Introduction
Colorectal adenocarcinoma (CRC) is the third most
common type of cancer worldwide accounting for 8.9–9.4% of all
cancer cases (1,2). The 5-year survival rate for poorly
differentiatied CRC is 29% (3).
Development of human CRC is a multistep process,
involving numerous pathological changes in gene expression and
protein function. In a previous study, a cDNA subtraction library
was established and from this, 86 differentially expressed sequence
tags in human CRC were identified using the suppression subtractive
hybridization technique combined with cDNA microarray (4). Among these newly identified
differentially expressed tags, mRNA expression of ES274081 was
observed to be downregulated in human CRC by quantitative real
time-polymerase chain reaction (5), indicating a potential role in the
development of human CRC. In the present study, to explore the
function of NM_001013649, the corresponding protein, HCRCN81, was
identified using bioinformatic tools, mass spectrometry and western
blot analysis.
Materials and methods
Cloning of differentially expressed tags
in human CRC
Differentially expressed tags were identified from a
cDNA subtraction library, using the suppression subtractive
hybridization technique combined with cDNA microarray (GenBank
accession number, ES274081). Internet sources and bioinformatics
analysis software packages are shown in Table I.
 | Table IInternet sources. |
Bioinformatical analysis
Using the website, http://www.ncbi.nlm.nih.gov/unigene, a search was
performed on the accession number (ES274081) and the sequence
information of a 4,283-bp cDNA molecule (GenBank accession number,
NM_001013649) was obtained. Using the open reading frame (ORF)
finder tool provided by NCBI, the initiation codon was predicted
according to the principle that the initiation codon in the Kozak
sequence must be suitable for translation initiation (6). According to the Chou-Fasman
prediction method of the secondary structure of proteins, 41 amino
acids are capable of forming 2 helices. The hydrophobic portion of
NM_001013649 was analyzed with ProtScale and the α-helix and
β-sheet structures were predicted using PredictProtein. In
addition, the coded amino acid sequence was analyzed using BLAST,
CPHmodels, SMART, Pfam and Motif Scan.
Mass spectrometric analysis
Mass spectrometry (Proevolab, Beijing, China) was
used to confirm the predicted amino acid sequence of NM_001013649
(7). Briefly, the protein sample
was digested on ice with trypsin (0.01 μg/μl; 10 μl). Following
removal of trypsin, the digested sample was incubated with an
ammonium bicarbonate solution (5 μl; 25 mM) at 37°C overnight. The
incubated sample (2 μl) was mixed. The liquid gradient was set at
136 min and the MS acquisition was set at 110 min. For the
first-round scan, Fourier transform mass spectrometry was used with
a range of 400–1,500 Da. For the second-round scan, linear ion trap
quadrupole was used.
Western blot analysis
Tissue samples were obtained from two patients: a
74-year-old female patient with rectal tubular villous carcinoma,
with a complication of moderate epithelial hyperplasia and a
76-year-old male patient with rectal adenocarcinoma. Tumor and
normal tissue located 5 cm from the tumor tissues were collected to
detect the expression of the target protein using 12% SDS-PAGE. For
each reaction, 30 μg samples were used and the antibody serum was
diluted by 1:500. The antibody was made to order by CWBio, Beijing,
China (patent number: 201210445862.6). Informed consent was
obtained from the patients and the study was approved by the Ethics
Committee of Sichuan University.
Results
Bioinformatic analyses
Using the NCBI database, a 4,283 bp target
full-length cDNA located in chromosome 2, ORF 68 (2p11.2) was
obtained. An initiation codon (ATG) and a termination codon (TGA)
were located at the 73–75 and 571–573 nucleotides of this sequence,
respectively, which defined the longest ORF in this sequence,
representing 166 amino acids (Fig.
1).
The corresponding protein molecule contained 166
amino acids with a comparative MW of 18,750.9 kDa and a pI of 8.44.
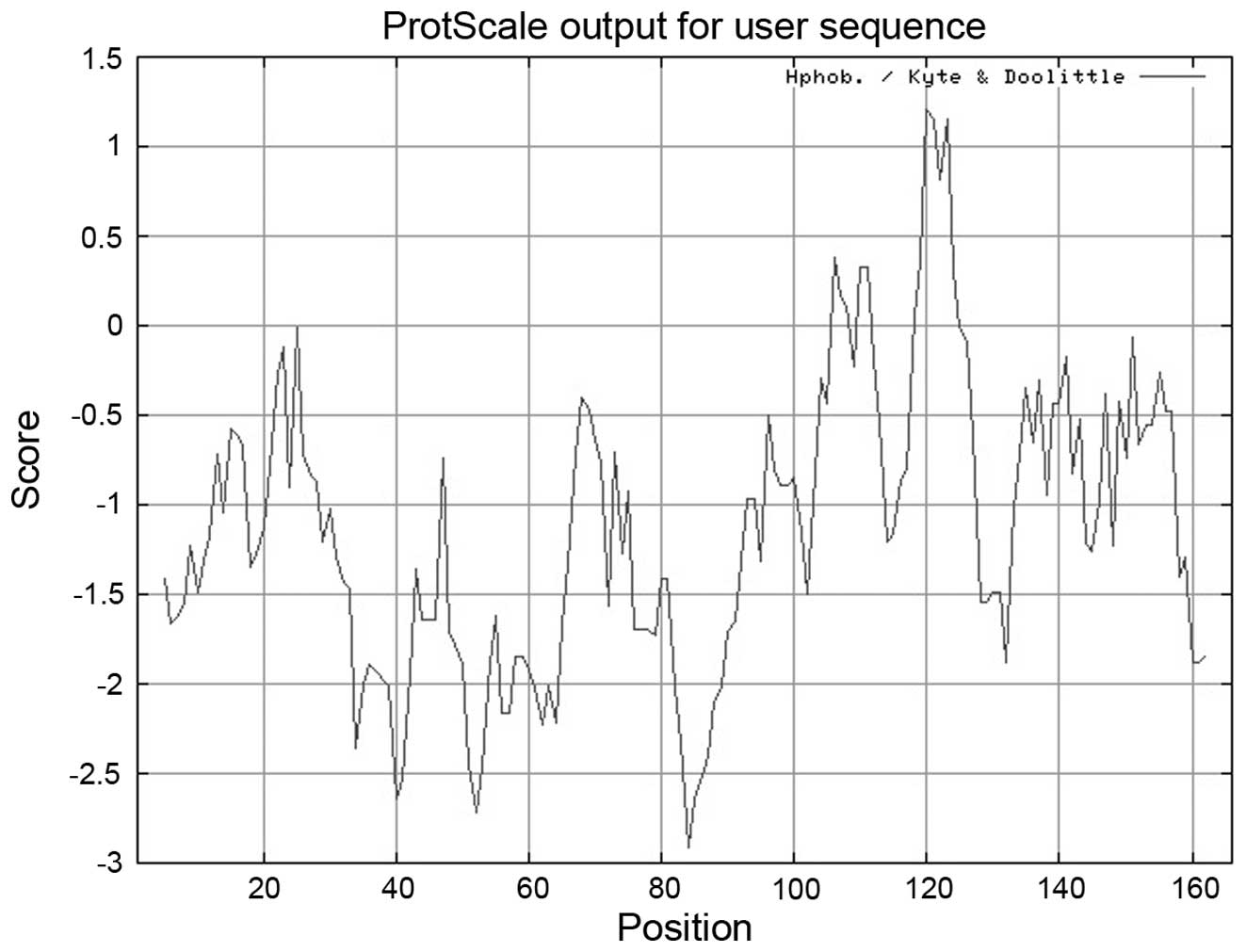
Protein hydrophilic analysis (Fig.
