Introduction
Angiogenesis is required for many physiological
processes, including embryogenesis and wound healing, and
neoangiogenesis is central to the development of various disorders,
particularly the rapid growth and metastasis of solid tumours
(1–5). For this reason, it is important to
study angiogenesis and neoangiogenesis, as such information may
ultimately provide treatments for these disorders. Hemangioma is a
benign vascular lesion and is the most common tumor of infancy.
However, the pathogenesis of hemangiomas is not well understood
(6).
1,2-Dimethylhydrazine (DMH) is a strong colon
carcinogen that induces colorectal tumors in experimental animals
(7,8) and induces intestinal tumors in rats
when given orally (9). DMH was
shown to cause proliferation of human umbilical vein endothelial
cells (HUVECs), and the sequential expression of growth factors was
demonstrated (10,11). In addition, we found that protein
kinase C (PKC) is highly associated with the abnormal growth of
HUVECs induced by DMH (12,13).
PKC is a well-known regulator of cell proliferation.
In particular, PKC can mediate G1-phase progression of the cell
cycle (14–16). Some studies have shown that
overexpression or inhibition of some PKC isoforms can enhance or
reduce tumor growth, respectively, in vivo (17,18).
In some human tumor tissues, PKC activity is higher than in normal
tissues, and elevated PKC activity was found to be associated with
increased metastatic or invasive potential in some human carcinoma
cells, including human bladder carcinoma cells (19,20).
11 PKC isoforms have been identified in mammalian cells, and these
isoforms have distinct cellular localizations and functions
(21).
In the current study, we examined the PKC isoforms
present in HUVECs and investigated the role of these PKC isoforms
in the abnormal proliferation of HUVECs induced by DMH.
Materials and methods
Reagents
DMH was obtained from Sigma Aldrich Chemical Co.
(Milwaukee, WI, USA). Calphostin C was purchased from BIOMOL
(Plymouth Meeting, PA, USA).
Cell culture
Vascular endothelial cells, specifically HUVECs,
were obtained from Modern Tissue Technologies, Inc., and in these
experiments, the cells were sub-cultured fewer than 8 times. The
HUVECs were maintained at 37°C in 5% CO2 in the control
medium, endothelial cell basal medium [EBM with 0.1% human
epidermal growth factor (hEGF), 0.1% hydrocortisone, 0.1% GA-1000,
0.4% bovine brain extract (BBE), and 2% FBS].
MTT assay (proliferation assay)
Proliferation was determined by the indirect
colorimetric MTT assay. The MTT assay is metabolized by
NAD-dependent dehydrogenase to form a colored reaction product, and
the amount of dye formed is directly correlated with the number of
cells. To determine the number of cells, HUVECs were seeded into a
24-well plate at 1×104 cells/well, cultured for 48 h in
EBM complete medium (with 0.1% hEGF, 0.1% hydrocortisone, 0.1%
GA-1000, 0.4% BBE, and 2% FBS), serum-starved for 24 h, and then
treated with or without various reagents for the indicated times.
For the MTT assay, the stock solution (5 mg/ml MTT) was added to
each well of the 24-well plates, which were seeded with HUVECs, and
then the plates were incubated at 37°C for 2 h. The formazan
granules generated by the live cells were dissolved in 100%
dimethyl sulfoxide (DMSO), and the absorbance at 570 nm was
monitored using a Power-Wavex microplate spectrophotometer (Bio-Tek
Instruments, Inc., Winooski, VT, USA).
Reverse-Transcription-Polymerase Chain
Reaction (RT-PCR) analysis
Total RNA was isolated from HUVECs using an RNeasy
mini kit (Qiagen, Valencia, CA, USA). Total RNA was
reverse-transcribed with AMV reverse transcriptase (Promega,
Madison, WI, USA). PCR amplification was performed using the primer
sets shown in Table I, as well as
5′-GACTATGACTTAGTTGCGTTA-3′ and 5′-GCCTTCATACATCTCAAGTTG-3′ for
β-actin. All of the primer sequences were generated using GenBank
sequences. PCR amplifications were performed in duplicate. β-actin
was used as control to assess PCR efficiency. PCR products were
electrophoresed on 2% agarose gel and visualized by ethidium
bromide staining.
 | Table ISpecific primer sequences of PKC
isoform. |
Table I
Specific primer sequences of PKC
isoform.
