogeneity was incorporated
into the random-effects model. Funnel plots did not show any
evidence of publication bias.
Discussion
Breast cancer is the most common cancer among women
and is the second leading cause of mortality worldwide (22). Its occurrence is associated with
genetic and environmental risk factors. CpG islands are located in
the promoter or first exon region of genes and are normally
unmethylated. However, in cancer cells, the hypermethylation of
these regions is associated with transcriptional silencing or a
decrease in expression. The promoter hypermethylation of tumor
suppressor genes, including p16, hMLH1 and VHL, has been
established as a common mechanism for tumor suppressor inactivation
in human cancer and has become a promising new molecular target for
its detection (8). Aberrant DNA
methylation has been increasingly recognized as a frequent
molecular alteration in breast cancer (11–14).
Genes involved in cell-cycle regulation (p16), cell adhesion
(CDH1), DNA repair (BRCA1) and cell signaling pathways (ER and
RARβ) have been reported to undergo hypermethylation (23,24).
The product of INK4A locus, p16, encodes a cyclin-dependent kinase
(CDK) inhibitor that functions as a negative regulator of
cyclin/CDK complexes. It binds preferentially to CDK4/6 and
prevents the association of CDK4/6 with D-type cyclins, thus
inhibiting pRB phosphorylation and progression through the cell
cycle (25,26). p16 also plays a significant role in
the maintenance of normal cellular properties and prevention of
centrosome dysfunction and genomic instability (27). p16 inactivation occurs at the early
stage of carcinogenesis (25,26)
and the loss of p16/Rb activity occurs through various mechanisms,
including the deletion, mutation and hypermethylation of p16
(27). p16 inactivation appears to
be crucial in the development of several tumors (13). However, its relevance to mammary
carcinogenesis remains unclear. Moreover, the homozygous deletions
of p16 are observed in half of the breast cancer cell lines and
neither homozygous deletions nor point mutations are frequently
observed in primary breast cancers, suggesting that these
alterations were acquired in the cultures (13). Aberrant hypermethylation has been
suggested to be a useful biomarker, with implications for breast
cancer etiology, diagnosis and management. Gene-specific promoter
alterations are common epigenetic aberrations in human breast
cancer. However, the epigenetic changes in p16 gene
hypermethylation specific to breast cancer etiology remain elusive.
The current pooled analysis comprehensively assessed the
correlation between p16 gene hypermethylation and the incidence of
breast cancer based on eight studies, which included 691 breast
cancer cases and 525 controls. Using the pooled crude ORs from
studies, we identified that p16 gene hypermethylation is associated
to 6.58-fold increased risks of breast cancer compared with the
control group. Moreover, no significant association was identified
among ER, PR and methylation status in breast cancer.
The current study has several potential limitations.
First, the possibility of information and selection biases and
unidentified confounders cannot be completely excluded as all
studies were observational. Second, the searching strategy was
restricted to English articles and thus, non-English articles with
potentially high-quality data were not included due to anticipated
difficulties in obtaining accurate medical translation. Third, most
of the studies included in this meta-analysis were conducted in
various ethnicities. Fourth, the results of the current study were
not further validated.
In conclusion, despite the discrepancy in the p16
hypermethylation in the breast cancer and control groups, we found
that p16 hypermethylation is associated with increased risk of
breast cancer. Jing et al(11) demonstrated that p16 methylation is
associated with the PR status. Tao et al(2) did not identify any significant
differences in p16 methylation patterns relevant to the ER and PR
status of breast cancer for postmenopausal women. The authors
identified that p16 hypermethylation tended to be more frequent
among ER-negative cases than ER-positive cases (OR, 1.51; 95% CI,
1.01–2.32). Various studies (2,11,13,21)
did not identify any significant differences in tumor-related gene
methylation patterns relevant to the ER and PR status of breast
tumors. This meta-analysis indicates that the ER, PR and
methylation status of the p16 gene in breast cancer are not
significantly associated with one another. p16 hypermethylation,
which mediates the inactivation of the p16 gene, plays a
significant role in breast carcinogenesis. Therefore, it may have
useful clinical applications in the detection of breast cancer at
the early stages. In addition, current cancer therapies use DNA
methylation inhibitors, including 5-aza-2′-deoxycytidine
(5-Aza-CdR), to reactivate the repressed p16 gene. The reactivation
of the p16 gene in cells with methylated genes restores normal cell
growth control (28–30). According to the current study, the
p16 promoter methylation status could be used for breast cancer
diagnostic and therapeutic purposes.
Acknowledgements
This project was supported by grants from National
Natural Science Foundation of China (81141094, 30870962, to X.
Guan), Natural Science Foundation of Jiangsu Province (BK2011656,
to X. Guan).
References
|
1
|
DeSantis C, Siegel R, Bandi P and Jemal A:
Breast cancer statistics, 2011. CA Cancer J Clin. 61:409–418. 2011.
View Article : Google Scholar
|
|
2
|
Tao MH, Shields PG, Nie J, et al: DNA
hypermethylation and clinicopathological features in breast cancer:
the Western New York Exposures and Breast Cancer (WEB) Study.
Breast Cancer Res Treat. 114:559–568. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holst CR, Nuovo GJ, Esteller M, et al:
Methylation of p16(INK4a) promoters occurs in vivo in
histologically normal human mammary epithelia. Cancer Res.