2) revealed the presence of a hydrophilic fragment, in the
region of 80–90 amino acids and a hydrophobic fragment in the
region of 115–125 amino acids. Using the tool NetNGlyc, no
O- or N-linkage glycosylation locus was observed in
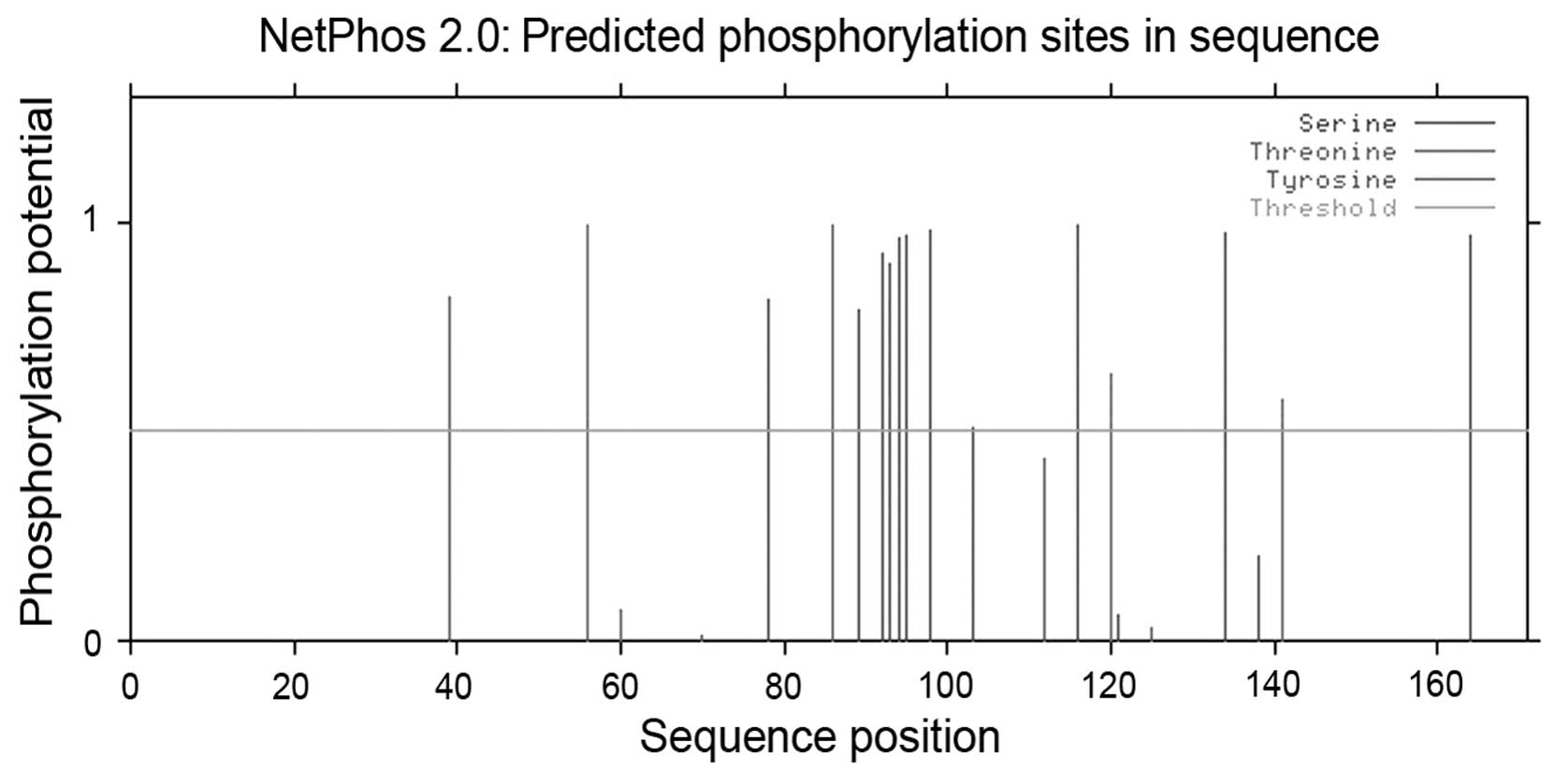
this sequence. Using NetPhos, potential phosphorylation sites were
observed at the following amino acid residues: 39, 56, 78, 86, 89,
92, 93, 94, 95, 98, 103, 116, 120, 134, 138, 141 and 164 (Fig. 3). In addition, no transmembrane
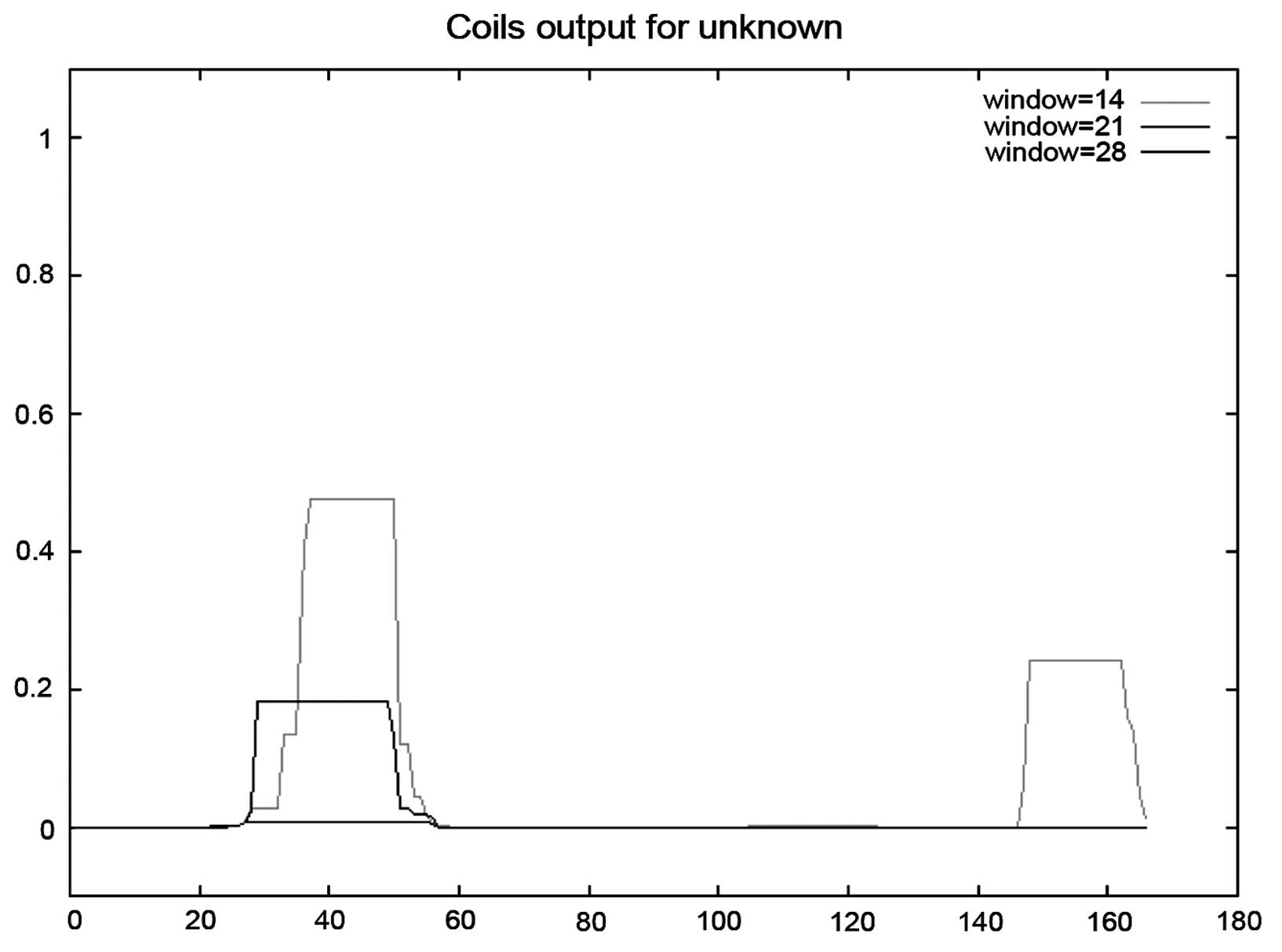
region or orientation was identifed in this sequence. The results