| Name | | Sequence |
|---|
| PKCα; | Sense | 5′-cgg aag ccc cac
ctt ctg c |
| Antisense | 5′-ctt tgt tgc cag
cag ggc |
| PKCβI | Sense | 5′-cgt gat gaa tgt
tcc cag c |
| Antisense | 5′-cgc agt tct tca
ttg gc |
| PKCβII | Sense | 5′-cgc tga caa ggg
tcc agc |
| Antisense | 5′-cca atc cca aat
ctc tac |
| PKCγ | Sense | 5′-gca gcc cca cct
tct gcg |
| Antisense | 5′-gcc ccc atg aag
tcg ttg cg |
| PKCδ | Sense | 5′-gca gat gca ct gca
ccg |
| Antisense | 5′-gcc cac gac tgt
gaa cg |
| PKCɛ | Sense | 5′-gac ag aac tat ctt
gag |
| Antisense | 5′-agt tgt cct gta
gga aag |
| PKCζ | Sense | 5′-gcg ctt taa cag
gag agc gt |
| Antisense | 5′-gct tct ctg tct
gta ccc ag |
| PKCη | Sense | 5′-gct gct gcg cac
gac cgg cg |
| Antisense | 5′-gcc acg ttc gct
tgc cat cg |
| PKCθ | Sense | 5′-gca ggc aaa ggt
cca cca cg |
| Antisense | 5′-gcc acc tta atc
atg gcc ag |
| PKCι | Sense | 5′-cgg gtg aac gcc
tac tac c |
| Antisense | 5′-cgc ctg ttg aaa
cgc ttg gc |
| PKCμ | Sense | 5′-gct gtg ggg gct
ggt acg |
| Antisense | 5′-gca tct cgc cac
tgt cg |
Real-Time PCR
Total RNA was extracted, and the expression of
β-actin and PCK mRNA were evaluated by real-time RT-PCR. The primer
sequences used in the experiment were as shown in Table I, and 5′-GACTATGACTTAGTTGCGTTA-3′
and 5′-GCCTTCATACATCTCAAGTTG-3′ for β-actin. Real-time quantitation
was based on the LightCycler assay, using a fluorogenic SYBR Green
I reaction mixture for the PCR and the LightCycler Instrument
(Roche). The amplification program consisted of 1 cycle of 95°C
with a 60-s hold (‘hot start’), followed by 45 cycled of 95°C with
0-s hold, specified annealing temperature with a 5-s hold, 72°C
with 12-s hold, and specified acquisition temperature with 2-s
hold. All experiments were conducted 3 times, and included both
negative and positive controls. β-actin mRNA was amplified as an
internal control. LightCycler software version 3.3 (Roche
Diagnostics) was used to analyze the PCR kinetics and calculate
quantitative data. A standard curve generated in a separate run was
loaded into the runs of each sample (without standard curves). Each
run included 1 sample of known concentration, which was in the
range covered by the standard curve, thus allowing for estimation
of the exact copy number by the second derivative maximum method.
For each sample, the copy number of target gene mRNA was divided by
that of β-actin mRNA to normalize for target gene mRNA expression
and avoid sample-to-sample differences in RNA quantity.
Small interfering RNA (siRNA)
transfection
Small interfering RNA (siRNA) duplex oligo
(on-TARGET plus SMART pool Dharmacon, Lafayette, CO, USA) targeting
PKCμ mRNA or non-targeting duplex oligo (on-TARGET plus siCONTROL,
Dharmacon) as a negative control was transfected using DharmaFECT
Transfection Reagent (Dharmacon, CO, USA).
Western blot analysis
Confluent serum-starved HUVECs were treated with the
appropriate conditions, washed with ice-cold PBS, and then lysed in
total cell lysis buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris,
pH 7.5, and 1 mM EDTA). Protein content was determined with a
protein assay kit (Bio-Rad Laboratories, Richmond, CA, USA).
Proteins were loaded on a 10% SDS-polyacrylamide gel,
electrotransferred to nitrocellulose membranes (Hybond-ECL;
Amersham Pharmacia Biotech, Piscataway, NJ, USA), and then probed
with monoclonal antibodies (GAPDH Cell Signaling Technology,
Beverly, MA, USA; PKCμ Abcam, Cambridge, UK). Immunoreactive bands
were detected using anti-mouse and anti-rabbit
peroxidase-conjugated secondary antibodies (Amersham Pharmacia
Biotech) and visualized by enhanced chemiluminescence (ECL
detection kit; Amersham Pharmacia Biotech).