63:1596–1601. 2003.PubMed/NCBI
|
|
4
|
Tlsty TD, Crawford YG, Holst CR, et al:
Genetic and epigenetic changes in mammary epithelial cells may
mimic early events in carcinogenesis. J Mammary Gland Biol
Neoplasia. 9:263–274. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wajed SA, Laird PW and DeMeester TR: DNA
methylation: An alternative pathway to cancer. Annals Surg.
234:10–20. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Baylin SB and Herman JG: DNA
hypermethylation in tumorigenesis - epigenetics joins genetics.
Trends Genet. 16:168–174. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
8
|
Di Vinci A, Perdelli L, Banelli B, et al:
p16(INK4a) promoter methylation and protein expression in breast
fibroadenoma and carcinoma. Int J Cancer. 114:414–421.
2005.PubMed/NCBI
|
|
9
|
Lehmann U, Langer F, Feist H, Glockner S,
Hasemeier B and Kreipe H: Quantitative assessment of promoter
hypermethylation during breast cancer development. Am J Pathol.
160:605–612. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cavusoglu AC, Sevinc AI, Saydam S, et al:
Promoter methylation and expression changes of CDH1 and P16 genes
in invasive breast cancer and adjacent normal breast tissue.
Neoplasma. 57:465–472. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jing F, Jun L, Yong Z, et al: Multigene
methylation in serum of sporadic Chinese female breast cancer
patients as a prognostic biomarker. Oncology. 75:60–66. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jing F, Yuping W, Yong C, et al: CpG
island methylator phenotype of multigene in serum of sporadic
breast carcinoma. Tumour Biol. 31:321–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Raish M, Dhillon VS, Ahmad A, et al:
Promoter hypermethylation in tumor suppressing genes p16 and FHIT
and their relationship with estrogen receptor and progesterone
receptor status in breast cancer patients from Northern India.
Transl Oncol. 2:264–270. 2009. View Article : Google Scholar
|
|
14
|
Vallian S, Sedaghat M, Nassiri I and
Frazmand A: Methylation status of p16 (INK4A) tumor suppressor gene
in Iranian patients with sporadic breast cancer. J Cancer Res Clin
Oncol. 135:991–996. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Zee KJ, Calvano JE and Bisogna M:
Hypomethylation and increased gene expression of p16(INK4a) in
primary and metastatic breast carcinoma as compared to normal
breast tissue. Oncogene. 16:2723–2727. 1998.PubMed/NCBI
|
|
16
|
Woodcock DM, Linsenmeyer ME, Doherty JP
and Warren WD: DNA methylation in the promoter region of the p16
(CDKN2/MTS-1/INK4A) gene in human breast tumours. Br J Cancer.
79:251–256. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Moher D, Liberati A, Tetzlaff J, Altman DG
and Grp P: Preferred reporting items for systematic reviews and
meta-analyses: The PRISMA statement (Reprinted from Annals of
Internal Medicine). Physical Therapy. 89:873–880. 2009.
|
|
18
|
Bero L and Rennie D: The Cochrane
Collaboration. Preparing, maintaining, and disseminating systematic
reviews of the effects of health-care. JAMA. 274:1935–1938. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Higgins JPT, Thompson SG, Deeks JJ and
Altman DG: Measuring inconsistency in meta-analyses. BMJ.
327:557–560. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jing F, Zhang J, Tao J, et al:
Hypermethylation of tumor suppressor genes BRCA1, p16 and 14-3-3
sigma in serum of sporadic breast cancer patients. Onkologie.
30:14–19. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li SY, Rong MN and Iacopetta B: DNA
hypermethylation in breast cancer and its association with
clinicopathological features. Cancer Lett. 237:272–280. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
23
|
Yang X, Yan L and Davidson NE: DNA
methylation in breast cancer. Endocr Relat Cancer. 8:115–127. 2001.
View Article : Google Scholar
|
|
24
|
Szyf M, Pakneshan P and Rabbani SA: DNA
methylation and breast cancer. Biochem Pharmacol. 68:1187–1197.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Rocco JW and Sidransky D:
p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp Cell Res.
264:42–55. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sherr CJ and McCormick F: The RB and p53
pathways in cancer. Cancer Cell. 2:103–112. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McDermott KM, Zhang JM, Holst CR,
Kozakiewicz BK, Singla V and Tlsty TD: p16(INK4a) prevents
centrosome dysfunction and genomic instability in primary cells.
PLoS Biol. 4:350–365. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bender CM, Pao MM and Jones PA: Inhibition
of DNA methylation by 5-aza-2′-deoxycytidine suppresses the growth
of human tumor cell lines. Cancer Res. 58:95–101. 1998.
|
|
29
|
Zhang B, Huang T, Liu K, Chen J and Wang
G: Effects of 5-Aza-CdR on cell proliferation of breast cancer cell
line MDA-MB-435S and expression of maspin gene. J Huazhong Univ of
Sci Technology Med Sci. 27:543–546. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
David GL, Yegnasubramanian S, Kumar A, et
al: MDR1 promoter hypermethylation in MCF7 human breast cancer
cells - Changes in chromatin structure induced by treatment with
5-aza-cytidine. Cancer Biol Ther. 3:540–548. 2004. View Article : Google Scholar : PubMed/NCBI
|