Introduction
Acute ischemic stroke, the most common form of
stroke, can produce an irreversibly injured core, in which cell
death is rapid and not salvageable, and an ischemic penumbra, where
tissue is damaged but potentially salvageable (1,2). The
penumbra has a limited life span and appears to undergo
irreversible damage within a few hours unless reperfusion is
initiated and/or neuroprotective therapy is administered (3). Therefore, the early rapid recovery of
cerebral blood flow and effective neuroprotective treatment in
cerebral ischemia remains vital. However, to date, there remains no
established treatment for lessening ischemic brain injury.
Brain injury after focal cerebral ischemia develops
from a series of pathological processes, including excitotoxicity,
peri-infarct depolarizations, ionic imbalance, oxidative stresses
and apoptosis (1,2,4,5).
Although these mechanisms have been implicated in ischemic neuronal
death, Ca2+ overload remains the central focus. Cerebral
ischemia causes significant glutamate release and exposure to high
levels of glutamate leads to the overactivation of
N-methyl-D-aspartate receptors (NMDARs), causing Ca2+
overload, which leads to calpain activation (5–7). The
activation of calpain leads to proteolysis of transient receptor
potential canonical (subtype) 6 (TRPC6) channels. TRPC6 channels
play a critical role in promoting neuronal survival against focal
cerebral ischemia (8). TRPC6
activates cAMP-response element binding protein (CREB) through the
Ras/MEK/ERK pathway, and contributes to TRPC6-mediated CREB
activation, resulting in neuronal survival (9). Activation of calpain leads to TRPC6
degradation and contributes to neuronal damage in ischemia
(8). Therefore, inhibition of
TRPC6 degradation to preserve neuronal survival may be a new
therapeutic strategy against ischemic brain damage.
NPD1, a stereospecific derivative of docosahexaenoic
acid (DHA) formed through a lipoxygenase enzyme that acts on free
DHA (10,11), reduces infarct volume at 48 h after
reperfusion (12). However, the
precise mechanism responsible for the neuroprotective activity of
NPD1 has yet to be fully elucidated. Potential mechanisms
explaining how NPD1 may serve an endogenous neuroprotective role
include reducing apoptosis, inhibiting leukocyte infiltration and
pro-inflammatory gene expression, and binding toxic peroxides
(12–16). However, previous studies have not
unequivocally confirmed the effects of intracerebroventricular
(ICV) injection of NPD1 on TRPC6/CREB-mediated neuroprotection.
The present study was performed to investigate
whether ICV injection of NPD1 at 2 h after reperfusion has a
neuroprotective effect, and to verify whether NPD1 improves
neurological status through inhibition of calpain proteolysis of
TRPC6, subsequently inducing CREB activation via the Ras/MEK/ERK
pathway.
Materials and methods
Animals and surgical procedures
Male Sprague-Dawley rats, weighing 200–250 g, were
purchased from Hunan weasleyg scene of experimental animals Co.,
Ltd. Experimental protocols were approved by the committee of
experimental animals of Tongji Medical College and conformed to
internationally accepted ethical standards (Guide for the care and
use of laboratory animals; NIH Publication 80–23, revised 1978).
The animals were anesthetized with 10% chloral hydrate (400 mg/kg,
i.p.). Transient focal cerebral ischemia was produced by
intraluminal occlusion of the right middle cerebral artery (MCA)
for 2 h. Briefly, the right carotid artery was exposed to separate
the external carotid artery and the internal carotid artery. The
external carotid artery was occluded at the level at which the MCA
branches out and a 4-0 monofilament nylon suture (Beijing Sunbio
Biotech Co. Ltd., Beijing, China) with a rounded tip was introduced
through the internal carotid artery until mild resistance was felt.
Two hours later, the filament was gently removed for the
reperfusion (reperfusion confirmed by laser Doppler). Sham surgery
rats were treated similarly, although the filament was not advanced
to the origin of the MCA. The body temperature was maintained at
37.5±0.5°C with a temperature-controlled heating pad attached to a
rectal probe during surgery. Continuous laser-Doppler flowmetry
(Perimed PF5000, Stockholm, Sweden) was used to monitor regional
cerebral blood flow (rCBF) in the cortex supplied by the MCA to
ensure accurate occlusion and reperfusion. Animals that showed a
CBF reduction <70% were excluded from the experimental group, as
well as animals that died after ischemia induction. In a separate
experiment, physiological parameters (cranial temperature, arterial
pH, PaCO2 and PaO2) were monitored and
analyzed (n=6). Arterial blood samples were obtained 5 min prior to
ischemia (baseline), 60 min following ischemia, and 12, 24 and 48 h
following reperfusion for blood gas analysis.
Animal groups and treatments
Rat ICV injection was performed under anesthesia
with a stereotaxic instrument using a microsyringe pump. A scalp
incision was made and a burr hole was opened in the right parietal
skull, 1.8 mm lateral and 1.0 mm posterior to the bregma. A syringe
was inserted into the brain to a depth of 4.2 mm below the cortical
surface. Drugs or vehicle were injected slowly (0.5 μl/min) into
the right ventricle.
All treatments were administered in a blinded
manner. The rats were randomly divided into four groups, and each
group was again divided into three subgroups (n=12 per subgroup)
according to the time of reperfusion (12, 24 and 48 h after
reperfusion). The experimental groups and subgroups were as
follows: i) Sham surgery (Group S; subgroup S12, S24 and S48); ii)
middle cerebral artery occlusion (MCAO; Group I; subgroup I12, I24
and I48); iii) ischemia combined with NPD1 treatment (Group N;
subgroup N12, N24 and N48) and iv) ischemia combined with NPD1 plus
PD98059 (MEK inhibitor) treatment (Group P; subgroup P12, P24 and
P48). Another 27 rats were randomly divided into three groups: i)
Sham surgery (Group S); ii) MCAO (Group I); iii) ischemia combined
with PD98059 treatment (Group M).