PKC activity assay
Separated proteins were analyzed using the Protein
Kinase C assay system according to the manufacturer’s protocol
(#V5330 Promega, Madison, USA). To compare the reaction to the PKC
negative control, PKC Activator 5× buffer, Pep Tag PKC Reaction 5×
buffer, Pep Tag C1 Peptide, and Peptide Protection solution were
added. In the PKC positive control, these same 4 solutions, which
were added to the negative control, were also added. In addition,
purified PKC was also added. The third sample, the PKC standard,
also had the same 4 solutions with the same amount of protein. All
3 samples were reacted at 30°C for 30 min, and then incubated at
95°C for 10 min to stop the reaction. Then, 80% glycerol was added
to the reactions, which were electrophoresed in a 0.8% agarose gel
containing 50 mM Tris-HCl at 100 V for 15 min. Phosphorylated PKC
moved to the + charge while non-phosphorylated PKC moved to the –
charge therefore, PKC was separated into 2 bands. The gel
containing the phosphorylated protein was cut out and placed in 1.5
ml tube. Then, gel-solubilization solution was added and the tube
was heated to 95°C until the gel melted completely. This solution
was transferred to another 1.5 ml tube containing
gel-solubilization solution and glacial acetic acid. The solution
was then transferred to a 96-well plate, which was placed in a
scanning multi well spectrophotometer (ELISA reader Molecular
Devices, Menlo Park, CA, USA) to measure the OD at 570 nm.
Statistical analysis
All results are presented as the means ± standard
error of the mean. Comparisons between groups were analyzed using
the t-test (two-sided) or analysis of variance. Post hoc range
tests and pairwise multiple comparisons were conducted using the
t-test (two-sided) with Bonferroni adjustments. P values of
<0.05 were considered statistically significant.
Results
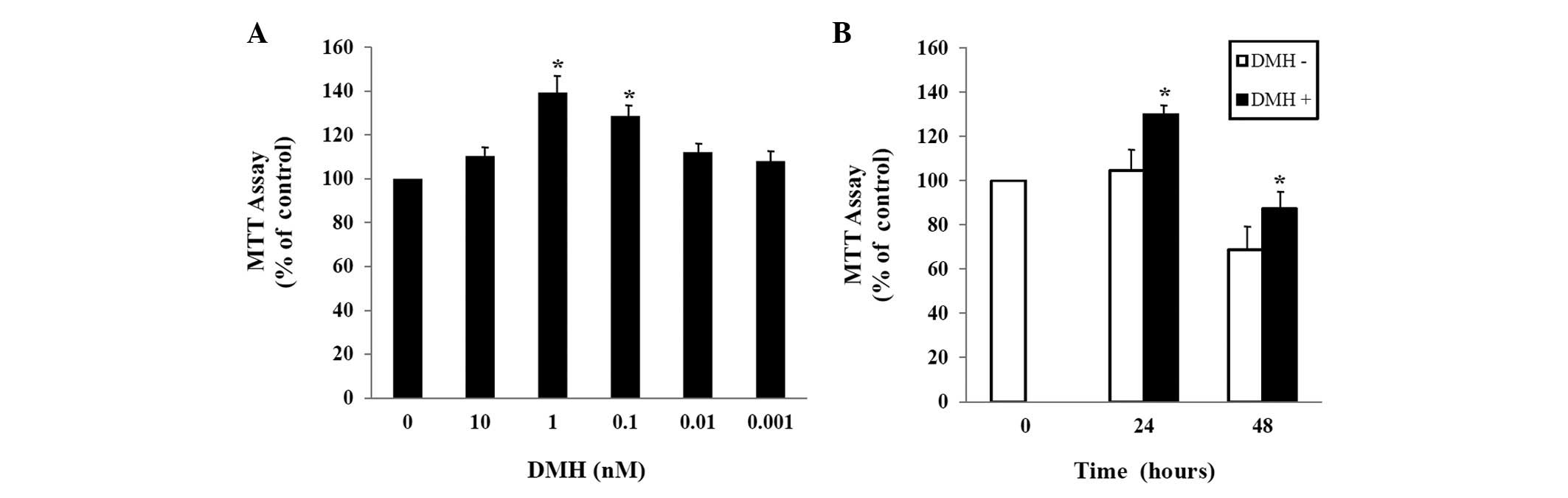
DMH induces abnormal proliferation of
HUVECs
To explore the effect of DMH on the growth of
HUVECs, HUVECs were exposed to the indicated concentration of DMH
for 48 h, and proliferation was determined using the MTT assay. As
shown in Fig. 1A, treatment of
HUVECs with DMH for 48 h increased cell proliferation
dose-dependently, with maximal stimulation at 1 nM. The DMH-induced
abnormal proliferation was observed 24 h after exposure at
1×10−9 M DMH (Fig.
