Introduction
Crigler-Najjar (CN) syndrome is a rare autosomal
recessive inherited disorder characterized by non-hemolytic,
unconjugated hyperbilirubinemia (1). The disease is caused by defective
activity of the hepatic enzyme bilirubin uridine 5′-diphosphate
(UDP)-glucuronosyltransferase. The levels of serum bilirubin and
the response to phenobarbital treatment have been used to classify
CN syndrome into two types: CN I and II. CN type I is characterized
by severe unconjugated hyperbilirubinemia (≥340 μmol/l) from birth,
owing to the absence of bilirubin-UDP-glucuronosyltransferase (UGT)
activity. CN I patients are refractory to phenobarbital
administration. Unless treated with liver transplantation, CN I is
lethal during the neonatal period or in infancy. CN type II occurs
due to a severe, but incomplete deficiency of UGT hepatic activity.
It is characterized by chronic unconjugated hyperbilirubinemia
which is partially responsive to phenobarbital induction (2). Most of the UGT enzymatic activity
results from the expression of the UGT1A1 gene. Mutations of
the UGT1A1 gene have been found to be responsible for cases
of CN syndrome (3,4).
In the present study, we investigated an unusual CN
syndrome patient with extremely high levels of unconjugated
hyperbilirubin. The patient recovered well from the illness when
treated with phototherapy and phenobarbital, and a molecular
analysis of the UGT1A1 gene was performed.
Case report
The patient was a 4-month-old Chinese boy. He was
born at term (weight 3,500 g) by normal vaginal delivery following
a pregnancy of normal duration. The patient was O Rhesus-positive
and was breast-fed. The family noted his icterus when he was 3 days
old. He developed severe jaundice and suffered from kernicterus
(spasm period). He was first admitted to hospital when he was 7
days old; his serum total and indirect bilirubin concentrations
were 686.58 μmol/l (40.4 mg/dl) and 666.82 μmol/l (39.2 mg/dl),
respectively. He showed no signs of hemolysis, infection or liver
dysfunction. The patient was treated with phototherapy (66 h) for 7
days and phenobarbital (5 mg/kg/day) for 3 days. Following this
treatment, his elevated total serum bilirubin levels decreased to
91.8 μmol/l (5.4 mg/dl) and his psychomotor development recovered
to normal. He passed hearing screening in the ears by examination
with transiently evoked otoacoustic emission when he was 12 days
old. The patient received no treatment after he was 14 days old. At
4 months of age, he had a return visit, during which he exhibited
no jaundice or mental or growth retardation. The patient was born
to non-consanguineous parents without icteric family history.
Mutation analysis
A blood sample was collected from the patient and
genomic DNA was isolated from peripheral blood lymphocytes using
standard procedures. All coding exons, exon-intron boundaries and
the promoter region of UGT1A1 were amplified by the
polymerase chain reaction (PCR) using standard primers and
amplification conditions as described previously (5,6). PCR
was performed in a 50-μl reaction system containing 10X PCR buffer,
0.1 mmol/l deoxyribonucleotide triphosphates (dNTPs), 20 mmol/l
Mg2+, 200 ng genomic DNA, 10 pmol/μl of each sense and
antisense primer and 1.25 units Taq DNA polymerase. The PCR
products were subjected to gel electrophoresis for purification and
sequencing using an ABI Prism 3730XL® sequencer (Applied
Biosystems, Carlsbad, CA, USA). The study was performed according
to the Declaration of Helsinki and approved by the Ethics Committee
of the Affiliated Hospital of Guiyang Medical College. Informed
consent was obtained from the patient’s parents.