NPD1 (100 ng/μl; Cayman Chemical Company, Ann Arbor,
MI, USA) was dissolved in ethanol. PD98059 (1.5 mg/ml; Sigma, St.
Louis, MO, USA) was prepared in 1% DMSO (Sigma). NPD1 (5 μl) or
ethanol (5 μl) was injected slowly (0.5 μl/min) into the right
ventricle at 2 h after reperfusion. PD98059 (0.5 ml, i.p.) or DMSO
(0.5 ml, i.p.) was also administered to rats 20 min prior to the
surgery.
Measurement of infarct volume
The extent of infarction was measured with
2,3,5-triphenyl-tetrazolium chloride (TTC). At 48 h after
reperfusion, rats were deeply anesthetized with 10% chloral
hydrate, and the brains were rapidly removed, washed in
phosphate-buffered saline (PBS) at room temperature and frozen at
-20°C for 10 min. Brain tissue from an area 4 mm anterior and 6 mm
posterior to the bregma was cut into five serial 2 mm coronal
sections. The sliced brain tissues were stained with 2% TTC
(Amresco, Solon, OH, USA) for 30 min at 37°C in the dark followed
by overnight immersion in 4% paraformaldehyde in 0.1 M PBS, pH 7.4,
at 4°C. The infarcted tissue remained unstained (white), whereas
normal tissue was stained red. The extent of ischemic infarction
was traced and the integrated volume was calculated using Image J
software (NIH Image). Infarct volume was calculated by adding the
infarction areas of all sections and multiplying by slice
thickness. To compensate for the effect of brain edema, the
corrected infarct volume was calculated as follows: percentage of
corrected infarct volume = {[total lesion volume − (ipsilateral
hemisphere volume − contralateral hemisphere volume)]/contralateral
hemisphere volume} × 100.
Neurological test
Neurological evaluation of motor sensory functions
was carried out at 48 h after reperfusion. An 18-point scale of
neurologic deficit scores was used for evaluation of neurologic
behavior (17). The scores were
assessed in a blinded fashion. The scale was based on the following
six tests: i) spontaneous activity; ii) symmetry in the movement of
four limbs; iii) forepaw outstretching; iv) climbing; v) body
proprioception; and vi) response to vibrissae touch. The score
assigned to each rat at completion of the evaluation equaled the
sum of all six test scores. The final minimum score was 3 and the
maximum was 18.
Lysis and protein content
determination
All the rats were sacrificed by decapitation at 12,
24 and 48 h after reperfusion. Slices containing maximal ischemic
damage were selected (from an area between 3 and 6 mm posterior to
the frontal pole). The tissues were immediately frozen in liquid
nitrogen and stored at −80°C. Total protein extraction was
performed using a commercially available kit (KGP250; Nanjing
Keygen Biotech Co. Ltd., Nanjing, China). Nuclear protein
extraction was performed using the ProteoJET cytoplasmic and
nuclear protein extraction kit (Fermentas International, Glen
Burnie, MD, USA). Protein concentrations were determined using the
BCA protein assay kit (Beyotime, Jiangsu, China).
Western blot analysis for aII-spectrin,
TRPC6 and p-CREB
Protein samples from total or nuclear fractions were
boiled for 10 min in 1X sample buffer (Beyotime) prior to loading
onto a Tris-HCl gel. Equal amounts of total protein extracts or
nuclear protein extracts were separated by SDS-PAGE and transferred
onto polyvinylidene difluoride membranes by electrophoresis, and
membranes were blocked with 5% non-fat milk in TBST (0.1% Tween-20
in TBS) for 1 h at room temperature. Membranes were then incubated
overnight at 4°C with either a mouse monoclonal anti-aII-spectrin
(1:1000; Enzo Biochem, New York, USA), rabbit polyclonal anti-TRPC6
(1:1000; Abcam, Cambridge, MA, USA), rabbit polyclonal anti-p-CREB
(1:1000; Cell Signaling Technology, Beverly, MA, USA), rabbit
polyclonal anti-Lamin B1 (1:500; Bioworld Technology Inc., St.
Louis, MN, USA) or mouse monoclonal anti-GAPDH antibody (1:100;
Proteintech Group, Inc., Chicago, IL, USA) followed by horse radish
peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (1:3000;
Proteintech Group, Inc.) or anti-rabbit IgG antibody (1:5000;
Proteintech Group, Inc.). Labeled proteins were detected using the
Chemi-Doc Imaging System (Bio-Rad, Hercules, CA, USA). Protein
bands were quantified by Image Lab™ image acquisition and analysis
software (Bio-Rad). Western blots were repeated three times using
samples prepared from three different rats for each experimental
condition studied.
Quantum dot-based immunofluorescence
At 12, 24 and 48 h after reperfusion, rats (n=3 for
each group) were anesthetized with 10% chloral hydrate and infused
through the left ventricle with cold saline as a vascular rinse
followed by a fixing solution containing 4% paraformaldehyde in
PBS. The brains were removed and fixed overnight in 4%
paraformaldehyde in PBS at 4°C. The brains were then blocked and
embedded in paraffin. Paraffin-embedded brains were cut into 4-μm
sections according to standard procedures. Paraffin sections (n=3
for each group) were incubated overnight with antibodies against
TRPC6 (1:100; Abcam) and p-CREB (1:100; Cell Signaling Technology)
at 4°C after being blocked with 2% bovine serum albumin (BSA). The
samples were then incubated with a biotinylated secondary antibody
at 37°C for 30 min. Paraffin sections were then incubated with
streptavidin-conjugated QDs605 (1:100, Wuhan Jiayuan Quantum Dot
Co., Ltd., Wuhan, China) after being blocked with 2% BSA. TRPC6-
and p-CREB-positive cells were measured at ×200 magnification per
visual field in the cortex, three visual fields per section in
three brain sections. Fluorescent signals were detected with a
fluorescence microscope (BX51; Olympus, Tokyo, Japan). The
acquisition and quantitative analysis of images was performed with
a multi-spectral imaging system (Nuance Fx; CRi, Hopkinton, MA,
USA).