1B).
DMH-induced abnormal proliferation of
HUVECs is mediated by activation of PKC
We previously reported that DMH treatment of HUVECs
increased expression of PKC (12).
In this study, we confirmed the effect of Calphostin C, an
inhibitor of PKC, on the proliferation of DMH-treated HUVECs. As
shown in Fig. 2A, treatment of
HUVECs with 1×10−9 M DMH for 24 h induced PKC
activation, while co-treatment with 5×10−9 M Calphostin
C prevented DMH-induced PKC activation. Consistent with the result
shown in the PKC activity assay, DMH-induced abnormal proliferation
of HUVECs was completely inhibited by treatment with Calphostin C.
The PKC inhibitor blocked the DMH-induced increases in both HUVEC
proliferation and PKC activity (Fig.
2B).
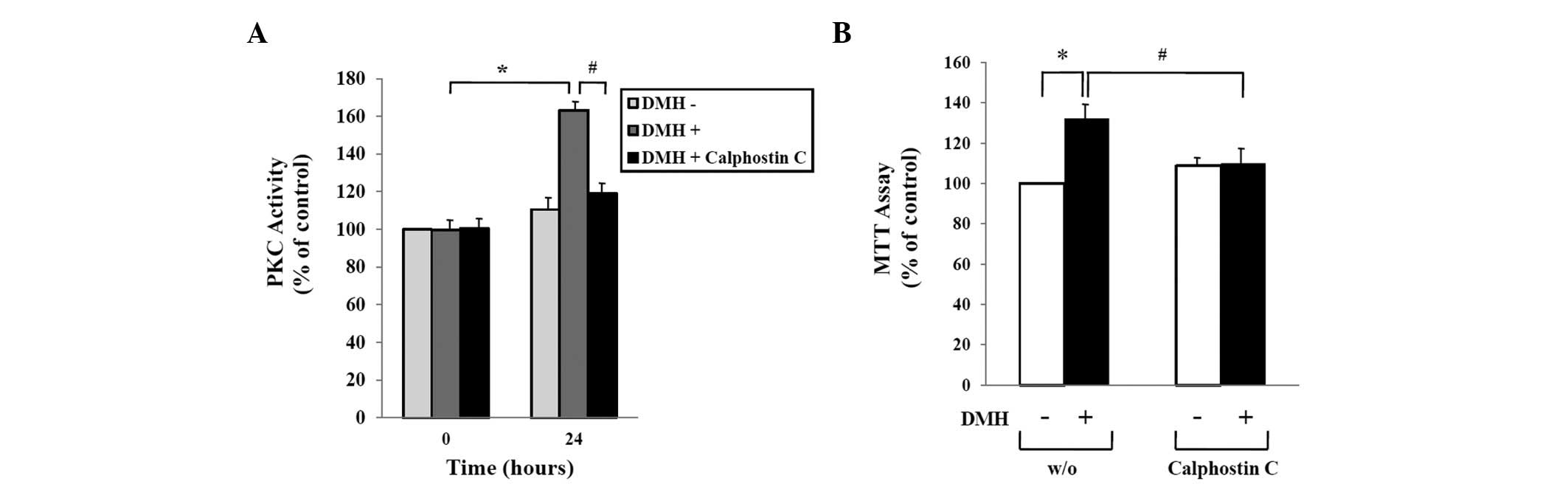 | Figure 2Role of PKC activation in DMH-induced
abnormal proliferation of HUVECs. (A) HUVECs were seeded into
6-well culture plates at a density of 1×105 cells/well,
cultured for 48 h in growth medium, serum-starved for 24 h, and
then treated with vehicle, 1×10−9 M DMH, or
5×10−9 M Calphostin C, a DMH inhibitor for 24 h as
indicated. PKC activity in the supernatant was measured by ELISA as
described in Materials and methods. (B) Serum-starved HUVECs were
treated with vehicle, 1×10−9 M DMH, and
5×10−9 M Calphostin C, a DMH inhibitor, for 24 h as
indicated. Cell proliferation was determined by the MTT assay and
expressed as a percentage of the control (mock-incubated HUVECs).
The data shown represent the mean ± SEM of 4 different experiments.
