Introduction
The liver is one of the most iron-rich organs of the
body. Approximately 20–30% of the body’s iron is stored in
hepatocytes and reticuloendothelial macrophages, thus excessive
iron accumulation is clearly observed in the liver. Numerous
studies have indicated that dietary iron overload enhances hepatic
fibrosis and even induces neoplastic transformation (1,2).
These phenomena may be associated with the role of iron in
triggering oxidative stress (3).
However, the molecular mechanism of iron-induced hepatic damage is
poorly understood.
Mitochondria are a potential target of iron-mediated
injury, due to the fact that they are intrinsically rich in iron
(4). Numerous data have confirmed
that mitochondrial dysfunction increases reactive oxygen species
(ROS) production, with serious consequences, not only for
respiratory function, but also for mitochondrial DNA transcription
(5,6). The role of ROS in determining cell
fate during exposure to excessive iron is already known; however,
the mechanism whereby the burst of ROS is induced by iron overload
is not yet understood. The mitochondrial production of ROS may be
involved in signal transduction pathways where an initial oxidative
stress signal originating at various cell sites is amplified by the
mitochondria, the phenomenon of which is termed ROS-induced ROS
release (RIRR).
RIRR is generated by circuits requiring
mitochondrial membrane channels, including the mitochondrial
permeability transition pore (mPTP) (7). mPTPs are multi-protein complexes that
are capable of forming large, non-selective pores in the inner
mitochondrial membrane (8). They
are directly stimulated by environmental factors, such as ROS and
injury (9). When the mPTP is
continuously open, it causes mitochondrial swelling, which may
result in rupture of the outer mitochondrial membrane (10). Generated ROS may subsequently be
released into the cytosol and trigger RIRR in the neighboring
mitochondria. Thus, mitochondrion-to-mitochondrion RIRR constitutes
a positive feedback mechanism for enhanced ROS production that
potentially causes hepatic damage.
Due to the fact that iron is a catalyst in the
Haber-Weiss reaction and is involved in the initiation of oxygen
radical formation (11), we
hypothesized that the toxicity of iron overload is associated with
mPTP-mediated RIRR. To test this hypothesis, cyclosporin A (CsA)
was used to inhibit mPTP opening, in order to explore the
underlying mechanisms of hepatic damage induced by iron
overload.
Materials and methods
Materials
Ferrocene and CsA were obtained from Sigma (St.
Louis, MO, USA). AIN-93G was purchased from Dyets, Inc. (Bethlehem,
PA, USA), and 2′,7′-dichlorfluorescein-diacetate (DCFH-DA) and
rhodomine 123 were purchased from Molecular Probes (Montluçon,
France). William’s medium E (WME) and fetal calf serum (FCS) were
obtained from Gibco-BRL (Paisley, Scotland), while collagenase D
was purchased from Boehringer Mannheim (Mannheim, Germany). The
remaining chemicals were purchased from the local market. Kunming
mice were purchased from Tongji Medical School, Huazhong University
of Science and Technology (Wuhan, China). The animals were cared
for in accordance with the Guide for the Care and Use of Laboratory
Animals. The use of animals was reviewed and approved by the
Nanchang University Medical College Animal Care Review
Committee.
Animal model for dietary iron
overload
Thirty-six male Kunming mice, initially weighing
14.7±0.7 g, were used in the present study. Mice were randomly
divided into 3 groups designated as control, iron-overloaded and
CsA + iron-overloaded. The animal model for dietary iron overload
in this study was similar to that described previously (12). Briefly, the iron-overloaded mice
were fed for 4 months on a pellet diet (AIN-93G) supplemented with
iron in the form of ferrocene, while the CsA + iron-overload group
was fed the AIN-93G diet supplemented, not only with iron, but also
with CsA (10 mg/kg). Control mice were fed the AIN-93G diet without
iron and CsA. Possible differences in dietary consumption among the
3 groups were controlled for; the control animals received an
amount of food that was equal to that which the respective treated
animals consumed each day. The proportion of iron in the diet was
maintained at 0.2% (w/w) for 90 days and then decreased to 0.4%
(w/w) for the remaining 30 days. All the groups were kept at 23±2°C
under a 12-h dark/light cycle. Animal care in this study conformed
to the National Institutes of Health (NIH) Guide for Care and Use
of Laboratory Animals (NIH publication 86-23, revised 1986).
Following chronic feeding, mice were euthanized by cervical
dislocation, and blood was collected by cardiac puncture. The liver
was immediately excised, weighed and divided for analysis as
described below.
Determination of serum and liver iron
concentrations
Serum iron concentration was determined using the
assay based on the generation of an iron-ferrozine complex, as
described previously by Galleano and Puntarulo (13). Iron concentration in the digested
liver sample was measured spectrophotometrically at 535 nm,
following reaction with 2 mM bathophenanthroline disulfonic acid
(14).
Determination of serum aspartate
transaminase (AST) and alanine transaminase (ALT) levels
Serum AST and ALT levels were measured using an
autoanalyser (Cobas Integra 400; Holliston, MA, USA) and an ALT/AST
reagent kit from Roche Diagnostics (Indianapolis, IN, USA).
Preparation of the mitochondrial
fraction
Mitochondria were isolated by conventional
differential centrifugation from the liver of mice that had been
starved overnight. The livers were homogenized in 250 mM sucrose, 1
mM EGTA and 10 mM HEPES buffer (pH 7.2). The mitochondrial
suspension was washed twice in the same medium containing 0.1 mM
EGTA, and the final pellet was resuspended in 250 mM sucrose to a
final protein concentration of 80–100 mg/ml, measured using the
Biuret method, with bovine serum albumin (BSA) as the protein
standard.
Mitochondrial swelling
The swelling experiments were conducted according to
the procedure performed by Beavis et al(15), at 25°C in a standard medium
containing 125 mM sucrose, 10 mM HEPES buffer (pH 7.2), 2.5 mM
succinate and 4.0 mM rotenone. The final volume used was 1.0 ml,
and the protein concentration was ~0.5 mg/ml. Absorbance changes at
520 nm were monitored in a thermostatically controlled Hitachi
U-2000 spectrophotometer.