Statistical analysis
For all quantitative analysis of data, measurements
were made with the experimenter blinded to the treatment group.
GraphPad Prism (version 5 for Windows; GraphPad Software, La Jolla,
CA, USA) software was used for all statistical analyses. Results
are presented as the means ± SEM. The neurological score data
comparison was analyzed using the Kruskal-Wallis test followed by
the post hoc Dunn’s test. For all other measurements, one-way
analysis of variance (ANOVA) followed by Newman-Keuls multiple
comparison test was used. P<0.05 was considered to indicate a
statistically significant difference.
Results
Physiological parameters
No statistical significance was noted among
different time points for any of the physiological parameters,
including cranial temperature and blood gas (Table I).
 | Table IPhysiological parameters. |
Table I
Physiological parameters.
| Time point | Temperature (°C) | PaO2
(mmHg) | PaCO2
(mmHg) | Arterial pH |
|---|
| Baseline | 37.2±0.2 | 96.6±5.4 | 38.6±5.9 | 7.37±0.08 |
| Ischemia, 60 min | 37.7±0.3 | 92.1±6.4 | 37.3±3.8 | 7.36±0.05 |
| Reperfusion, 12
h | 37.5±0.2 | 98.9±5.1 | 38.1±4.1 | 7.35±0.10 |
| Reperfusion, 24
h | 37.1±0.1 | 94.8±5.7 | 35.6±4.3 | 7.38±0.12 |
| Reperfusion, 48
h | 37.3±0.2 | 91.7±7.2 | 37.8±5.2 | 7.39±0.13 |
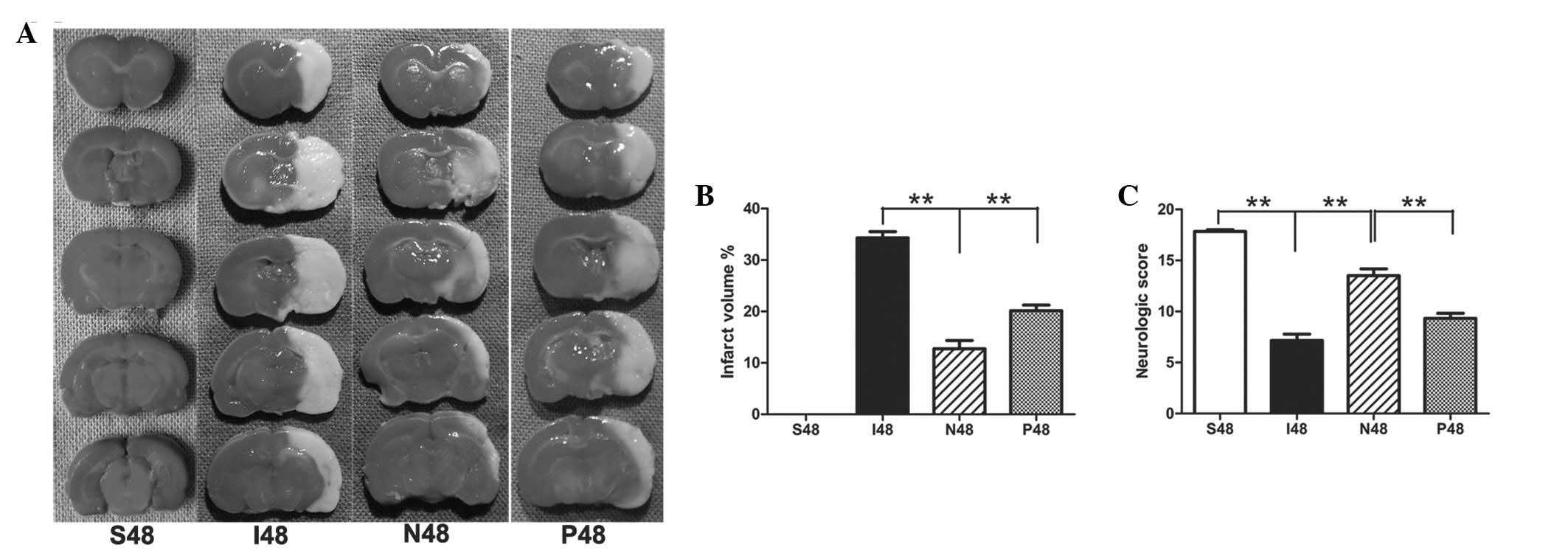
NPD1 significantly reduces infarct
volumes in ipsilateral ischemic hemispheres at 48 h after
reperfusion
After ischemic/reperfusion injury, the white-stained
infarct area was prominent in the MCAO group, and almost the entire
MCA area appeared infarcted. By contrast, NPD1-treated rats had
significantly reduced infarct volumes compared with the MCAO group
(P<0.01). After application of PD98059, the infarct volume was
significantly increased compared with the NPD1-treated group
(Fig. 1A and B; P<0.01).
NPD1 promotes functional recovery at 48 h
after reperfusion
Sham surgery rats did not have any deficits.
Statistical analysis confirmed that NPD1-treated animals had
significantly greater neurological scores than the MCAO group
(P<0.01). After treatment with PD98059, the neurological scores
were significantly decreased compared with the NPD1-treated group
(Fig. 1C; P<0.01).