*p<0.05 compared to the control;
#p<0.05 compared to DMH only-treated HUVECs for 24
h. |
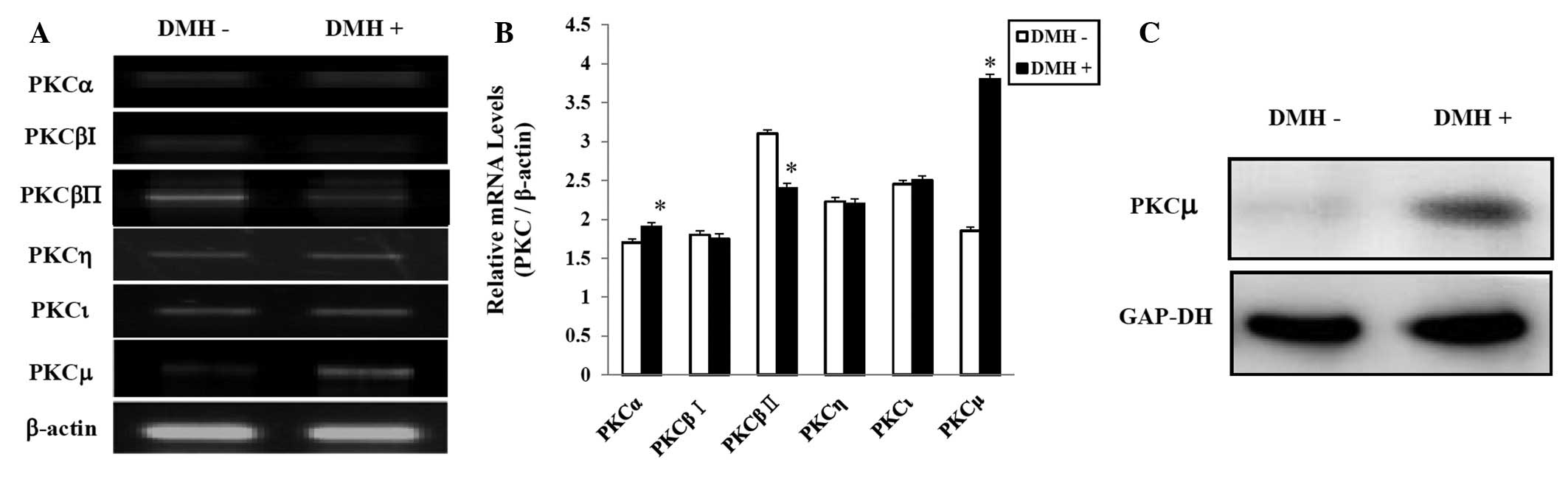
DMH increases the expression of PKCμ in
HUVECs
We showed that PKC activation affects the
DMH-induced abnormal proliferation of HUVECs. Therefore, we next
determined whether DMH regulates the expression of various PKC
isoforms. We examined the mRNA levels of the various PKC isoforms
by RT-PCR and real-time PCR. According to the results of both
assays, 6 out of 11 isoforms were expressed. Statistical assessment
showed higher PKCα;, PKCμ, and PKCι expression in the DMH-treated
group. As shown in Fig. 3A and B,
exposure of the cells to DMH significantly increased the level of
PKCμ mRNA. The increased PKCμ at the protein level was confirmed by
western blot using a specific antibody. RT-PCR and western blot
analysis showed that DMH stimulated the expression of PKCμ
(Fig. 3C).