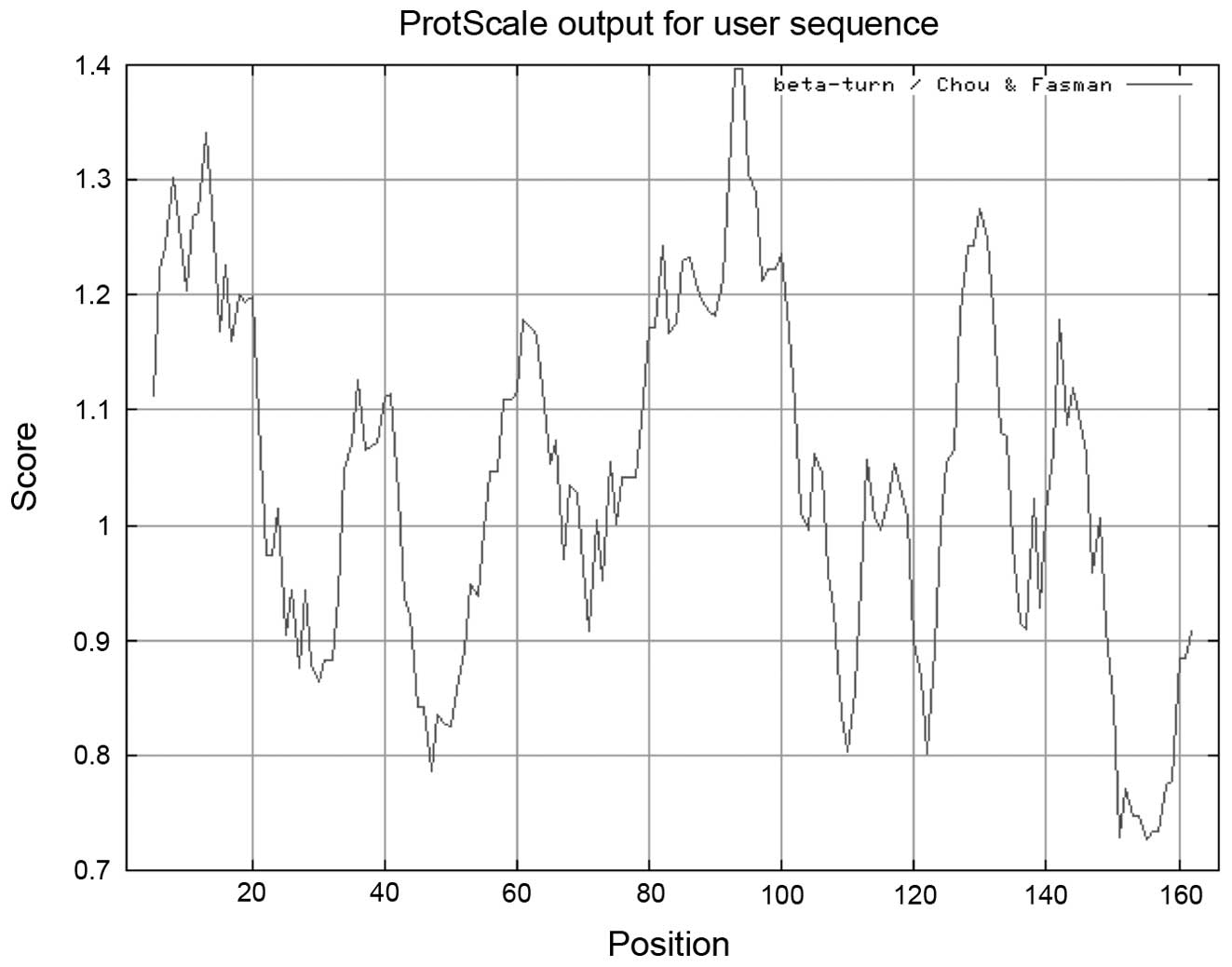
of the coiled-coil analysis is shown in Fig. 4. β-turn analysis indicated that a
β-turn may be present near amino acid residues 6, 13, 60, 93, 131
and 145 (Fig. 5).
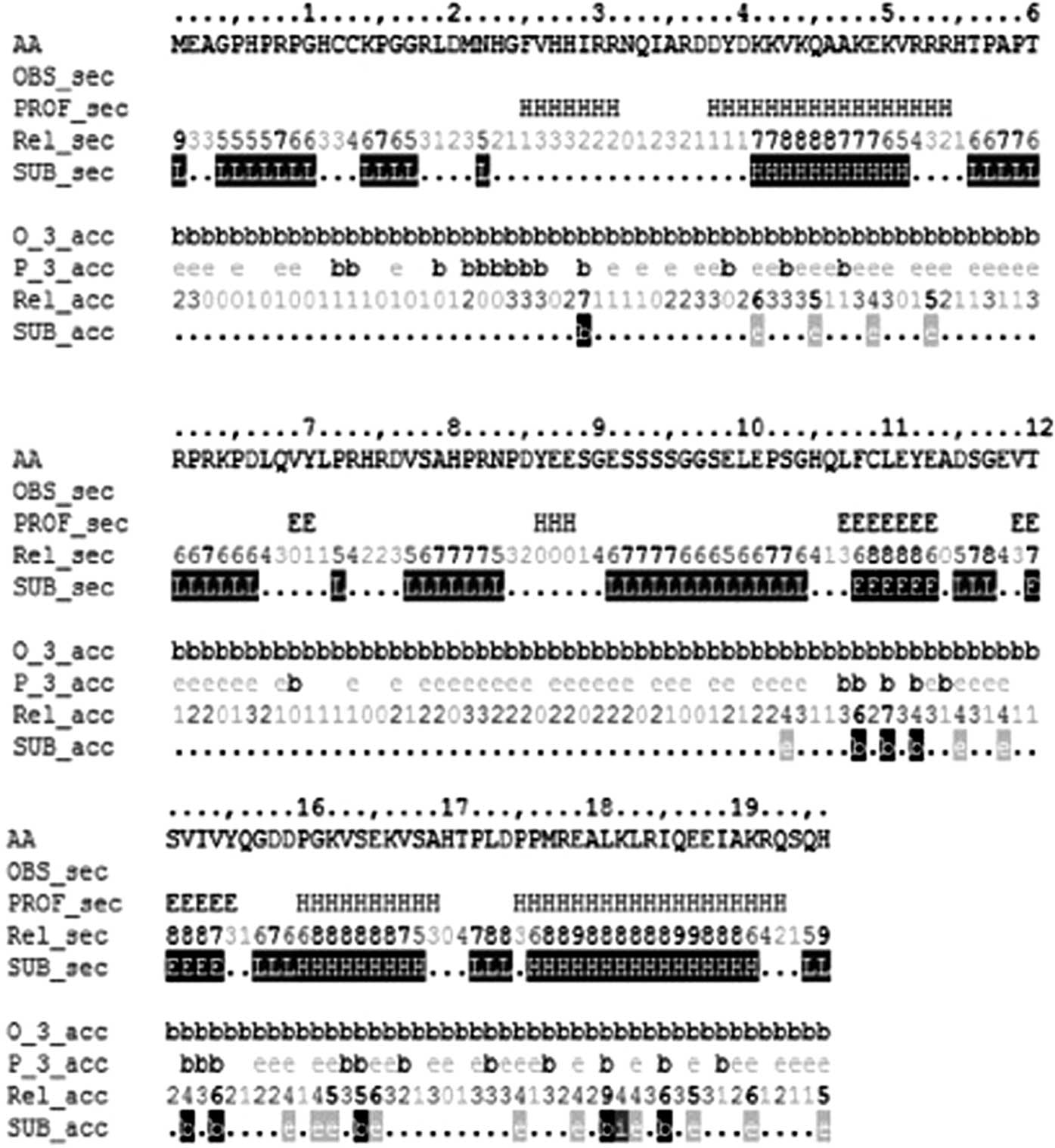
HCRCN81
In the predicted secondary structure of the HCRCN81
protein, 56 amino acid residues were involved in the formation of
α-helices, including the 25–31, 38–54, 86–88, 130–139 and 145–163
residues, accounting for 33.73% of the entire protein sequence. By
contrast, 16 amino acids were involved in the formation of