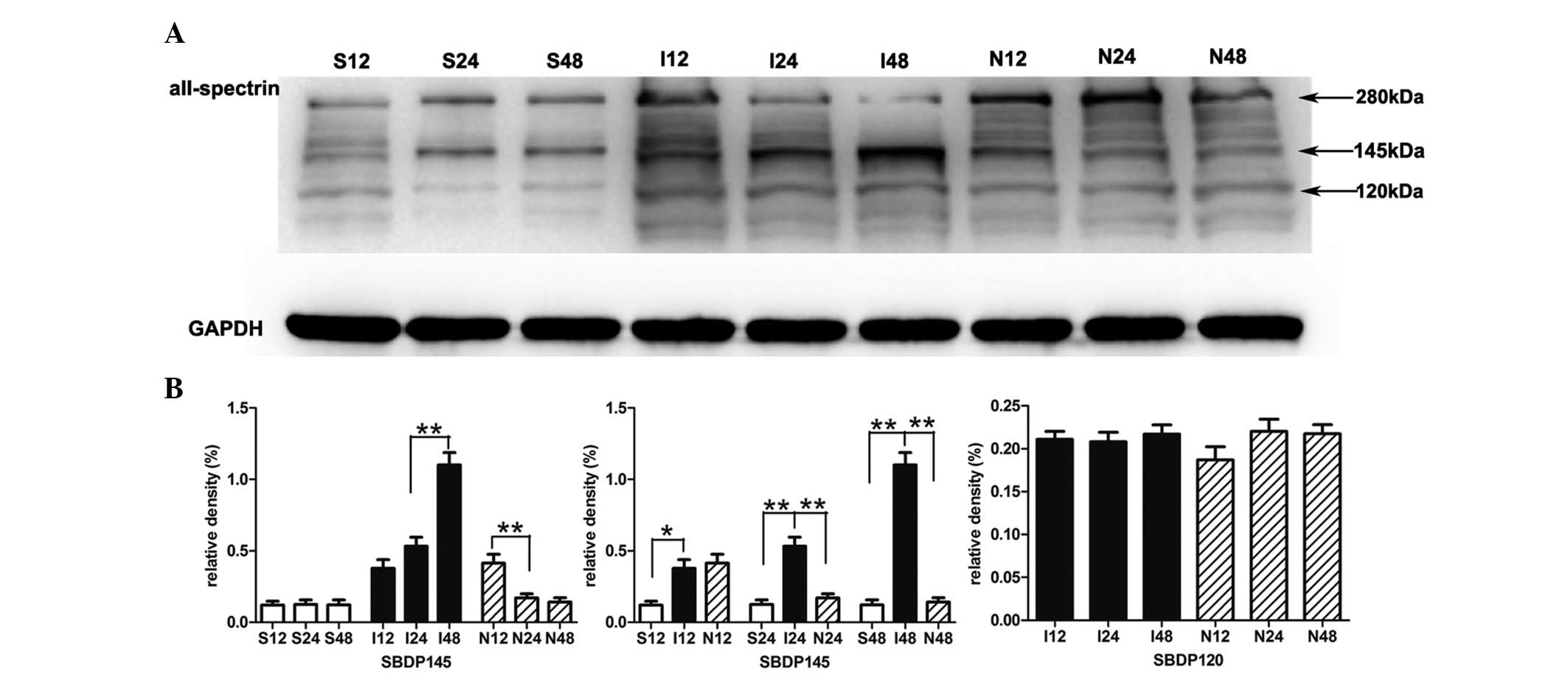
NPD1 inhibits the formation of
calpain-specific aII-spectrin breakdown products (SBDP145)
The sham surgery group presented little SBDP145.
Quantitative analysis confirmed that the protein levels of SBDP145
in the MCAO group were gradually increased during the experimental
time course. Compared with the sham surgery group, the protein
levels of SBDP145 in the MCAO group were significantly increased as
early as 12 h after reperfusion indicating early calpain activity
(P<0.05); this significant increase was also present at 24 h
(P<0.01) and 48 h (P<0.01). When MCAO rats were treated with
NPD1, the protein levels of SBDP145 were significantly decreased at
24 h (P<0.01) and 48 h (P<0.01). In addition, NPD1 attenuated
the calpain-specific fragment of aII-spectrin, but had no effect on