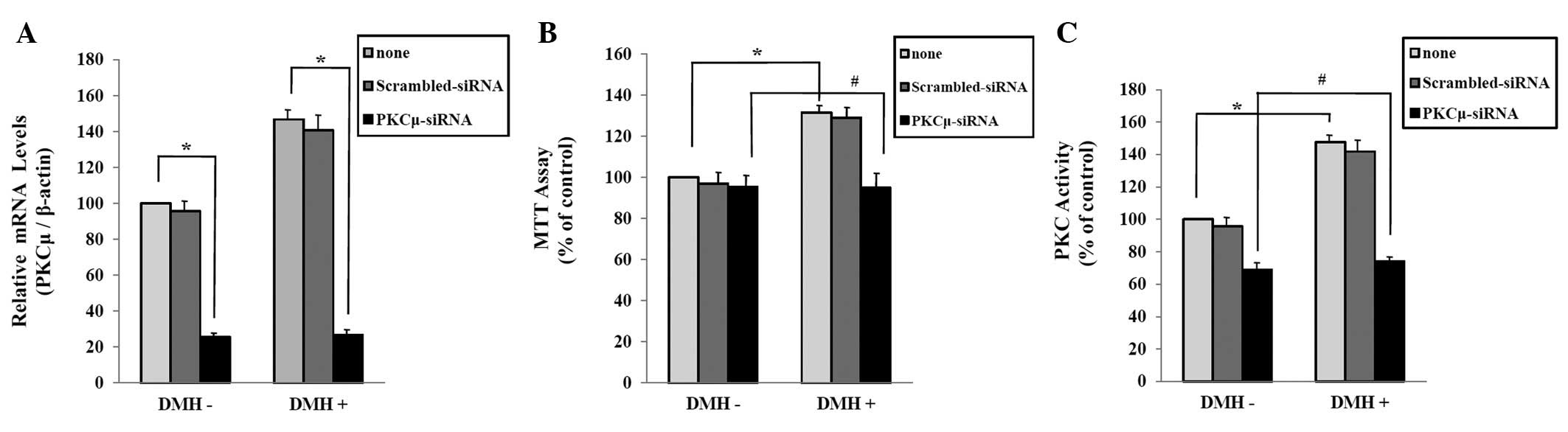
PKCμ is a key factor in DMH-induced
abnormal proliferation of HUVECs
To determine the role of PKCμ in the DMH-induced
abnormal HUVECs proliferation, we examined the effect of PKCμ
down-regulation by interference technique using siRNA transfection.
Real-time PCR analysis showed that the introduction of PKCμ siRNA
oligonucleotides effectively inhibited the expression of PKCμ in
HUVECs, and that PKCμ siRNA effectively inhibited DMH-induced
expression of PKCμ (Fig. 4A). In
the MTT assay, cell proliferation was increased in DMH-treated
HUVECs. In contrast, down-regulation of PKCμ expression by the
transfection of PKCμ siRNA decreased DMH-induced abnormal HUVECs
proliferation (Fig. 4B). Next, we
confirmed that whether PKCμ regulates DMH-induced PKC activation in
HUVECs. The PKC activity assay showed that siRNA-induced
down-regulation of PKCμ decreased the DMH-induced PKC activation in
HUVECs (Fig. 4C).
Discussion
It has proceed the study that angiogenesis is
critical to several physiological and disease processes, and
understanding the mechanism underlying new blood vessel formation
is required to develop treatments for blood vessel abnormalities,
such as hemangioma, in the field of plastic surgery (22,23).
For the present study, we thought that the best way to study
abnormalities in blood vessel proliferation, such as hemangiomas,
was to evaluate the mechanism of blood vessel formation. First,
abnormal proliferation of HUVECs was induced in vitro by
DMH, which is known to induce hemangiomas in vivo. We
determined the specific concentration needed to induce the maximum
effect of abnormal HUVEC proliferation. We also completed the
experiment model as expressed growth factor in this process was
confirmed by RT-PCR (10).
The mechanisms underlying angiogenesis and
revascularization of blood vessel formation include the vascular
endothelial cells and related mediators, such as interaction growth
factor with delivery substance, and several signal transduction
pathways. In this complex process, there are several enzymes, such
as protein tyrosine kinase (PTK), PKC, and oxidase (24–27),
which are important mediator molecules of signal transduction
pathways. Because in abnormal proliferation, the process of blood
vessel formation is in similar pathway, to explore the signal
transduction process of abnormal cell proliferation, we examined
the distribution of the signal transduction channel through use of
an inhibitor. We found that signal transduction of PKC is
important. We also confirmed that PKC plays an important role in
the process of DMH-induced abnormal HUVECs proliferation by
directly showing the activity of PKC through an enzymological
approach.
In mammal cells, there are 11 known PKC isoforms.
They are divided into 3 groups based on their structure and
function: classic PKCs or cPKCs (α;, βI, βII, and γ), novel PKCs or
nPKCs (δ, ɛ, η, θ, and μ), and atypical PKCs or a PKCs (ζ and ι)
(21). PKCα;, βI, βII, δ, ɛ, and ζ
exist mostly in the brain, lung and spleen (28,29).
PKCα; and ɛ facilitate proliferation of NIH3T3 cells, and these
factors have been reported to be very important in cornea
endothelial cells (30,31). The ones identified in this study
are primarily found in liver and kidney tissue (28,32).
In liver tissue, mostly PKC β and βII are present, but PKC α; and ɛ
are not. In contrast, PKC β and βII are not found in kidney, but
PKC α;, δ, ɛ, and ζ are. Based on this, the function of the
isoforms is predicted to be different in each organ therefore, it
is very important for us to predict their location or function so
that they can be used in preliminary research of various
diseases.