Hepatocyte preparation
Hepatocytes were isolated by a two-step collagenase
perfusion method. Following mechanical disruption of the liver
capsule, the liver cells were collected in WME and serially
filtered through 30-, 50- and 80-mesh filters in an 85-ml Cellector
tissue sieve (Bellco Biotechnology, Vineland, NJ, USA). Typically,
10–25×106 cells were obtained from one mouse liver.
Measurements of intracellular Δψ
The mitochondrial membrane potential (Δψ) was
measured by flow cytometry using rhodomine 123, a fluorescent dye
that has been demonstrated to selectively accumulate in the
mitochondria of living hepatocytes by a mechanism that is dependent
on the Δψ (16). The hepatocytes
were resuspended in 0.5 ml of 10 μg/ml rhodomine for 15 min at
37°C, and were immediately submitted for flow analysis.
Measurement of intracellular ROS
production
To assess the intracellular ROS levels, flow
cytometric analyses were performed using the oxidative-sensitive
fluorescent probe, DCFH-DA, as previously described (17). Hepatocytes were incubated with 10
μM DCFH-DA for 30 min at 37°C. Formation of
2′,7′-dichlorofluorescein (DCF) was then detected using a
FACSCalibur (Becton-Dickinson, Mountain View, CA, USA) equipped
with an argon laser (488 nm) in the FL1 channel.
Determination of oxidative stress
parameters and lipid peroxidation
MDA
The level of 3,4-methylenedioxyamphetamine (MDA) was
determined according to the procedure employed by Okhawa et
al(18). Briefly, 0.5 ml
supernatant was mixed with 1.5 ml thiobarbituric acid, 1.5 ml
acetic acid (pH 3.5), 0.2 ml sodium dodecyl sulfate and 0.5 ml
distilled water. Following mixing, the samples and standards were
heated at 100°C for 1 h. The absorbance was recorded at 532 nm and
compared with that of the MDA standards.
SOD
Superoxide dismutase (SOD) activity was determined
according to the method utilised by Beauchamp and Fridovich
(19). The reaction mixture
consisted of 100 μmol/l xanthine, 100 μmol/l EDTA, 25 μmol/l NBT
and 50 mmol/l Na2CO3 (pH 10.2). The reaction
was initiated by the addition of xanthine oxidase, and then the
absorbance at 560 nm was read every 30 sec for 5 min. SOD activity
was assayed spectrophotometrically as the inhibition of the
photochemical reduction of NBT at 560 nm.
GSH-Px
Glutathione peroxidase (GSH-Px) activity was
measured according to the method employed by Lawrence and Burk
(20). The assay reaction
comprised 50 mmol/l K2HPO4 buffer, 1 mmol/l
EDTA, 1 mmol/l NaN3, 1 mmol/l reduced glutathione, 0.2
mmol/l NADPH, 0.25 mmol/l H2O2 and 1 U/ml
glutathione reductase. GSH-Px activity was assayed by monitoring
NADPH oxidation at 340 nm, by measuring the absorbance every 15 sec
for 5 min. The activity was calculated using a molar extinction
coefficient for NADPH of 6.22 mM−1cm−1 at 340
nm.
Catalase
Catalase activity in the liver homogenate was
assayed using a modification of the procedure described by
Pedraza-Chaverri et al(21). The catalase activity of hepatic
homogenates was assayed at 25°C, based on the disappearance of 10
mM H2O2 at 240 nm. The results are expressed
as U/mg protein.
TUNEL assay
The terminal deoxynucleotidyl transferase-mediated
nick-end labeling (TUNEL) assay was performed to detect hepatocyte
apoptosis. The hepatocytes were plated on glass Lab-Tek Chamber
slides (Sigma) and washed with PBS, then fixed in 1%
paraformaldehyde for 10 min. These were then postfixed in
pre-cooled ethanol-acetic acid (2:1) for a further 5 min at −20°C.
Following washing with PBS, the cells were incubated with a TUNEL
reaction buffer at 37°C for 1 h in a humidified chamber. As a
positive control, cells were treated with DNase I (1.0 mg/ml,
Sigma) for 10 min, to introduce nicks into the genomic DNA. The
percentage of cardiomyocytes with DNA nick-end labeling was
determined by counting the number of cells exhibiting brown nuclei
among 1,000 cells in duplicate plates.
Statistical analysis
Values were expressed as the mean ± standard
deviation from ≥12 independent experiments. Each treatment was
performed in triplicate culture wells. The differences in the means
between each group were tested by one-way ANOVA followed by the
Student-Newman-Keuls test (comparison between multiple groups).
P<0.05 was considered to indicate a statistically significant
difference.