caspase-3 and its cleavage activity on aII-spectrin (Fig. 2).
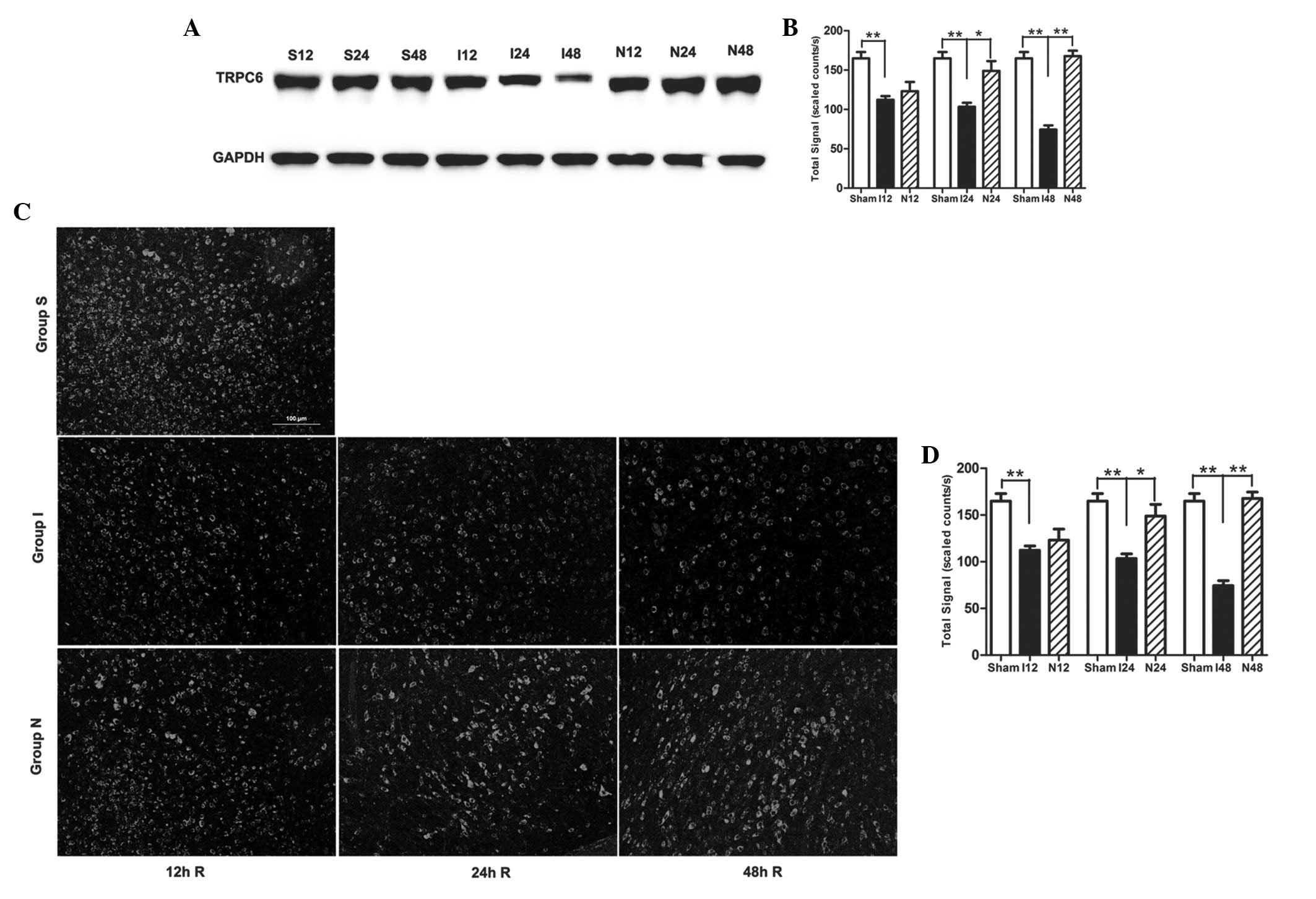
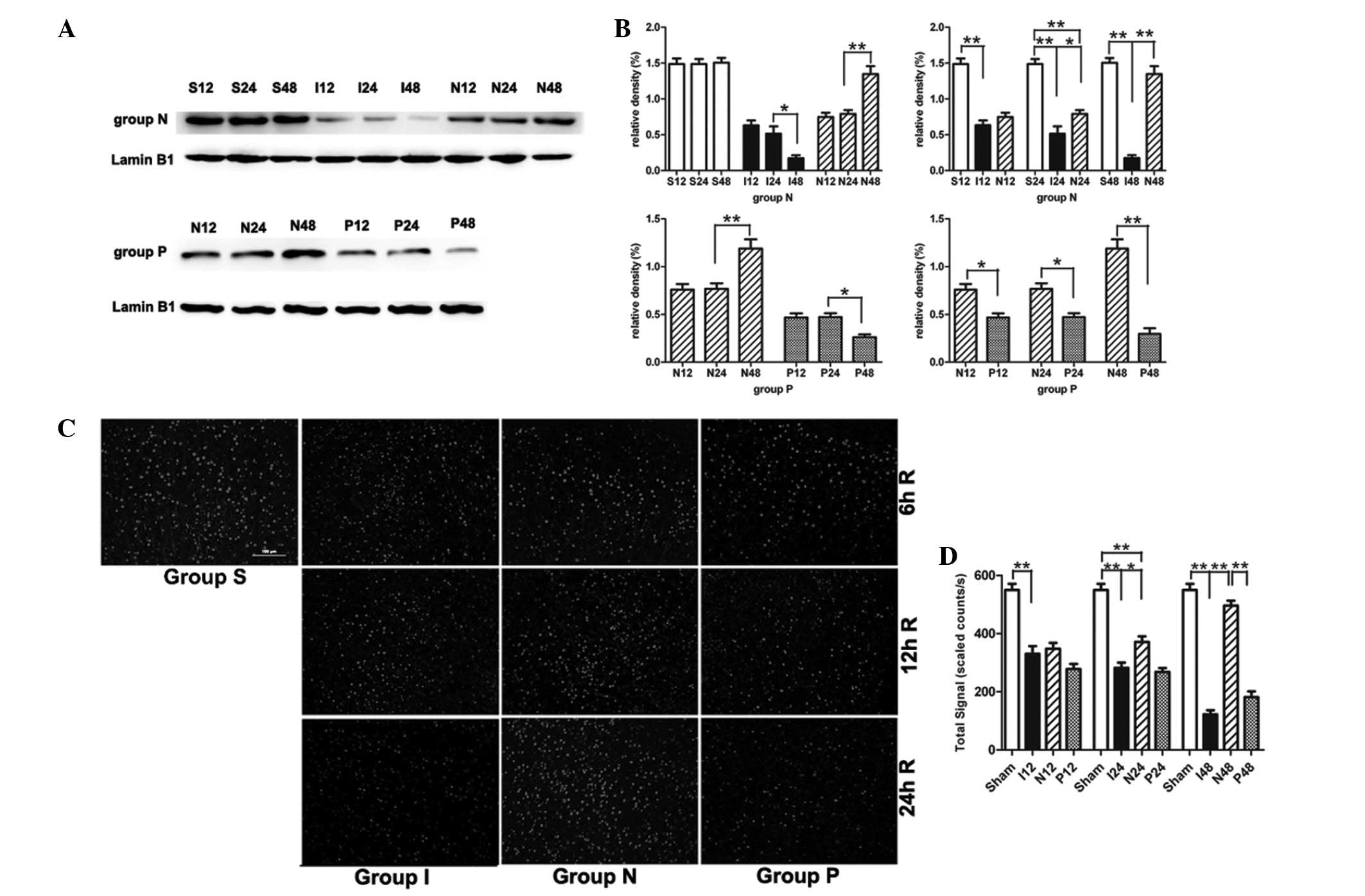
NPD1 inhibits calpain-mediated TRPC6
channel degradation
In the MCAO group, the TRPC6 protein level was also
gradually decreased during the experimental time course (Fig. 3A and B). Compared with the sham
surgery group, the TRPC6 level in the MCAO group was significantly
decreased at 12 h (P<0.01), 24 h (P<0.01) and 48 h
(P<0.01). When MCAO rats were treated with NPD1, the protein
levels of TRPC6 were significantly increased at 24 h (P<0.05)
and 48 h (P<0.01). Immunofluorescence analysis showed the
cytomembrane staining pattern of TRPC6 in neurons of the cerebral
cortex and the immunofluorescence analysis obtained similar results
as the western blot analysis (Fig. 3C
and D).