According to the results of PKC isoforms expression
by RT-PCR during DMH-induced abnormal HUVECs growth, among the 11
PKC isoforms, 6 kinds of PKC which is PKCα;, βI, βII, η, ι, and μ
has been verified in the control and DMH-treatment groups. Among
these 6 PKC isoforms, PKCμ had the highest expression in the
DMH-treated group compared to the control. Therefore, it is thought
that PKCμ plays an important role in DMH-induced HUVECs
over-growth.
In this study, we found that PKCμ may play a major
role in the abnormal proliferation of HUVECs induced by DMH
treatment through siRNA experiment. The study of PKC isoforms will
allow us to manipulate gene expression in signal transduction
leading to cell proliferation and differentiation. In addition, it
will lead to a better understanding of abnormal angiogenesis and
embryologic mechanisms, so that it can be used as preliminary
study.
Acknowledgements
This study was supported by a Medical Research
Institute Grant (2007–31) Pusan National University.
References
|
1
|
Klagsbrun M and D’Amore PA: Regulators of
angiogenesis. Annu Rev Physiol. 53:217–239. 1991. View Article : Google Scholar
|
|
2
|
Folkman J and Shing Y: Angiogenesis. J
Biol Chem. 267:10931–11934. 1992.
|
|
3
|
Folkman J: What is the evidence that
tumors are angiogenesis dependent? J Natl Cancer Inst. 82:4–6.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ingber D, Fujita T, Kishimoto S, Sudo K,
Kanamaru T, Brem H and Folkman J: Synthetic analogues of fumagillin
that inhibit angiogenesis and suppress tumour growth. Nature.
348:555–557. 1990. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Weidner N, Semple JP, Welch WR and Folkman
J: Tumor angiogenesis and metastasis-correlation in invasive breast
carcinoma. N Engl J Med. 324:1–8. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mulliken JB: Pathogenesis of hemangiomas.
Vascular Birthmarks: Hemangiomas and Malformations. Mulliken JB and
Young AE: Saunders; Philadelphia: pp. 63–76. 1988
|
|
7
|
Druckrey H: Production of colonic
carcinomas by 1,2-dialkylhydrazines and azoalkanes. Carcinoma of
the Colon and Antecedent Epithelium. Burdette WJ: Sprinpfield, III:
Charles C Thomas; pp. 267–279. 1970
|
|
8
|
Druckrey H, Preussmann R, Matzkies F and
Ivankovic S: Selective production of intestinal cancer in rats by
1,2-dimethylhydrazine. Naturwissenschaften. 54:285–286.
1967.PubMed/NCBI
|
|
9
|
Wiebecke B, Löhrs U, Gimmy J and Eder M:
Production of tumors in the intestines of mice by
1,2-dimethylhydrazine. Z Gesamte Exp Med. 149:277–278.
1969.PubMed/NCBI
|
|
10
|
Kim HO, Kang YS, Bae YC, Park SY, Hwang SM
and Nam SB: Gene expression profiling of
1,2-Dimethylhydrazine-stimulated human umbilical vein endothelial
cells. J Korean Soc Plast Reconstr Surg. 31:858–864. 2004.
|
|
11
|
Kim SH, Kang YS, Bae YC, Park SY and Nam
SB: RT-PCR of Up-Regulated Factors in Abnormally Proliferated
Vascular Endothelial Cells by 1,2-Dimethylhydrazine. J Korean Soc
Plast Reconstr Surg. 32:689–698. 2005.
|
|
12
|
Bae YC, Park SY, Nam SB, Herh JY and Kang
YS: A study for the mechanism of abnormal proliferation in vascular
endothelial cells using inhibitors to the signal transduction
pathway. J Korean Soc Plast Reconstr Surg. 33:5–12. 2006.
|
|
13
|
Lee J, Bae YC, Park SY, Moon JS and Nam
SB: Role of protein kinase C in abnormal proliferation of vascular
endothelial cell induced by 1,2-dimethylhydrazine; analysis of
isoform. J Korean Soc Plast Reconstr Surg. 34:8–12. 2007.(In
Korean).