Results
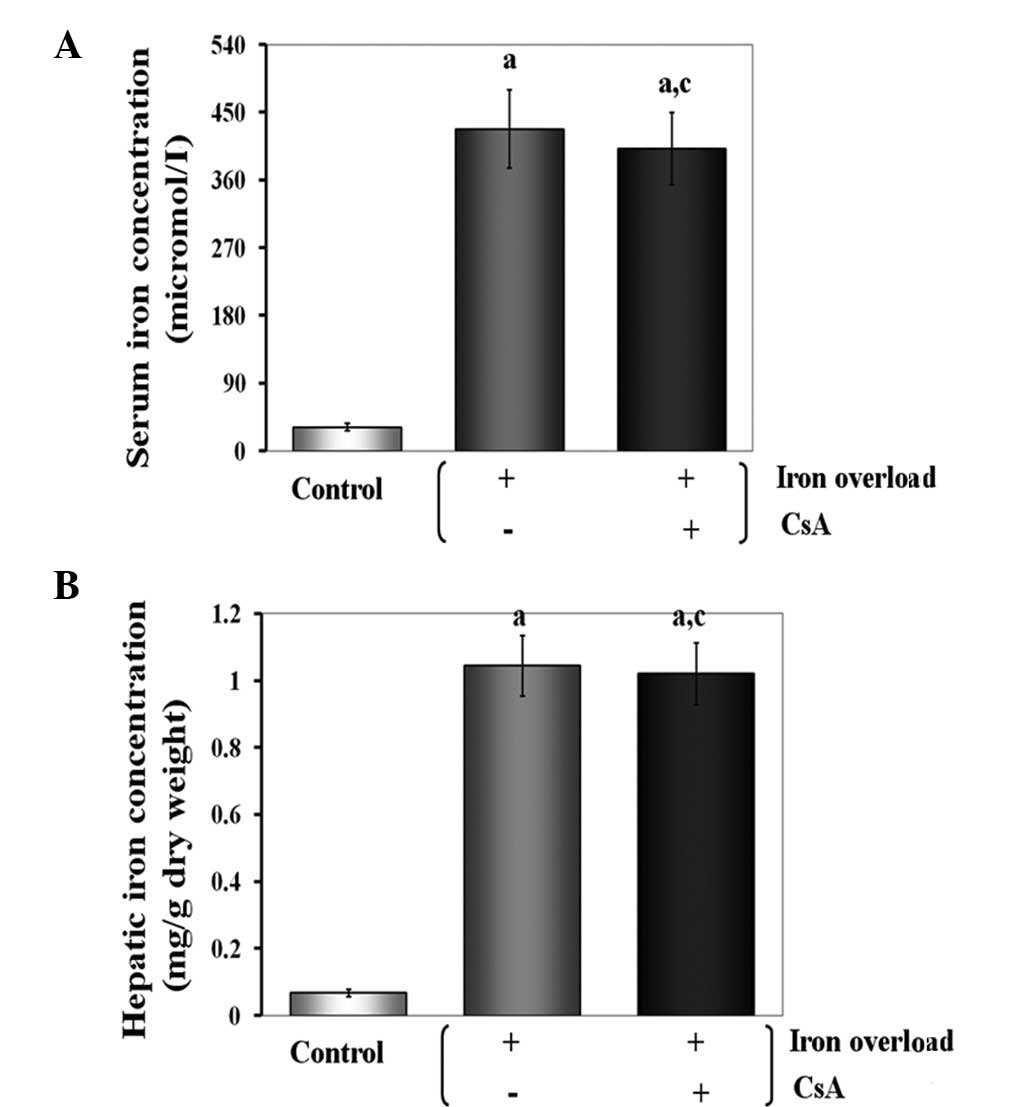
Effects of CsA administration on serum
and hepatic iron concentrations in iron-overloaded mice
As expected, the serum and hepatic iron
concentrations were significantly increased in all treated animals.
When mice were supplemented with CsA, the serum and hepatic iron
concentrations were not significantly different compared with those
of the iron-overloaded group. This observation suggests that the
severe iron loading caused by a continuous iron-supplemented diet
was not alleviated by CsA aministration (Fig. 1).
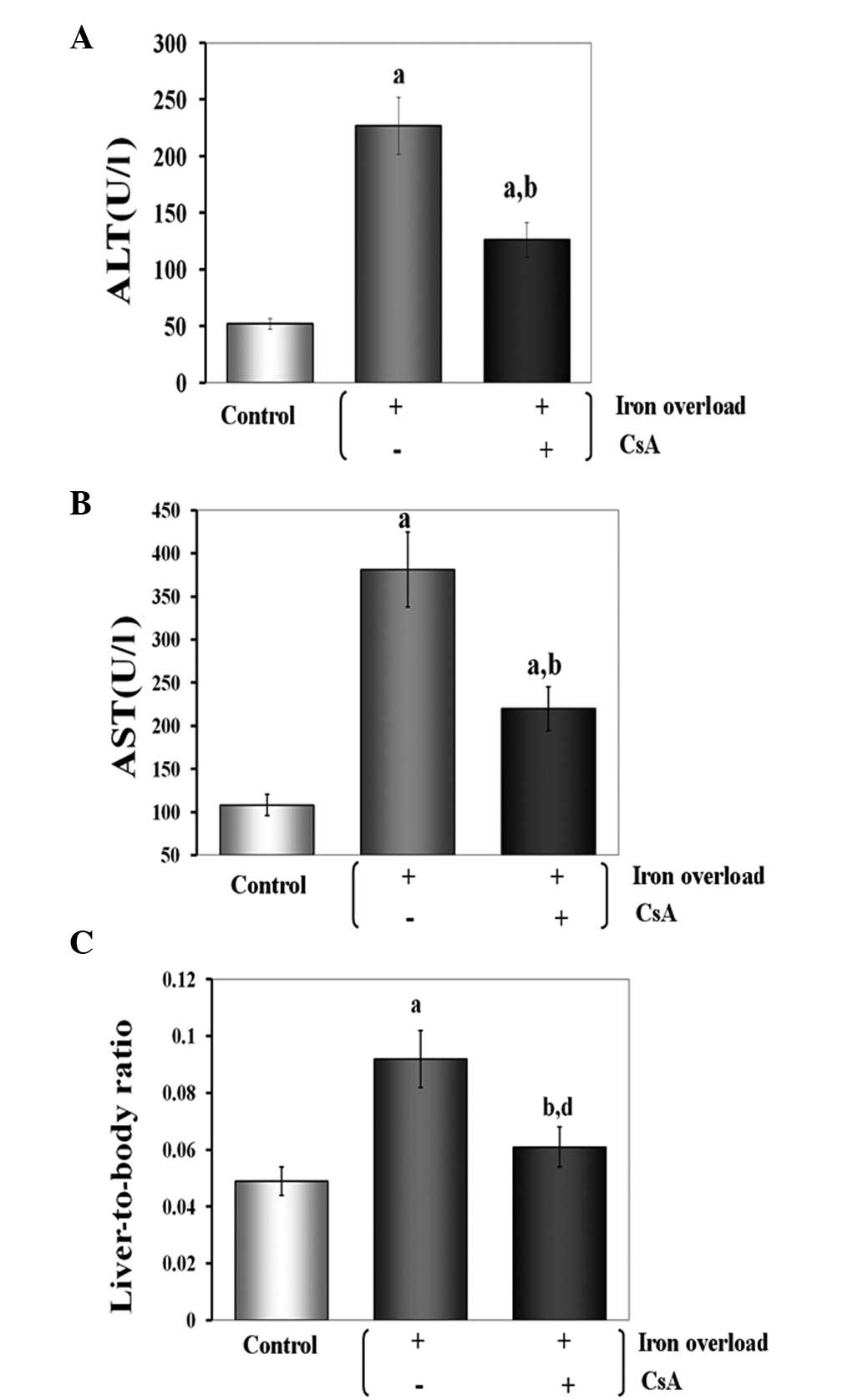
Effects of CsA administration on
liver-to-body weight ratio (%) and serum levels of transaminases in
iron-overloaded mice
There were no obvious health abnormalities in any of
the groups, but the liver-to-body weight ratio was significantly
increased in all the treated animals. Compared with the
iron-overloaded group, CsA showed significant protection against
iron overload in the CsA + iron-overloaded group (P<0.01).