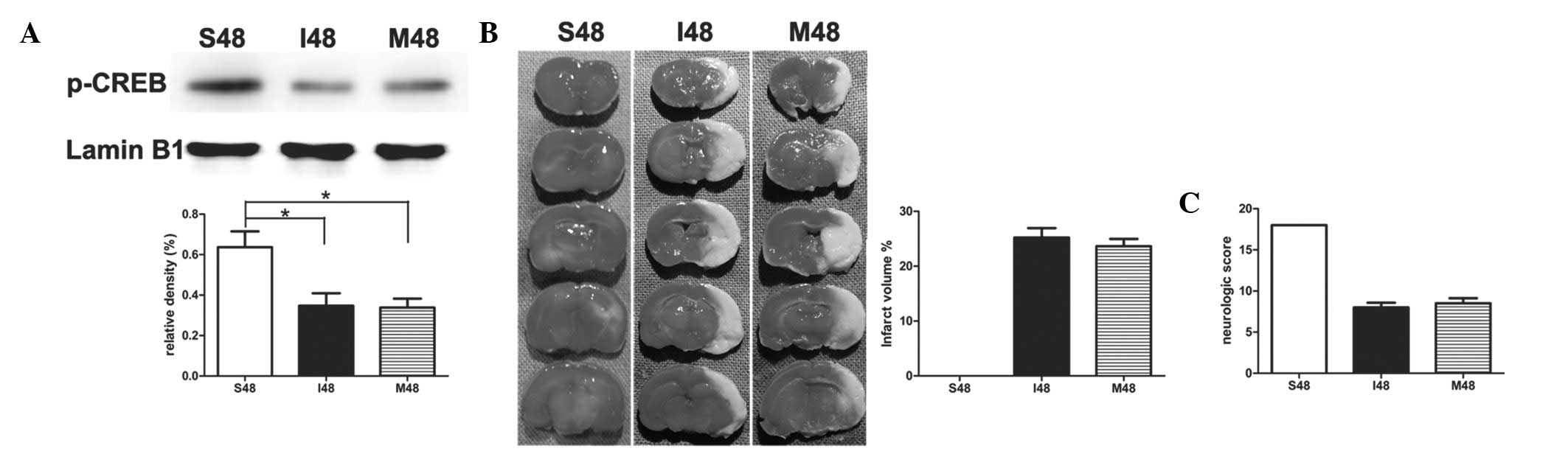
PD98059 exerts no effect on ischemic
stroke in rats at 24 h after reperfusion
To study the effect of PD98059 in stroke-induced
rats, PD98059 was administered 20 min prior to surgery. Notably,
after application of PD98059, no statistical significance was noted
in the protein levels of p-CREB between group I and group M
(Fig. 4A). There was no
significant difference between the two groups during the whole
process of ischemia and reperfusion by measuring the infarct
volumes and neurological scores (Fig.
4B and C).
NPD1 maintains phosphorylation of CREB
through inhibition of TRPC6 degradation
In the MCAO group, p-CREB was gradually decreased
during the experimental time course (Fig. 5A and B). Compared with the sham
surgery group, p-CREB in the MCAO group was significantly decreased
at 12 h (P<0.01), 24 h (P<0.01) and 48 h (P<0.01).
Compared with the MCAO group, p-CREB in the NPD1-treated group was
significantly higher than that at 24 h (P<0.05) and 48 h
(P<0.01). As expected, administration of PD98059 20 min prior to
surgery leads to a significantly decreased p-CREB level compared
with the NPD1-treated group at 12 h (P<0.05), 24 h (P<0.05)
and 48 h (P<0.01). Immunofluorescence staining also showed the
same results, and immunoreactivity appeared as nucleus labeling,
with no labeling within the cytoplasm or cell membrane (Fig. 5C and D).
Discussion
Our results strongly demonstrate that NPD1, when
applied by the ICV route at 2 h after reperfusion, significantly
reduced infarct volumes measured by TTC staining. We observed that
the decreased infarct volumes obtained with NPD1 were mirrored by
enhanced functional recovery. These protective effects are
comparable with the observations of Marcheselli et
al(12), who revealed that
NPD1 administered continuously by ICV perfusion reduced the infarct
volume by 50% at 48 h after reperfusion. In our study, application
of NPD1 with PD98059 20 min prior to surgery led to infarct volumes
that were significantly increased, and the neurological scores were
significantly decreased. These results demonstrated that ICV
injection of NPD1, at very low doses (500 ng, ICV), effectively
reduced cerebral ischemic injury in rat models.
There are several mechanisms, including
excitotoxicity, ionic imbalance, peri-infarct depolarizations,
oxidative stresses and apoptosis (1,2,5) that
have been implicated in ischemic neuronal death. Ca2+
overload remains the most critical mechanism. The NMDA receptor is
an important excitatory neurotransmitter receptor in the brain,
which has been reported as the pivotal player for Ca2+
overload in response to cerebral ischemia. A large number of in
vitro and in vivo studies have suggested that NMDA
receptor antagonists are effective in ischemic neuronal death.
Pharmacological agents that block glutamate release or
glutamate-mediated postsynaptic excitability may reduce neural
degeneration in stroke rats (18,19).
However, clinical trials examining the treatment of stroke using
NMDA antagonists have all failed and have caused severe side
effects (20).
Calpains are intracellular calcium-dependent
cysteine endopeptidases, which are activated by NMDARs-mediated
cytosolic Ca2+ overload (8,21).