β-sheets, including the 69–70, 107–113 and 119–125 amino acid
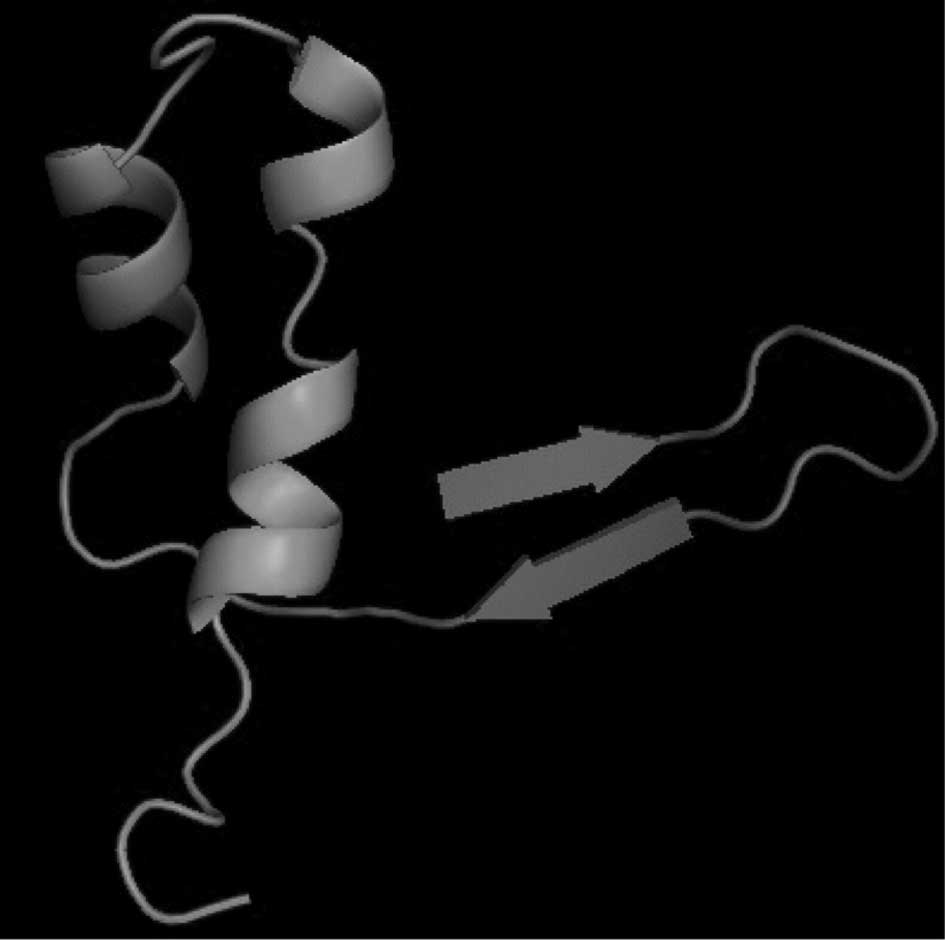
residues, accounting for 9.64% of the protein sequence (Fig. 6). Using the tool CPHmodels
(http://www.cbs.dtu.dk/services/CPHmodels/) provided by
the Center for Biological Sequence Analysis (Lyngby, Denmark) the
tertiary structure of this protein was predicted (Fig. 7).
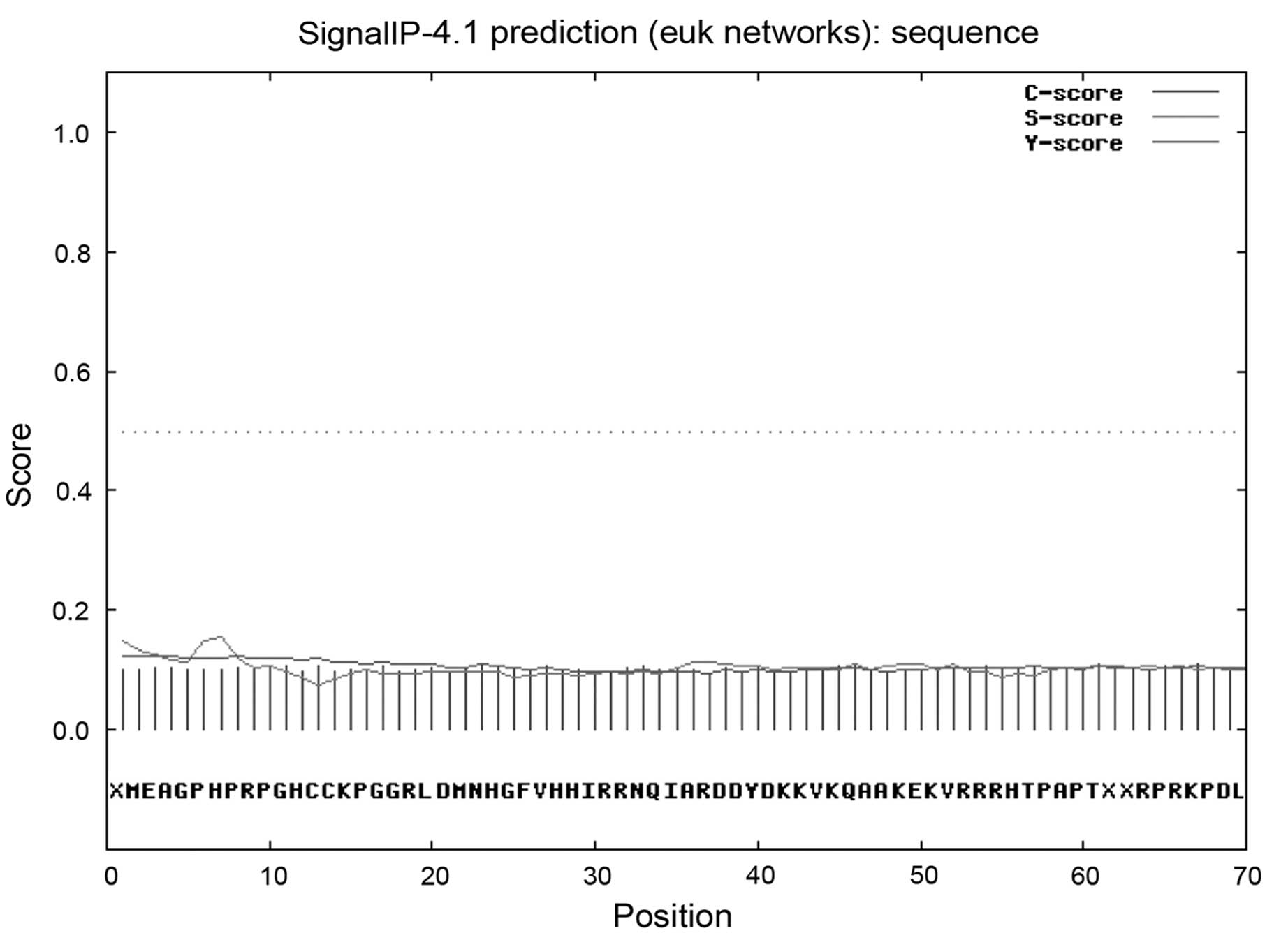
In addition, the localization pattern of the
protein, HCRCN81, was explored. Using SignalP, no signal peptide of
1–70 amino acids was identified (Fig.