Results and discussion
The patient affected with CN syndrome underwent
mutation analysis by sequencing of the PCR-amplified DNA fragments,
including all coding exons, exon-intron boundaries and promoter
region sequences, of the UGT1A1 gene. Sequencing revealed
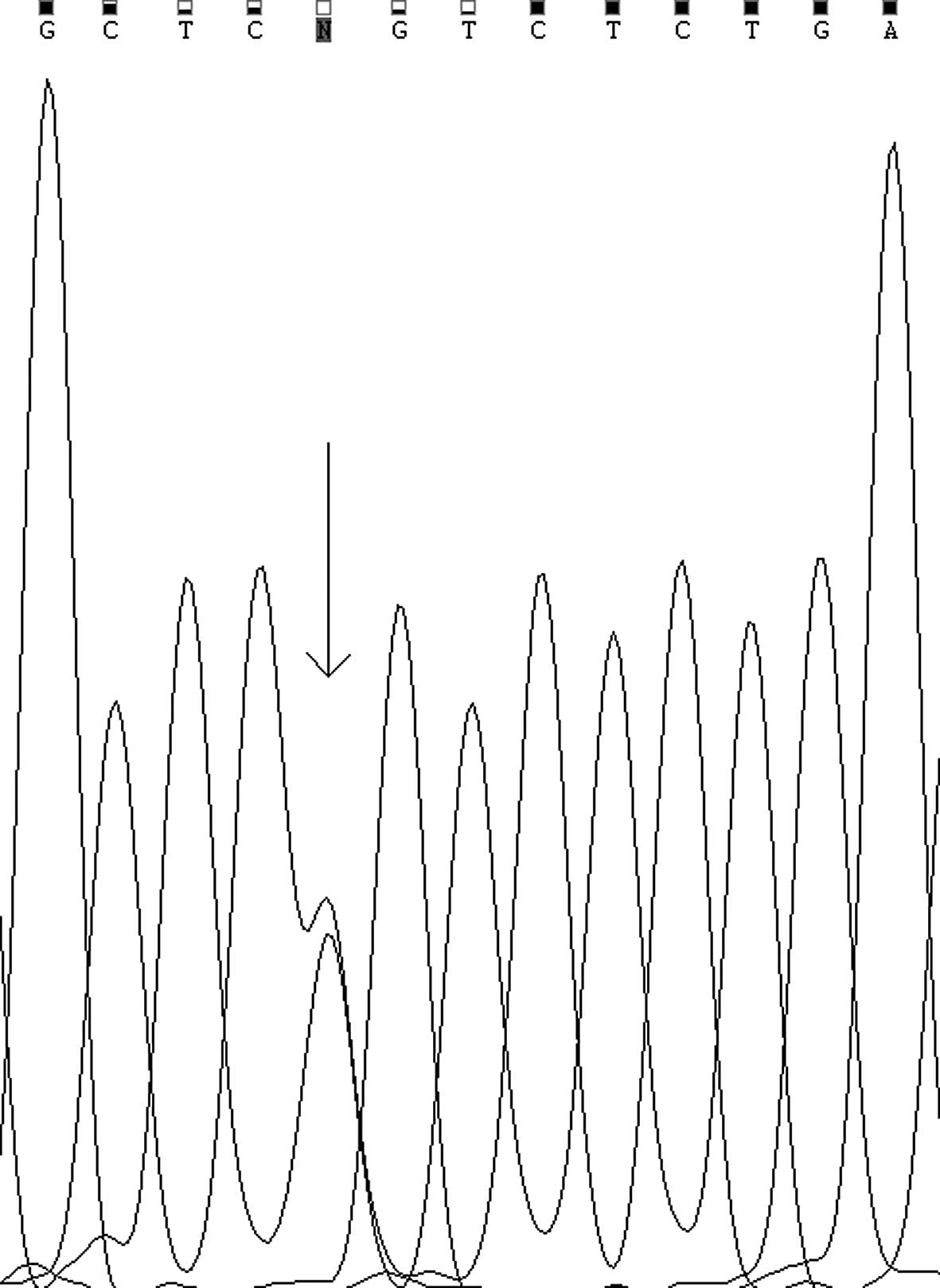
compound heterozygous mutations, a missense mutation c.211G>A
(p.G71R) in the first exon (Fig.