Serum levels of transaminases (ALT and AST) were
used as indicators to evaluate the involvement of CsA in the
structural damage to the liver. In this experiment, the enzyme
assays of serum transaminases demonstrated that the iron overload
significantly raised the levels of ALT and AST to 227.1 and 381.3
U/l, respectively (P<0.01 for both). CsA was capable of
effectively inhibiting the enzyme activity. The levels of ALT and
AST were reduced to 125.5 and 220.1 U/l, respectively (P<0.01
for both) when CsA was administered. This result revealed that the
negative effect of iron overload in mice may be alleviated by CsA
administration (Fig. 2).
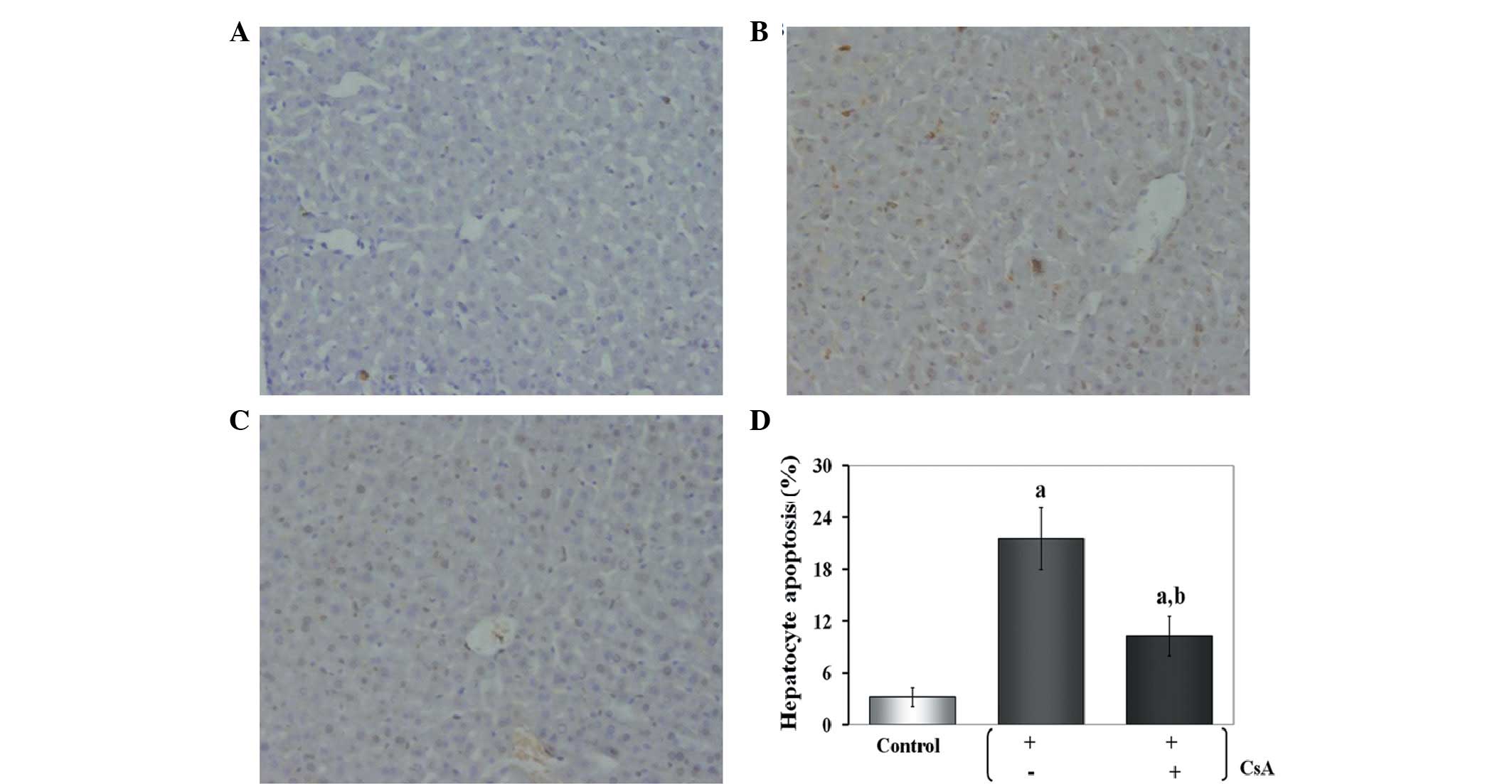
Effects of CsA administration on
hepatocyte apoptosis in iron-overloaded mice
In the control group, a limited number of
TUNEL-positive hepatocytes were detected. By contrast, numerous
hepatocytes in the iron-overloaded group presented as positive for
TUNEL, and this number was significantly greater than that of the
control group (P<0.01). The number of TUNEL-positive cells was
significantly reduced (10.4±2.1%) compared with the iron-overloaded
group when CsA was administered (P<0.01) (Fig. 3).
Effects of CsA administration on the
oxidative parameters of iron-overloaded mice
The effects of CsA on the oxidative stress of
iron-overloaded mice (n=12) were estimated by determining the
activities of MDA, SOD, GSH-Px and catalase in the liver tissue.