8). Utilizing TargetP, it was noted that there was a reduced
possibility of the protein, HCRCN81, to be localized in
mitochondria, endoplasmic reticulum or other components of the
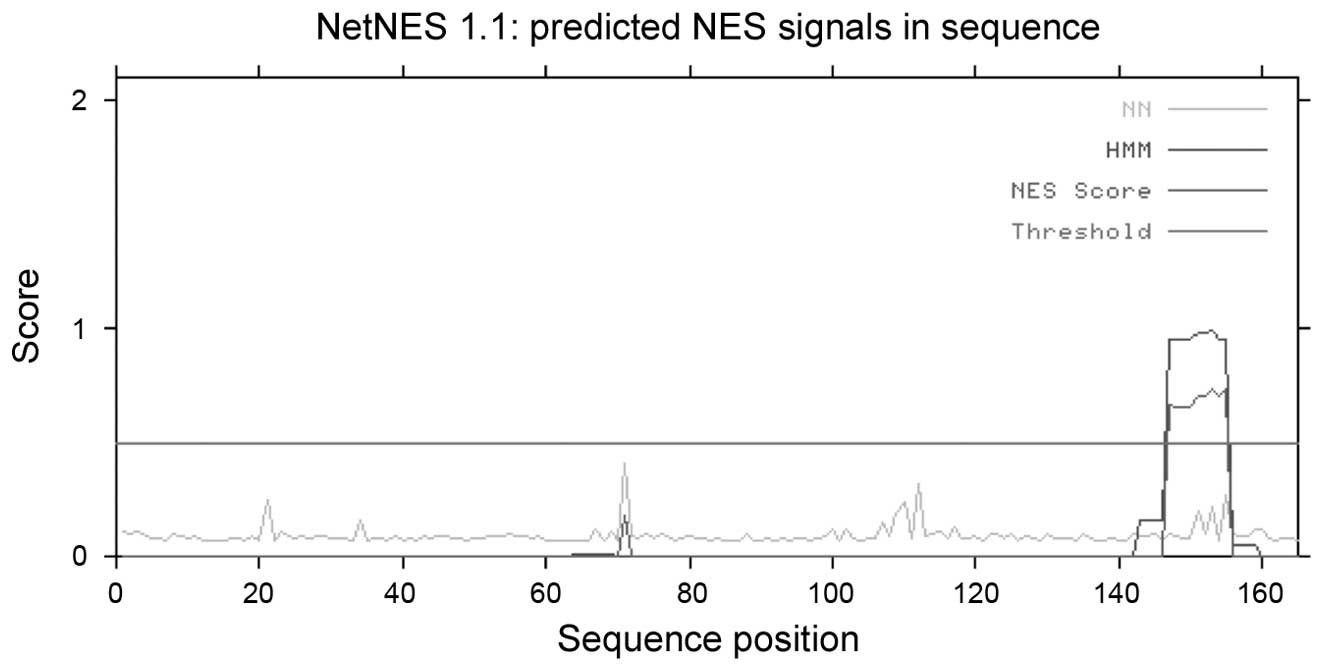
secretory pathway. Analysis using the tool NetNES revealed the
presence of a leucine-rich nuclear export signal (NES) at amino
acid residues 147–155 of the protein, HCRCN81 (Fig. 9).
Analysis of the protein sequence using SMART and
Pfam (8), revealed two areas with
low complexity at residues 52–65 and 87–104 of HCRCN81. This
protein is a member of the UPF0561 family and shares a domain
containing 1–126 amino acids with the UPF0561 family members. Using
SMART, three regions in HCRCN81 that may be homologs of other known
structures were identified: i) 11–39 amino acids shared with SCOP:
d1fn9a (E-value 2.20e+00), which was present in the outer capsid
protein σ3 (9); ii) 90–108 amino
acids shared with SCOP: d1eg3a2 (E-value 7.30e+00), which was
present in the EF-hand domain (10); iii) 110–145 amino acids shared with
SCOPL: d1qlma_(E-value 1.10e+00), which was present in
methenyltetrahydromethanopterin cyclohydrolase (11).
