Introduction
Alzheimer’s disease (AD) is one of the most common
diseases among the elderly. AD is the primary cause of dementia in
old age (1). Over the last two
decades, several hypotheses have been proposed to explain AD
pathogenesis. One such hypothesis is the amyloid hypothesis, which
states that β-amyloid (Aβ) peptide deposits are the fundamental
cause of the disease (2).
The accumulation of Aβ in cerebral senile plaques is
a major pathological hallmark of AD. Therefore, Aβ peptides are
central to the pathogenesis of AD. Despite the genetic and cell
biological evidence that supports the amyloid hypothesis, it is
becoming increasingly clear that AD etiology is complex, and that
Aβ alone is unable to account for all aspects of AD. In 2000,
evidence strongly suggested that advanced glycation end products
(AGEs) have an important toxic role in AD pathogenesis (3). In vitro experiments
demonstrated that AGEs and Aβ are co-localized in the core of
senile plaques, and that they are able to attract and cause the
aggregation of soluble Aβ.
Aβ is a pleiotropic peptide and is capable of
binding to receptors at several different membrane locations
(4). The receptor for AGEs (RAGE),
a multi-ligand receptor of the immunoglobulin superfamily of cell
surface molecules (5), possesses a
cell surface binding site for Aβ peptides (4) and is expressed at higher levels when
stimulated by excessive levels of Aβ (6). RAGE has been extensively studied for
its roles in the migration and differentiation of neuronal cells
during development, the perturbation of neuronal cells by Aβ and
the inflammatory response (3,7).
Induced expression of RAGE is frequently correlated
with pathological stages such as diabetic endothelial damage and AD
(8,9). It has been proposed that RAGE is
responsible for Aβ neurotoxicity (10). However, the mechanism whereby RAGE
is able to recognize AGEs and Aβ, if these molecules are not
glycated, remains unclear. Studies have suggested that AGEs are
inducers of chronic inflammation and acute-phase responses in a
variety of diseases (4,11). However, this hypothesis is not
clear in the brains of patients with AD.
In the present study, we aimed to observe the
effects of AGEs on PC12 cells pretreated with Aβ25–35, to explore
whether the mechanism of action is associated with oxidative
stress, and to study whether inhibiting the activity of RAGE
attenuates the toxic effect of AGEs.
Materials and methods
Production of AGEs and fibrillar Aβ
AGEs were produced by incubation of 1 mM bovine
serum albumin (BSA; Roce, Indianapolis, IN, USA) with 1 M glucose
at 50°C in phosphate-buffered saline (PBS) at pH 7.4 for 60 days.
Samples were initially filtered through a 0.2-μm filter and kept
sterile during the incubations. A slightly elevated temperature was
used to accelerate the reaction and avoid bacterial contamination.
Unbound sugars were removed through extensive dialysis using
distilled water. AGEs were lyophilized and resuspended in PBS. The
controlled BSA was incubated under the same conditions, except that
glucose was omitted. AGE purity was assessed with fluorometry and
chromatographic analysis exploiting the selective fluorescence of
AGEs at an optical density of 400 nm (the excitation wavelength was
370 nm) (12). Fibrillar Aβ25–35
(13) was produced by incubating
the peptide (1 mM) in 10 mM PBS under sterile conditions for 7 days
at 37°C.
Cell culture
PC-12 cells (ATCC, Manassas, VA, USA) were seeded
into 96-well flat bottom tissue culture plates (Corning, USA) at a
density of 3×105 cells/ml for the MTT assay, and into
24-well plates at a density of 6×105 cells/ml for the
remaining assays. Cells were grown in Dulbecco’s Modified Eagle’s
Medium (DMEM; Life Technologies, Carlsbad, CA, USA) supplemented
with 10% fetal calf serum, streptomycin and penicillin (100 mg/ml
and 100 U/ml), and 2 mM L-glutamine at 37°C in a humidified
atmosphere containing 5% CO2 and 95% air.
Grouping
Aβ-PC12 cells were PC-12 cells pretreated with
Aβ25–35. Aβ-PC12 cells were treated with different levels of AGEs.