The MDA level is a key marker of endogenous lipid peroxidation. In
the iron-overloaded group, the MDA level increased significantly in
the liver compared with the control group (P<0.01). By contrast,
the MDA level in the CsA-treated group decreased significantly
compared with the ferrocene-treated group (P<0.01). This
revealed that CsA was able to successfully block lipid
peroxidation. SOD, GSH-Px and catalase are intracellular
antioxidant enzymes that protect against oxidative processes. As
shown in Table I, iron overload
induced severe oxidative damage and the SOD, GSH-Px and catalase
levels decreased markedly, while CsA effectively normalized the
enzyme activities.
 | Table IEffects of CsA on oxidative stress
parameters in iron-overloaded mice. |
Table I
Effects of CsA on oxidative stress
parameters in iron-overloaded mice.
| Index | Control |
Iron-overloaded | CsA +
iron-overloaded |
|---|
| SOD activity (U/mg
protein) | 382.2±15.5 | 120.1±5.0a | 302.6±13.0b |
| GSH-Px activity
(mU/mg protein) | 248.4±8.9.. | 108.6±4.0a | 160.3±6.2a,b |
| Catalase activity
(U/mg protein) | 316.2±14.5 | 186.3±6.5a | 271.6±8.7b.. |
| MDA content
(pmol/mg protein) | 130.4±4.4.. | 518.5±21.1a | 281.2±10.3a,b |
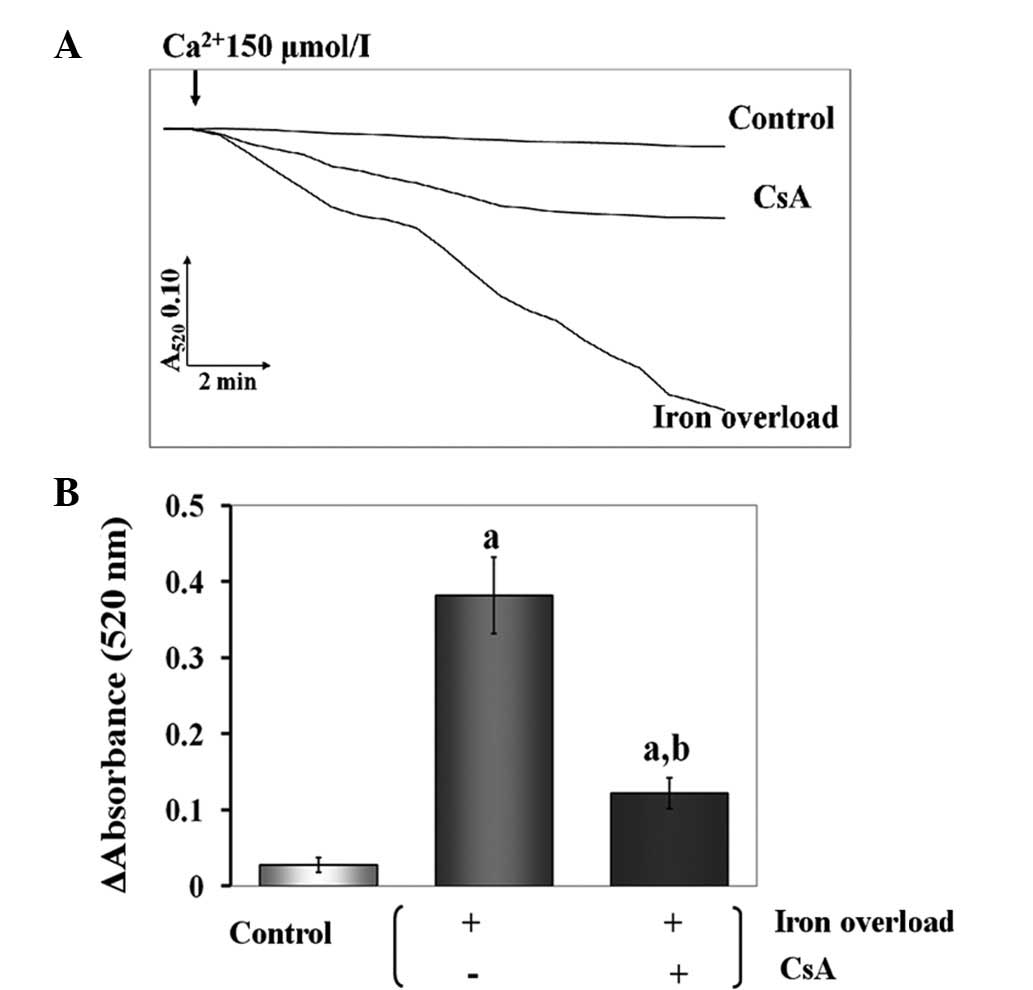
Effects of CsA administration on
mitochondrial swelling in iron-overloaded mice
Iron-induced damage to the inner mitochondrial
membrane may be assessed by the classic swelling techniques, which
monitor the net influx of the osmotic support that is associated
with a non-specific increase in membrane permeability. It was
demonstrated that iron-dextran induced mitochondrial swelling, as
revealed by the large decrease in the absorbance of the
mitochondrial suspension at 520 nm. However, CsA inhibited the
swelling process (P<0.01) (Fig.
4).