Analysis using Motif Scan revealed cAMP- and
cGMP-dependent protein kinase phosphorylation sites at residues
53–56 and 161–164; casein kinase II phosphorylation sites at
residues 98–101 and 141–144; protein kinase C phosphorylation sites
at residues 134–136; tyrosine kinase phosphorylation sites at
residues 63–70 and bipartite nuclear localization signal at
residues 30–44.
Using the tool, PeptideMass, it was observed that
HCRCN81 may be digested into peptides, the of which sequences are
shown in Fig. 10 (selected
500–3,000 kDa). Mass spectrometric analysis revealed that the
protein molecule contained 165 amino acids, had lost a histidine in
the terminal region, had a monoisotopic MW of 18.6033 and a pI of
8.43. The sequences are shown in Fig.
11, which covered 81.3% of the entire protein molecule. Taking
the remaining the peptide sequences into consideration, results of
mass spectrometry were considered to be accurate.
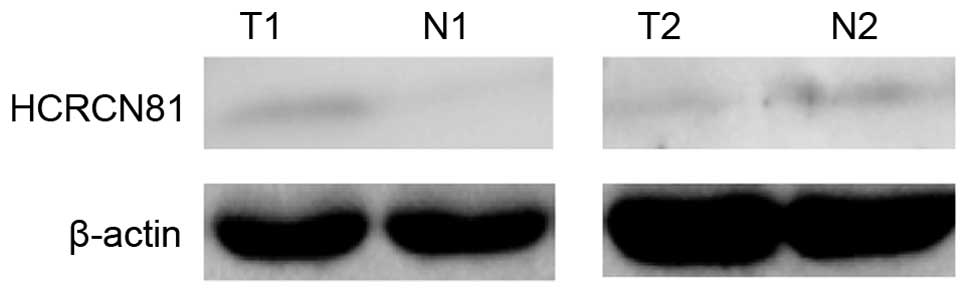
Western blot analysis demonstrated a clear band at
the position of 18 kDa (Fig.
12).
Discussion
Following completion of the Human Genome Project and
current progression of the post-Human Genome Project, a large
amount of genomic information is available at present. To improve
understanding of the significant amount of the notable increase of
biological information, bioinformatics has become a useful tool for
biological and biomedical studies. In the early stages of the Human
Genome Project, small clones were prioritized for full-length
sequencing (12). A number of
software and network tools have been significant in cloning novel
genes. In particular, a large amount of information on expressed
sequence tags (ESTs) has led to marked changes in the methods used
for identifying and cloning novel genes. A series of gene
expression analyses (13) and
large-scale sequencing studies of ORF tags have made a significant
contribution to the definition of the transcriptome and the
improvement of genome annotation (14).
In the present study, a number of network tools and
software sets were used to analyze the target sequence, which was
beneficial in studying the function of the HCRCN81 protein. More
specifically, the full-length cDNA sequence from the NCBI database
was used, BLAST was used to search the ESTs and DNAstar was
employed for in silico cloning. An overlap of two EST alkali
bases of >40 bp, with a shared similarity of 95% in the
overlapping region, confirmed an accurate match in sequences
(15). Following this, the EST was
extended for as long as possible and the matching sequences were
assembled together forming a new EST for BLAST analysis to identify
more matching sequences. This approach was repeated until no more
matching sequences were found. Following this, the obtained
full-length cDNA sequence was analyzed using bioinformatic tools
and databases from the internet (16) and a novel protein, HCRCN81,
involved in human CRC, was identified. For this identification, the
first step was to translate the cDNA sequence into an amino acid
sequence. Initially, numerous initiation codons were identified in
NM_001013649. Following this, ORF was used for analysis and it was
noted that the majority of these initiation codons resulted in
sequences that were too short for a protein molecule. Therefore,
the longest peptide sequence was determined as the protein
molecule. This result was validated by uniGene in NCBI. The
full-length amino acid sequence was then obtained and the physical
and chemical characteristics of the protein molecule were analyzed
using multiple tools. In particular, ProtParam and ProtScale
provided by ExPASy were used to determine the MW and theoretical
pI. Additional software sets and tools were used to determine the
hydrophobic region, phosphorylation sites and O- and
N-linkage glycosylation sites within the sequence. Following
this, the protein structure was predicted. Jpred and PredictProtein
were used to analyze the secondary structure. Results of the two
approaches were consistent. To predict the tertiary structure of
the target protein, the sequence was analyzed using SWISS-MODEL and
CBS. The SWISS-MODEL did not yield a result, however, CBS analysis
revealed a partial structure of the protein between 108–166 amino
acids.
In addition, the function of this protein was
investigated. Since NLS and NES were predicted to be present in
this protein, it was speculated that the protein may be involved in
nucleic function. The outer capsid protein σ3 is capable of
stimulating translation by blocking the activation of the
dsRNA-dependent protein kinase, EIF2AK2/PKR (17). The function of
methenyltetrahydromethanopterin cyclohydrolase is to catalyze the
reversible interconversion from 5-formyl-H(4)MPT to methenyl-H(4)MPT(+), by performing the following
catalytic reaction: 5,10-methenyl-5,6,7,8-tetrahydromethanopterin +
H2O = 5-formyl-5,6,7,8-tetrahydromethanopterin. In
addition, casein kinase II activation has been reported as a
downstream event of Wnt signaling activation (18) and tyrosine kinases have been found
to transport into the nuclear regions where gene expression may be
modified (19). Considering the
presence of numerous potential phosphorylation sites, particularly
kinase phosphorylation sites in the EF-hand domain, it was
hypothesized that, following synthesis, this target protein may be
transported into the nucleus, due to the presence of an NLS, which
may be phosphorylated, leading to the exposure of its functional
regions. It may then bind to Ca2+ through the EF-hand
domain (10), exposing the NES
region, which, in turn, may lead to its transportation out of the
nucleus. Consistent with this hypothesis, it has been reported that
the EF-hand is directly associated with chronic inflammatory
disorders and cancer (15).