Under physiological conditions, calpain activity is likely to be
stimulated by transient localized increases in cytosolic
Ca2+ and tightly regulated by the presence of an
endogenous inhibitor calpastatin. By contrast, the increase in
cytosolic Ca2+ during cerebral ischemia overwhelms
endogenous regulatory systems resulting in pathological calpain
activity (6,22). Calpain inhibitors provide varying
degrees of neuroprotection in animal models (22,23).
aII-Spectrin, the most well-studied target of calpain and caspase,
is an abundant cytoskeletal protein that is specifically cleaved by
calpain into 150/145-kDa, and is also specifically cleaved by
caspase-3 into 150/120-kDa fragments. These characteristics make
aII-spectrin cleavage a useful tool to evaluate the activity of
calpains and caspase-3 (24,25).
In our study, brain samples from sham surgery rats presented very
little SBDP145, whereas MCAO rats had elevated levels of SBDP145 in
the cortical regions of the ipsilateral hemisphere in the first 48
h post-injury. NPD1 treatment significantly reduced SBDP145
formation and made it recover to basal levels at 24 h. However,
NPD1 treatment had no effect on the formation of caspase-3-specific
aII-spectrin breakdown products of 120kDa (SBDP120) in cerebral
ischemia. Therefore, our results strongly demonstrate that NPD1,
when applied at 2 h after reperfusion, specifically inhibited
calpain (not caspase) activation, which induced resistance to
ischemia and reperfusion injuries.
The transient receptor potential (TRP) channel was
first identified in Drosophila melanogaster(26) and is a subfamily of the
nonselective cation channels permeable to Ca2+. TRPC6
channels are present in numerous cell types, including neurons
(27,28). TRPC6 protein in neurons in ischemia
was specifically downregulated by calpain proteolysis (8). Channels formed by the TRP family of
proteins have a variety of biological functions. For example, TRPC3
and TRPC6 are involved in brain-derived neurotrophic factor
(BDNF)-mediated growth cone turning, neuron survival and spine
formation (9,29). TRPC6 also promoted dendritic growth
via the CaMKIV-CREB-dependent pathway (30). A previous study provided evidence
that TRPC6 was specifically degraded in transient ischemia and this
degradation occurred prior to and during neuronal cell death, and
that increases in its protein level or activity prevented neuronal
death. Therefore, the conventional conception about treatment of
ischemic brain damage with NMDA receptor antagonists may have to be
renovated. However, inhibition of calpain proteolysis of TRPC6 may
protect animals from ischemic brain damage (8). TRPC6 channels play a critical role in
promoting neuronal survival against focal cerebral ischemia and
calpain-mediated downregulation of TRPC6 contributes to ischemic
brain injury (8). In our study,
the levels of TRPC6 proteins in the MCAO group were greatly
decreased at 12 h after reperfusion and the reduction in TRPC6
protein levels remained prominent at 24 and 48 h, in support of the
observations of Du et al(8). NPD1 treatment significantly enhanced
the protein levels of TRPC6 at 24 and 48 h. In addition, NPD1
significantly reduced infarct volumes and enhanced functional
recovery at 48 h. Therefore, our results indicated that inhibition
of calpain proteolysis of TRPC6 by NPD1 protects rats from ischemic
brain damage.
In cortical neurons, entry of Ca2+
results in calcium-dependent activation of ERK, which in turn
activates CREB transcriptional pathways to support neuronal
survival (9,30,34).
Phosphorylation of serine-133 in CREB allows it to contact its
co-activator, CREB-binding protein/p300, and is necessary for its
activation. The CREB activation is a critical event in
neuroprotection against ischemic injury (35,36). Overexpressing
TRPC6 markedly increased CREB phosphorylation and CREB-dependent
transcription (9). Blocking TRPC6
degradation maintained phosphorylation of CREB and greatly
prevented ischemic brain damage. In our study, the protein levels
of p-CREB significantly increased in the NPD1-treated group at 24 h
and recovered to the level of the sham surgery group at 48 h. When
MEK activity was specifically inhibited by PD98059, the
neuroprotective effect of NPD1 was attenuated and correlated with
decreased CREB levels. These results clearly demonstrated that the
activation of CREB through the MEK pathway is a pivotal downstream
effector for the neuronal protective effect of TRPC6. Taken
together, these results suggested that NPD1 blocked
calpain-mediated TRPC6 channel degradation and stimulates the
Ras/MEK/ERK pathway that converges on CREB activation, and
contributed to neuroprotection.
Unlike the intravenous and intraperitoneal routes,
the ICV route of administration is a useful experimental method to
study the effects of chemicals or cellular grafts in the
ventricular compartment of the brain following focal ischemia
(37,38). In the present study, it is noteworthy that ICV injection
of NPD1 at 2 h after reperfusion very rapidly attenuated ischemic
cerebral injury within 12 h of reperfusion. This rapid effect may
be a result of the ICV route. The doses of NPD1 (500 ng)
administered into the lateral ventricle were very low, consistent
with one previous study (12),
suggesting that ICV injection of NPD1 is a cost-effective and
highly efficient method in cerebral ischemia.
In conclusion, our results suggest that ICV
injection of NPD1 at very low doses (500 ng) significantly reduces
calpain-mediated TRPC6 channel degradation, and stimulates the
Ras/MEK/ERK pathway that converges on CREB activation and rapidly
attenuates ischemic cerebral injury during the acute period of
ischemic stroke. Therefore, ICV administration of NPD1 following
cerebral ischemia as a neuroprotective treatment may confer clear
advantages and provides theoretical support for the use of NPD1 in
ischemic stroke management during the acute or subacute period.
References
|
1
|
Dirnagl U, Iadecola C and Moskowitz MA:
Pathobiology of ischaemic stroke: an integrated view. Trends
Neurosci. 22:391–397. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lo EH, Dalkara T and Moskowitz MA:
Mechanisms, challenges and opportunities in stroke. Nat Rev
Neurosci. 4:399–415. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lo EH: A new penumbra: transitioning from
injury into repair after stroke. Nat Med. 14:497–500. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Choi DW: Ischemia-induced neuronal
apoptosis. Curr Opin Neurobiol. 6:667–672. 1996. View Article : Google Scholar
|
|
5
|
Lipton P: Ischemic cell death in brain
neurons. Physiol Rev. 79:1431–1568. 1999.PubMed/NCBI
|
|
6
|
Bartus RT, Dean RL, Cavanaugh K, Eveleth
D, Carriero DL and Lynch G: Time-related neuronal changes following
middle cerebral artery occlusion: implications for therapeutic
intervention and the role of calpain. J Cereb Blood Flow Metab.