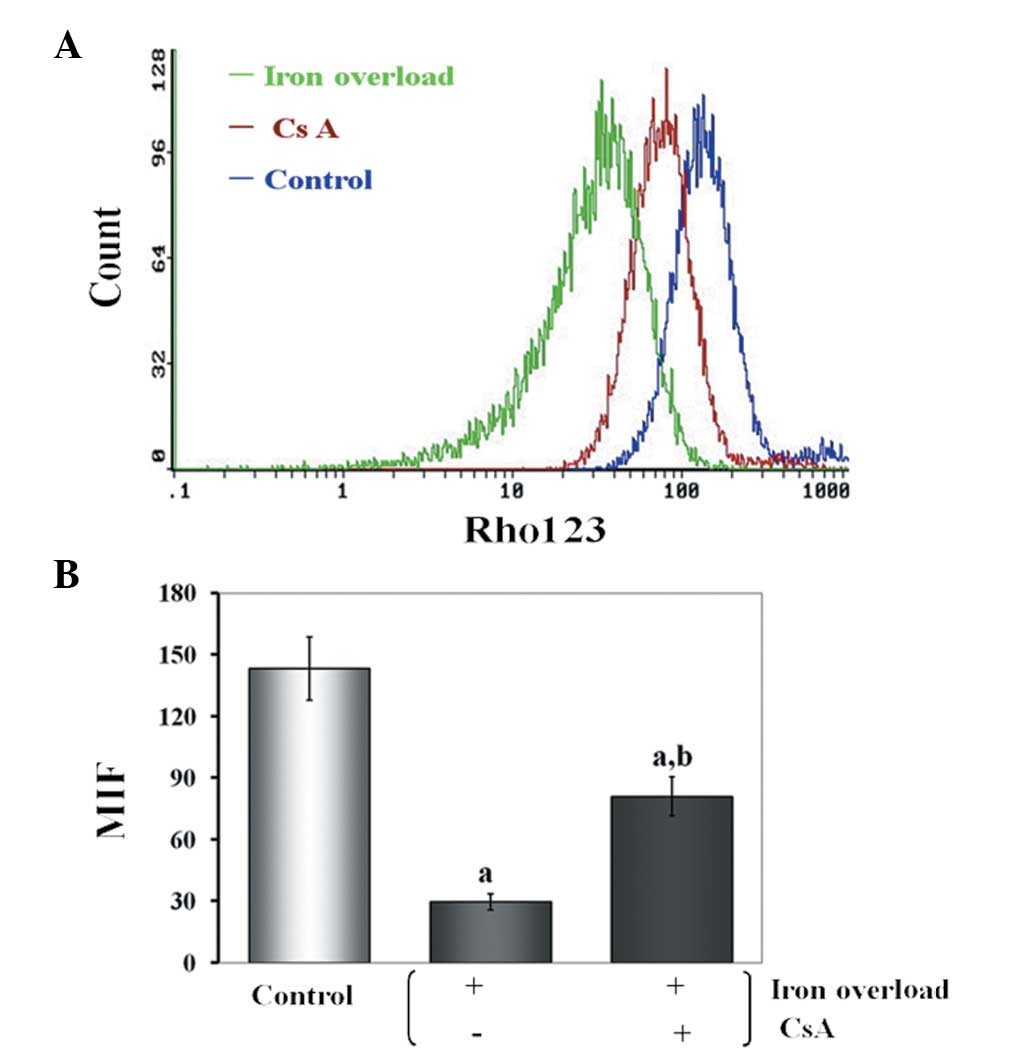
Effects of CsA administration on Δψ of
iron-overloaded mice
The Δψ was determined by the Δψ-sensitive
fluorescent probe, rhodimine 123. The findings revealed that iron
overload induced Δψ depolarization and CsA prevented Δψ dissipation
(Fig. 5).
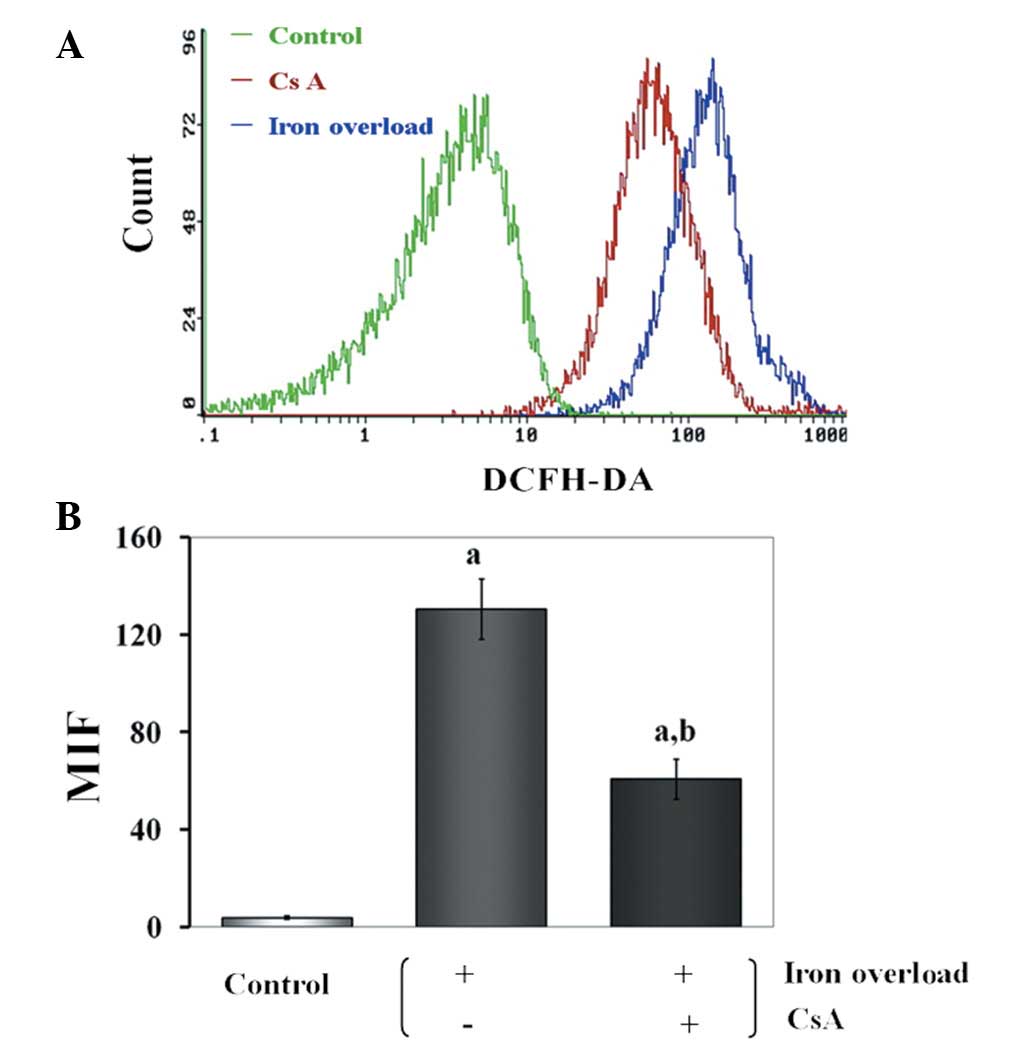
Effects of CsA administration on ROS
production in iron-overloaded mice
By flow cytometry, using the DCFH-DA fluorescent
probe, it was demonstrated that iron-dextran induced ROS
production. As expected, iron overload induced a ROS burst, while
ROS overproduction was prevented when CsA was added simultaneously
to iron (P<0.01) (Fig. 6).