1) in which glycine (G) is substituted for arginine (R) at
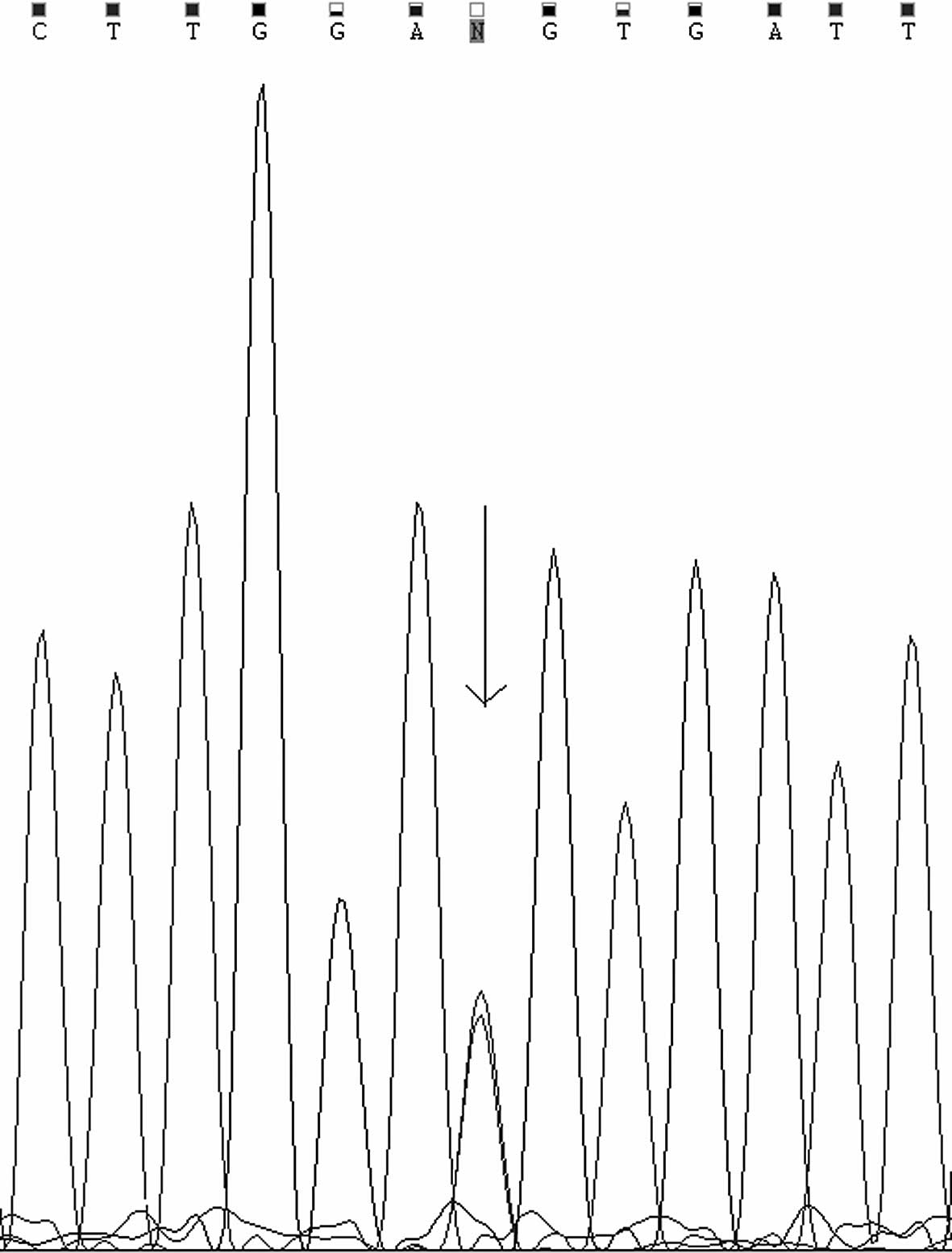
amino acid residue 71 of the UGT, and a synonymous mutation
c.1470C>T (p.D490D) in the fifth exon (Fig. 2), which has previously been
reported as a rare single-nucleotide polymorphism (SNP)
rs114123636, not the substitution of an aspartate (D) at amino acid
residue 490. The D490 residue is highly conserved among mammals.
Gene analysis also showed a normal homozygous TATA box (A[TA]6TAA)
in the promoter element of the patient.
Several authors have analysed genotype-phenotype
correlations in CN syndrome. As previously outlined, chain
terminating mutations are more commonly associated with a CN I
phenotype, whereas missense changes are more frequently observed
among less severely affected CN II patients (7). The serum bilirubin concentration of
the patient was consistent with the bilirubin levels of CN type I,
however, the therapeutic efficacy in the patient was superior to
that observed in CN type II disease. In the patient, two genetic
lesions coexisted in the UGT1A1 gene, the c.211G>A and
c.1470C>T mutations. c.211G>A (p.G71R) is the most prevalent
mutation in east Asian patients with CN II (8). A single heterozygous G71R missence
mutation appears to be insufficient for the severe phenotype in the
patient. Taking these findings into consideration, we propose that
a synergistic reaction may occur between the c.211G>A and
c.1470C>T mutations, leading to clearly reduced UGT enzymatic
activity. We speculate that the c.1470C>T synonymous mutation in
the individual may cause a reversible partial loss of UGT activity.