|
|
14
|
Livneh E and Fishman DD: Linking protein
kinase C to cell-cycle control. Eur J Biochem. 248:1–9. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishi K, Schnier JB and Bradbury EM: The
accumulation of cyclin-dependent kinase inhibitor p27kip1 is a
primary response to staurosporine and independent of G1 cell cycle
arrest. Exp Cell Res. 243:222–231. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fukumoto S, Nishizawa Y, Hosoi M, Koyama
H, Yamakawa K, Ohno S and Morii H: Protein kinase C delta inhibits
the proliferation of vascular smooth muscle cells by suppressing G1
cyclin expression. J Biol Chem. 272:13816–13822. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ways DK, Kukoly CA, DeVente J, Hooker JL,
Bryant WO, Posekany KJ, Fletcher DJ, Cook PP and Parker PJ: MCF-7
breast cancer cells transfected with protein kinase C-alpha exhibit
altered expression of other protein kinase C isoforms and display a
more aggressive neoplastic phenotype. J Clin Invest. 95:1906–1915.
1995. View Article : Google Scholar
|
|
18
|
Dean N, McKay R, Miraglia L, Howard R,
Cooper S, Giddings J, Nicklin P, Meister L, Ziel R, Geiger T,
Muller M and Fabbro D: Inhibition of growth of human tumor cell
lines in nude mice by an antisense of oligonucleotide inhibitor of
protein kinase C-alpha expression. Cancer Res. 56:3499–3507.
1996.PubMed/NCBI
|
|
19
|
O’Brian C, Vogel VG, Singletary SE and
Ward NE: Elevated protein kinase C expression in human breast tumor
biopsies relative to normal breast tissue. Cancer Res.
49:3215–3217. 1989.PubMed/NCBI
|
|
20
|
Schwartz GK, Redwood SM, Ohnuma T, Holland
JF, Droller MJ and Liu BC: Inhibition of invasion of invasive human
bladder carcinoma cells by protein kinase C inhibitor
staurosporine. J Natl Cancer Inst. 82:1753–1756. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Newton AC: Protein kinase C: structure,
function, and regulation. J Biol Chem. 270:28495–28498. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Gampper TJ and Morgan RF: Vascular
anomaly: Hemangiomas. Plast Reconstr Surg. 110:572–585. 2002.
View Article : Google Scholar
|
|
23
|
Mulliken JB and Glowacki J: Hemangiomas
and vascular malformations in infants and children: A
classification based on endothelial characteristics. Plast Reconstr
Surg. 69:412–422. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Clemens MJ, Trayner I and Menaya J: The
role of protein kinase C isoenzymes in regulation of cell
proliferation and differentiation. J Cell Sci. 103:881–887.
1992.PubMed/NCBI
|
|
25
|
Bischoff J: Perspectives series; cell
adhesion in vascular biology. J Clin Invest. 99:373–376. 1997.
|
|
26
|
Lee OH, Kim YM, Lee YM, Moon EJ, Lee DJ,
Kim JH, Kim KW and Kwon YG: Sphingosine1-phosphat induces
angiogenesis: its angiogenic action and signaling mechanism in
human umbilical vein endothelial cells. Biochem Biophys Res Commun.
264:743–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lutz MP, Esser IB, Flossmann-Kast BB,
Vogelmann R, Luhrs H, Friess H, Buchler MW and Adler G:
Overexpression and activation of the tyrosine kinase Src in human
pancreatic carcinoma. Biochem Biophys Res Commun. 243:503–508.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hug H: Protein kinase C isoenzymes
divergence in signal transduction? Biochem J. 291:329–343.
1993.PubMed/NCBI
|
|
29
|
Yang JH and Kodavinti PR: Possible
molecular targets of halogenated aromatic hydrobons in neural
cells. Biochem Biophys Res Commun. 280:1372–1377. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mischak H, Goodnight JA, Kolch W,
Martiny-Baron G, Schaechtle C, Kazanietz MG, Blumberg PM, Pierce JH
and Mushinski JF: Overexpression of prtein kinase C-δ and -ɛ in NIH
3T3 cells induces opposite effects on growth, morphology, anchorage
dependence, and tumorigeniccity. J Biol Chem. 268:6090–6096.
1993.
|
|
31
|
Grabam MA, Rawe I, Dartt DA and Joyce NC:
Protein kinase C refulation of corneal endothelial cell
proliferation and cell cycle. Invest Ophthalmol Vis Sci.
41:4124–4132. 2000.PubMed/NCBI
|
|
32
|
Farivar RS, Gardner-Thorpe J, Ito H,
Arshad H, Zinner MJ, Ashley SW and Whang EE: The efficacy of
tyrosine kinase inhibitors on human pancreatic cancer cell lines. J
Surg Res. 115:219–225. 2003. View Article : Google Scholar : PubMed/NCBI
|