Discussion
Iron is an essential micronutrient. The capacity of
readily exchanging electrons under aerobic conditions causes iron
to be essential for fundamental cell functions, such as DNA
synthesis, transport of oxygen and electrons, and cell respiration
(22). However, as humans have no
means to control their iron excretion, excess iron consumed in the
diet accumulates in parenchymal organs and threatens cell viability
(23). In the current study, in
mice fed a diet supplemented with ferrocene, severe iron overload
occurred and cell damage arose mainly in the liver (the body’s main
storage site for iron). In the present study, a specific inhibitor
of mPTP, CsA, was used (24).
Notably, CsA was unable to reduce iron accumulation in the liver,
but it was able to protect the liver from iron overload.
The mPTP is a voltage-dependent, high-conductance
channel located in the inner mitochondrial membrane, which has been
suggested to be formed by the interaction of several proteins that
connect the mitochondrial matrix to the cytosolic space (25). It has been proposed that the mPTP
is able to open in two modes: low and high conductance (26). When mPTPs are opened in the
long-lasting high-conductance mode, necrosis and apoptosis are
initiated (27). In the
physiological setting, ROS is the most important inducer of mPTP
opening (28).
The chemical structure of iron and its ability to
drive one-electron reactions causes iron to be key for the
production and metabolism of ROS in biological systems. A previous
study clearly demonstrated that these pathological events are
induced by iron-generated ROS (29). As demonstrated in the present
study, such an iron burden initiates significant oxidative stress.
We found that the production of ROS was greater in the
iron-overloaded group compared with the control group. ROS are
capable of causing oxidative damage to macromolecules, leading to
lipid peroxidation, oxidation of amino acid side chains
(particularly cysteine), formation of protein-protein cross-links
and oxidation of polypeptide backbones, resulting in protein
fragmentation, hepatic cell apoptosis and even necrosis (30).
Iron, as a transition metal catalyst, is essential
for the initiation step in the generation of ROS. Subsequently,
excess ROS are the major cause of liver damage. Nevertheless, the
mechanism of the burst of ROS induced by iron overload has not yet
been clarified. Recent data have confirmed that mitochondria are
also capable of producing a significant ROS release when the Δψ is
null following mPTP opening (31,32).
Additionally, ROS are the most important inducer of mPTP opening
(33). These transitions have been
described and occur via mechanisms involved in the process of RIRR
(31,32). Although the phenomenon of RIRR
initiated by ischemic reperfusion injury has been confirmed in
cardiomyocytes (34), RIRR
initiated by iron overload has not yet been demonstrated in
hepatocytes. We suggest that under conditions that lead to RIRR,
such as exposure to iron overload, the increase in ROS reaches a
threshold level triggering mPTP opening, which in turn leads to the
simultaneous collapse of Δψ and a transient increase in ROS
generation by the electron transfer chain (35). The release of this ROS burst into
the cytosol may potentially function as a second messenger to
activate RIRR in the neighboring mitochondria. Thus,
mitochondrion-to-mitochondrion RIRR constitutes a positive feedback
mechanism for enhanced ROS production, potentially leading to
significant mitochondrial and cell injury. RIRR is generated by
circuits requiring mitochondrial membrane channels, including the
mPTP. Therefore, the mPTP is important in RIRR. Our present results
have demonstrated that the large-amplitude ROS burst initiated by
iron overload was prevented by the administration of CsA. Moreover,
depolarization and apoptosis were prevented, while inhibition of
the ROS burst in mice was estimated by determining the activities
of MDA, SOD, GSH-Px, catalase, ALT and AST in serum and tissues.
CsA not only protected the liver from damage by efficiently
inhibiting MDA formation, and by reducing AST and ALT, but it also
enhanced the activities of the antioxidant enzyme system of the
host, including those of SOD, GSH-Px and catalase.
Similar to the propagation of mitochondrial
depolarization and ROS production demonstrated in isolated
cardiomyocytes by Zorov et al(34), it has been observed that
mPTP-mediated RIRR also exists in liver subjected to iron overload.
Iron overload initiates the generation of ROS, and ROS induce mPTP
opening. Extensive matrix swelling with long-lasting mPTP opening
may lead to the unfolding of cristae, causing the outer membrane to
rupture, irreversibly damaging the mitochondria, and consequently,
ROS are released from the mitochondrial matrix into the cytosol.
Thus, ROS may potentially function as a second messenger to
activate RIRR in the neighboring mitochondria, and the liver
overloaded with iron becomes damaged.
In conclusion, our results strongly support the
hypothesis that RIRR mediated by mPTP may generate a large number
of ROS, and provide a possible mechanism by which excess dietary
iron uptake results in liver damage in mice.
Acknowledgements
This study was supported by grants from the Natural
Scientific Foundations of China (grant nos. 30760075, 30860271 and
81100104).
References
|
1
|
Nahon P, Ganne-Carrié N, Trinchet JC and
Beaugrand M: Hepatic iron overload and risk of hepatocellular
carcinoma in cirrhosis. Gastroenterol Clin Biol. 34:1–7. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Allen KJ, Gurrin LC, Constantine CC, et
al: Iron-overload-related disease in HFE hereditary
hemochromatosis. N Engl J Med. 358:221–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Asare GA, Kew MC, Mossanda KS, Paterson
AC, Siziba K and Kahler-Venter CP: Effects of exogenous
antioxidants on dietary iron overload. J Clin Biochem Nutr.
44:85–94. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pardo Andreu GL, Inada NM, Vercesi AE and
Curti C: Uncoupling and oxidative stress in liver mitochondria
isolated from rats with acute iron overload. Arch Toxicol.