15:969–979. 1995. View Article : Google Scholar
|
|
7
|
Lipton SA and Rosenberg PA: Excitatory
amino acids as a final common pathway for neurologic disorders. N
Engl J Med. 330:613–622. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Du W, Huang J, Yao H, Zhou K, Duan B and
Wang Y: Inhibition of TRPC6 degradation suppresses ischemic brain
damage in rats. J Clin Invest. 120:3480–3492. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jia Y, Zhou J, Tai Y and Wang Y: TRPC
channels promote cerebellar granule neuron survival. Nat Neurosci.
10:559–567. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Simopoulos AP: Omega-3 fatty acids, the
brain and retina. Preface. World Rev Nutr Diet. 99:VII–XII.
2009.PubMed/NCBI
|
|
11
|
Niemoller TD, Stark DT and Bazan NG:
Omega-3 fatty acid docosahexaenoic acid is the precursor of
neuroprotectin D1 in the nervous system. World Rev Nutr Diet.
99:46–54. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Marcheselli VL, Hong S, Lukiw WJ, et al:
Novel docosanoids inhibit brain ischemia-reperfusion-mediated
leukocyte infiltration and pro-inflammatory gene expression. J Biol
Chem. 278:43807–43817. 2003. View Article : Google Scholar
|
|
13
|
Bazan NG: Cell survival matters:
docosahexaenoic acid signaling, neuroprotection and photoreceptors.
Trends Neurosci. 29:263–271. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Belayev L, Marcheselli VL, Khoutorova L,
et al: Docosahexaenoic acid complexed to albumin elicits high-grade
ischemic neuroprotection. Stroke. 36:118–123. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rodriguez DTE, Belayev L, Liu Y, et al:
Systemic fatty acid responses to transient focal cerebral ischemia:
influence of neuroprotectant therapy with human albumin. J
Neurochem. 83:515–524. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bazan NG: Homeostatic regulation of
photoreceptor cell integrity: significance of the potent mediator
neuroprotectin D1 biosynthesized from docosahexaenoic acid: the
Proctor Lecture. Invest Ophthalmol Vis Sci. 48:4866–4881.
4864–4865. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tsubokawa T, Jadhav V, Solaroglu I,
Shiokawa Y, Konishi Y and Zhang JH: Lecithinized superoxide
dismutase improves outcomes and attenuates focal cerebral ischemic
injury via antiapoptotic mechanisms in rats. Stroke. 38:1057–1062.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shen H, Chen GJ, Harvey BK, Bickford PC
and Wang Y: Inosine reduces ischemic brain injury in rats. Stroke.
36:654–659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shen H, Kuo CC, Chou J, et al: Astaxanthin
reduces ischemic brain injury in adult rats. Faseb J. 23:1958–1968.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hardingham GE, Fukunaga Y and Bading H:
Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB
shut-off and cell death pathways. Nat Neurosci. 5:405–414.
2002.PubMed/NCBI
|
|
21
|
Goll DE, Thompson VF, Li H, Wei W and Cong
J: The calpain system. Physiol Rev. 83:731–801. 2003. View Article : Google Scholar
|
|
22
|
Yao H, Ginsberg MD, Eveleth DD, et al:
Local cerebral glucose utilization and cytoskeletal proteolysis as
indices of evolving focal ischemic injury in core and penumbra. J
Cereb Blood Flow Metab. 15:398–408. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hong SC, Goto Y, Lanzino G, Soleau S,
Kassell NF and Lee KS: Neuroprotection with a calpain inhibitor in
a model of focal cerebral ischemia. Stroke. 25:663–669. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roberts-Lewis JM, Savage MJ, Marcy VR,
Pinsker LR and Siman R: Immunolocalization of calpain I-mediated
spectrin degradation to vulnerable neurons in the ischemic gerbil
brain. J Neurosci. 14:3934–3944. 1994.PubMed/NCBI
|
|
25
|
von Reyn CR, Spaethling JM, Mesfin MN, et
al: Calpain mediates proteolysis of the voltage-gated sodium
channel alpha-subunit. J Neurosci. 29:10350–10356. 2009.
|
|
26
|
Montell C, Jones K, Hafen E and Rubin G:
Rescue of the Drosophila phototransduction mutation trp by germline
transformation. Science. 230:1040–1043. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Harteneck C, Plant TD and Schultz G: From
worm to man: three subfamilies of TRP channels. Trends Neurosci.
23:159–166. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Montell C, Birnbaumer L and Flockerzi V:
The TRP channels, a remarkably functional family. Cell.
108:595–598. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Y, Jia YC, Cui K, et al: Essential role
of TRPC channels in the guidance of nerve growth cones by
brain-derived neurotrophic factor. Nature. 434:894–898. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tai Y, Feng S, Ge R, et al: TRPC6 channels
promote dendritic growth via the CaMKIV-CREB pathway. J Cell Sci.
121:2301–2307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sossin WS and Barker PA: Something old,
something new: BDNF-induced neuron survival requires TRPC channel
function. Nat Neurosci. 10:537–538. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Walton MR and Dragunow I: Is CREB a key to
neuronal survival? Trends Neurosci. 23:48–53. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Finkbeiner S: CREB couples neurotrophin
signals to survival messages. Neuron. 25:11–14. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen J, Li Y and Chopp M: Intracerebral
transplantation of bone marrow with BDNF after MCAo in rat.
Neuropharmacology. 39:711–716. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Modo M, Stroemer RP, Tang E, Patel S and
Hodges H: Effects of implantation site of dead stem cells in rats
with stroke damage. Neuroreport. 14:39–42. 2003. View Article : Google Scholar : PubMed/NCBI
|