During the translation of mRNA to protein, the c.1470C>T
mutation may lead to a decrease of corresponding tRNA for a period
of time, resulting in a loss or a decrease of UGT enzymatic
activity. The activity of UGT with the synonymous mutation may
increase to normal levels in the later stages of the neonatal
period, since the patient returned to normal without any treatment
after he was 14 days old. To the best of our knowledge, functional
analyses have not been performed to determine the molecular results
of the c.1470C>T synonymous mutation, but the clinical features
of this particular CN syndrome patient indicate that the synonymous
mutation is important for enhancing the effect of the heterozygous
coding mutation c.211G>A (p.G71R). The phenotype of this unusual
CN syndrome patient may be associated with the specific
genotype.
As previously reported, in order to detect any
genotype-phenotype correlation in CN syndrome patients it is
mandatory to evaluate the (TA)7 promoter of the UGT1A1 gene
(9). Our patient did not have the
polymorphism in the promoter region. The present study revealed
that the polymorphism of the coding region may affect the
expression of the UGT1A1 gene. In the present study, we
reported that this particular CN syndrome patient with the genotype
defined in this study had clinical features similar to CN type I,
but had a superior response to therapy than is usual for CN type
II. No similar case has been reported worldwide. Considering the
specific clinical features and therapeutic efficacy, a distinct
type of CN was suspected. The results of this study may be useful
in improving the precision of future genotype-phenotype correlation
studies.
Acknowledgements
The authors thank the patient and his family and the
clinicians who collaborated in this study. This study was supported
by the Section of Scientific Research, Science and Technology,
Department of Guizhou Province, China [code (2010)3173] and by the
key project of the Chinese Ministry of Education (No. 212155).
References
|
1
|
Crigler JF and Najjar VA: Congenital
familial non-hemolytic jaundice with kernicterus. Pediatrics.
10:169–179. 1952.PubMed/NCBI
|
|
2
|
Servedio V, d’Apolito M, Maiorano N, et
al: Spectrum of UGT1A1 mutations in Crigler-Najjar (CN) syndrome
patients: identification of twelve novel alleles and
genotype-phenotype correlation. Hum Mutat. 25:3252005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Aono S, Yamada Y, Keino H, et al:
Identification of defect in the genes for bilirubin
UDP-glucuronosyl-transferase in a patient with Crigler-Najjar
syndrome type II. Biochem Biophys Res Commun. 197:1239–1244. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Aono S, Yamada Y, Keino H, et al: A new
type of defect in the gene for bilirubinuridine
5′-diphosphate-glucuronosyltransferase in a patient with
Crigler-Najjar syndrome type I. Pediatr Res. 6:629–632. 1994.
|
|
5
|
Bosma PJ, Roy Chowdhury N, Goldhoorn BG,
et al: Sequence of exons and flanking regions of human bilirubin
UDP-glucuronosyltransferase gene complex and identification of a
genetic mutation in a patient with Crigler-Najjar syndrome type I.
Hepatology. 15:941–947. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ritter JK, Yeatman MT, Ferreira P, et al:
Identification of a genetic alteration in the code for bilirubin
UDP-glucuronosyltransferase in the UGT1 gene complex of a
Crigler-Najjar type I patient. J Clin Invest. 9:150–155. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kadakol A, Ghosh SS, Sappal BS, et al:
Genetic lesions of bilirubin-diphosphoglucuronate
glucosyltransferase (UGT1A1) causing Crigler-Najjar Gilbert
syndromes: correlation of genotype to phenotype. Hum Mutat.
4:297–306. 2000. View Article : Google Scholar
|
|
8
|
Akaba K, Kimura T, Sasaki A, et al:
Neonatal hyperbilirubinemia and mutation of the bilirubin uridine
diphosphate-glucuronosyltransferase gene: a common missense
mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int.
46:21–26. 1998.
|
|
9
|
Kakadol A, Sappal BS, Ghosh SS, et al:
Interaction of coding region mutations and the Gilbert-type
promoter abnormality of the UGT1A1 gene causes moderate degrees of
unconjugated hyperbilirubinaemia and may lead to neonatal
kernicterus. J Med Genet. 38:244–249. 2001. View Article : Google Scholar
|