83:47–53. 2009.PubMed/NCBI
|
|
5
|
Starkov AA: The role of mitochondria in
reactive oxygen species metabolism and signaling. Ann NY Acad Sci.
1147:37–52. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Brady NR, Elmore SP, van Beek JJ, Krab K,
Courtoy PJ, Hue L and Westerhoff HV: Coordinated behavior of
mitochondria in both space and time: a reactive oxygen
species-activated wave of mitochondrial depolarization. Biophys J.
87:2022–2034. 2004. View Article : Google Scholar
|
|
8
|
Gateau-Roesch O, Argaud L and Ovize M:
Mitochondrial permeability transition pore and postconditioning.
Cardiovasc Res. 70:264–273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Saotome M, Katoh H, Yaguchi Y, Tanaka T,
Urushida T, Satoh H and Hayashi H: Transient opening of
mitochondrial permeability transition pore by reactive oxygen
species protects myocardium from ischemia-reperfusion injury. Am J
Physiol Heart Circ Physiol. 296:H1125–H1132. 2009. View Article : Google Scholar
|
|
10
|
Halestrap AP: Calcium, mitochondria and
reperfusion injury: a pore way to die. Biochem Soc Trans.
34:232–237. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Galaris D, Skiada V and Barbouti A: Redox
signaling and cancer: the role of ‘labile’ iron. Cancer Lett.
266:21–29. 2008.
|
|
12
|
Tjalkens RB, Valerio LG Jr, Awasthi YC and
Petersen DR: Association of glutathione S-transferase
isozyme-specific induction and lipid peroxidation in two inbred
strains of mice subjected to chronic dietary iron overload. Toxicol
Appl Pharmacol. 151:174–181. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Galleano M and Puntarulo S: Hepatic
chemiluminescence and lipid peroxidation in mild iron overload.
Toxicology. 76:27–38. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brumby PE and Massey V: Determination of
nonheme iron, total iron, and copper. Methods Enzymol. 10:463–474.
1967. View Article : Google Scholar
|
|
15
|
Beavis AD, Brannan RD and Garlid KD:
Swelling and contraction of the mitochondrial matrix. I A
structural interpretation of the relationship between light
scattering and matrix volume. J Biol Chem. 260:13424–13433.
1985.PubMed/NCBI
|
|
16
|
Emaus RK, Grunwald R and Lemasters JJ:
Rhodamine 123 as a probe of transmembrane potential in isolated
rat-liver mitochondria: spectral and metabolic properties. Biochim
Biophys Acta. 850:436–448. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Desmots F, Rissel M, Pigeon C, Loyer P,
Loréal O and Guillouzo A: Differential effects of iron overload on
GST isoform expression in mouse liver and kidney and correlation
between GSTA4 induction and overproduction of free radicals. Free
Radic Biol Med. 32:93–101. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Okhawa H, Ohishi N and Yagi K: Assay for
lipid peroxides in animal tissues by thiobarbituric acid reaction.
Anal Biochem. 95:351–358. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Beauchamp C and Fridovich I: Superoxide
dismutase: improved assays and an assay applicable to acrylamide
gels. Anal Biochem. 44:276–287. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lawrence RA and Burk RF: Glutathione
peroxidase activity in selenium-deficient rat liver. Biochem
Biophys Res Commun. 71:952–958. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pedraza-Chaverri J, Granados-Silvestre MD,
Medina-Campos ON and Hernández-Pando R: Effect of the in vivo
catalase inhibition on aminonucleoside nephrosis. Free Radic Biol
Med. 27:245–253. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ramm GA and Ruddell RG: Iron homeostasis,
hepatocellular injury, and fibrogenesis in hemochromatosis: the
role of inflammation in a noninflammatory liver disease. Semin
Liver Dis. 30:271–287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen J and Chloupková M: Abnormal iron
uptake and liver cancer. Cancer Biol Ther. 8:1699–1708. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xie JR and Yu LN: Cardioprotective effects
of cyclosporine A in an in vivo model of myocardial ischemia and
reperfusion. Acta Anaesthesiol Scand. 51:909–913. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Halestrap AP, McStay GP and Clarke SJ: The
permeability transition pore complex: another view. Biochimie.
84:153–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Novgorodov SA and Gudz TI: Permeability
transition pore of the inner mitochondrial membrane can operate in
two open states with different selectivities. J Bioenerg Biomembr.
28:139–146. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Joza N, Susin SA, Daugas E, et al:
Essential role of the mitochondrial apoptosis-inducing factor in
programmed cell death. Nature. 410:549–554. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Halestrap AP: The mitochondrial
permeability transition - a‘pore way for the heart to die. J Clin
Basic Cardiol. 5:29–41. 2002.
|
|
29
|
Galaris D and Pantopoulos K: Oxidative
stress and iron homeostasis: mechanistic and health aspects. Crit
Rev Clin Lab Sci. 45:1–23. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Van HB, Woshner V and Santos JH: Role of
mitochondrial DNA in toxic responses to oxidative stress. DNA
Repair (Amst). 5:145–152. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brady NR, Hamacher-Brady A, Westerhoff HV
and Gottlieb RA: A wave of reactive oxygen species (ROS)-induced
ROS release in a sea of excitable mitochondria. Antioxid Redox
Signal. 8:1651–1665. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Serviddio G, Bellanti F, Sastre J,
Vendemiale G and Altomare E: Targeting mitochondria: a new
promising approach for the treatment of liver diseases. Curr Med
Chem. 17:2325–2337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zorov DB, Filburn CR, Klotz LO, Zweier JL
and Sollott SJ: Reactive oxygen species (ROS)-induced ROS release:
a new phenomenon accompanying induction of the mitochondrial
permeability transition in cardiac myocytes. J Exp Med.
192:1001–1014. 2000. View Article : Google Scholar
|
|
35
|
Batandier C, Leverve X and Fontaine E:
Opening of the mitochondrial permeability transition pore induces
reactive oxygen species production at the level of the respiratory
chain complex I. J Biol Chem. 279:17197–17204. 2004. View Article : Google Scholar : PubMed/NCBI
|