However, the function of the UPF0651 family remains unclear. A
previous study observed downregulation of the mRNA of this gene in
human CRC (5). Combined, these
observations indicate that this novel protein is potentially key to
the development of human CRC.
Mass spectrometry was used to confirm the
hypothesis. PeptideMass was used to predict the small peptide
sequences following trypsin treatment. By comparing the prediction
and the mass spectrometry results, it was confirmed that the
prediction of the amino acid sequence of the protein HCRCN81 was
accurate.
Western blot analysis, using antibody serum specific
for HCRCN81, revealed that HCRCN81 was expressed in tumor and
adjacent normal tissues. Previous studies (4,5) have
revealed that mRNA expression of NM_001013649 is downregulated in
human CRC tissue. Among 30 human CRC tissue samples, 5 revealed
upregulated NM_001013649 mRNA expression, whereas 25 demonstrated
downregulated NM_001013649 mRNA expression, accounting for 83% of
all tested samples. This observation indicated the potential
involvement of NM_001013649 in CRC pathogenesis. In addition,
NM_001013649 downregulation was detected in 91% of the moderately
differentiated samples (21/23) but only in 50% of the poorly
differentiated tissue samples (3/6) and the difference was
considered to be statistically significant (Fisher's exact
probability test, P<0.05). This observation reveals a possible
correlation between NM_001013649.3 transcriptional expression and
CRC tumor stage.
In the present study, using bioinformatic tools, the
structure of a novel protein, HCRCN81, translated from
NM_001013649.3 mRNA, was analyzed. Using western blot analysis,
protein expression of HCRCN81 in cell lines and colorectal tissue
samples was detected, with a MW of ~18 kDa, consistent with the
hypothesis. In addition, potential NLS and NES from the protein
sequence of HCRCN81 and the EF-hand domain were predicted. This
observation was in accordance with the hypothesis that HCRCN81 may
function as a cell cycle regulator in the nucleus, which, in turn,
may explain its possible role in CRC pathogenesis. Mass
spectrometry was used to determine the amino acid sequence of the
protein, the result of which was consistent with the prediction
using bioinformatic tools. The bioinformatic analysis of the
present study also indicated that HCRCN81 is a member of the
UPF0561 family. The protein function of the UPF0561 family has not
been well characterized. The current study indicates that proteins
of the UPF0561 family may have a similar function to HCRCN81 in
cell cycle regulation and additional studies on this family must be
performed.
Acknowledgements
This study was supported by a grant from the Sichuan
University for Stomatological Key Laboratories
(SKLODSCU20090021).
References
|
1
|
Pisani P, Parkin DM and Ferlay J:
Estimates of the worldwide incidence of eighteen major cancers in
1985. Implications for prevention and projections of future burden.
Int J Cancer. 55:891–903. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
3
|
Burton S, Norman AR, Brown G, Abulafi AM
and Swift RI: Predictive poor prognostic factors in colonic
carcinoma. Surg Oncol. 15:71–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen Y, Zhang YZ, Zhou ZG, Wang G and Yi
ZH: Identification of differently expressed genes in human
colorectal adenocarcinoma. World J Gastroenterol. 12:1025–1032.
2006.PubMed/NCBI
|
|
5
|
Jiang Q, Zhang C and Chen Y:
NM_001013649.3 gene is downregulated in human colorectal
adenocarcinoma. Mol Med Rep. 4:1279–1281. 2011.PubMed/NCBI
|
|
6
|
Zhang C and He F: Bioinformatics methods
and practices. Sci Press; pp. 64–142. 2002
|
|
7
|
Guthals A and Bandeira N: Peptide
identification by tandem mass spectrometry with alternate
fragmentation modes. Mol Cell Proteomics. 11:550–557. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schultz J, Milpetz F, Bork P and Ponting
CP: SMART, a simple modular architecture research tool:
Identification of signaling domains. Proc Natl Acad Sci USA.
95:5857–5864. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Schiff LA, Nibert ML, Co MS, et al:
Distinct binding sites for zinc and double-stranded RNA in the
reovirus outer capsid protein sigma 3. Mol Cell Biol. 8:273–283.
1988.PubMed/NCBI
|
|
10
|
Chazin WJ: Relating form and function of
EF-hand calcium binding proteins. Acc Chem Res. 44:171–179. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Upadhyay V, Demmer U, Warkentin E, et al:
Structure and catalytic mechanism of
N(5),N(10)-methenyl-tetrahydromethanopterin cyclohydrolase.
Biochemistry. 51:8435–8443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kawai J, Shinagawa A, Shibata K, et al:
The RIKEN Genome Exploration Research Group Phase II Team and the
FANTOM Consortium: Functional annotation of a full-length mouse
cDNA collection. Nature. 409:685–690. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Saha S, Sparks AB, Rago C, et al: Using
the transcriptome to annotate the genome. Nature Biotechnol.
20:508–512. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Camargo AA, Samaia HP, Dias-Neto E, et al:
The contribution of 700,000 ORF sequence tags to the definition of
the human transcriptome. Proc Natl Acad Sci USA. 98:12103–12108.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Radha V, Nambirajan S and Swarup G:
Association of Lyn tyrosine kinase with the nuclear matrix and
cell-cycle-dependent changes in matrix-associated tyrosine kinase
activity. Eur J Biochem. 236:352–359. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
General Higher Education Eleventh
Five-Year national planning materials. Genomics. Yang J: Higher
Education Press; China: pp. 74–88. 2002
|
|
17
|
Farsetta DL, Chandran K and Nibert ML:
Transcriptional activities of reovirus RNA polymerase in recoated
cores. Initiation and elongation are regulated by separate
mechanisms. J Biol Chem. 275:39693–39701. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gebhardt C, Németh J, Angel P and Hess J:
S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol.
72:1622–1631. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao Y and Wang HY: Casein kinase 2 is
activated and essential for Wnt/beta-catenin signaling. J Biol
Chem. 281:18394–18400. 2006. View Article : Google Scholar : PubMed/NCBI
|