PC-12 cells were divided into five groups: i) the control group:
PC12 cells were cultured with 12.5 μl of 30% BSA; ii) the Aβ group:
PC12 cells were cultured with 25 μmol/l Aβ25–35, and termed Aβ-PC12
cells; iii) the Aβ+L-AGE group: Aβ-PC12 cells were cultured with a
low volume of AGEs (12.5 μl); iv) the Aβ+M-AGE group: Aβ-PC12 cells
were cultured with a medium volume of AGEs (25 μl); and v) the
Aβ+H-AGE group: Aβ-PC12 cells were cultured with a high volume of
AGEs (50 μl).
RAGE was inhibited by trypsin (14) to determine whether RAGE was
involved in AGE and Aβ toxicity. In the trypsin group, PC12 cells
were pretreated with 1 mg/ml trypsin at 37°C for 30 min, and washed
three times with PBS. Subsequently, 25 μmol/l Aβ25–35 was added and
cells were incubated at 37°C for 24 h, before the addition of 50 μl
AGE. In the non-trypsin group, PC12 cells were not pretreated with
trypsin (Gibco, USA); however, 25 μmol/l Aβ25–35 was directly added
and the cells were incubated at 37°C for 24 h, which was followed
by the addition of 50 μl AGE.
MTT assay
Following incubation of the cells with AGEs and Aβ,
the medium was removed and the cells were washed with PBS.
Subsequently, 100 ml DMEM, without phenol red, and 25 ml of an MTT
solution (1.5 mg/ml in PBS) were added to each well, followed by
incubation for 4 h. The MTT solution was carefully removed from the
wells to avoid the loss of formazan crystals before they were
dissolved with 100 ml dimethyl sulfoxide/ethanol (1:1). Absorbance
was measured at 550 nm with the reference filter set to 630 nm. MTT
assays were performed in triplicate for each experiment.
Reverse transcription-polymerase chain
reaction (RT-PCR)
PC-12 cells were grown under the same conditions as
described previously. Following removal of the medium, cells were
washed with PBS and lysed with 1 ml TRIzol (Takara, Japan) for 5
min. Following centrifugation at 12,000 × g at 4°C for 10 min, the
supernatant was mixed with 200 μl chloroform and shaken for 30 sec.
Subsequently, 400 μl isopropanol was added to the aqueous phase,
and the mixture was allowed to stand for 10 min. Following
centrifugation at 12,000 × g at 4°C for 10 min, the obtained pellet
was rinsed with 75% ethanol, dried and then dissolved in diethyl
pyrocarbonate-treated water. Further RNA purification was performed
using the Qiagen RNeasy kit, according to the manufacturer’s
instructions (Qiagen, Germany). The Stratagene RT-PCR kit was used
for reverse transcription of total RNA (Table I).
 | Table IPrimers used in this study. |
Table I
Primers used in this study.
| Type | Primer sets | Length (bp) |
|---|
| RAGE |
5′-AGACCAAGTCCAACTACCGAG-3′ | 408 |
|
5′-CCTTCACAGATACTCCCTTCAT-3′ | |
| NF-κB |
5′-AGCACAGATACCACCAAGACCC-3′ | 198 |
|
5′-CCCACGCTGCTCTTCTATAGGAAC-3′ | |
| β-actin |
5′-CAATTCCATCATGAAGTGTGAC-3′ | 260 |
|
5′-CCACACAGAGTACTTGCGCTC-3′ | |
The PCR procedure was implemented as follows: RAGE:
1 cycle at 95°C for 4 min; 35 cycles at 95°C for 50 sec, 58°C for
50 sec and 72°C for 1 min; and 1 cycle at 72°C for 7 min; and
nuclear factor-κB (NF-κB): 1 cycle at 95°C for 1 min; 35 cycles at
95°C for 30 sec, 60°C for 30 sec and 68°C for 2 min; and 1 cycle at
65°C for 10 min.
PCR products were loaded and run on a 1.8% agarose
gel and visualized following ethidium bromide staining using a UV
transilluminator.
Apoptosis rate and flow cytometry (FCM)
analysis
PC12 cells were harvested by centrifugation at
12,000 × g for 5 min and then washed twice with cold PBS, before
being resuspended in 100 μl binding buffer with ~106
cells. Subsequently, 5 μl fluorescein isothiocyanate (FITC)-annexin
V and 5 μl propidium iodide (PI) were added. Cells were gently
oscillated and incubated in the dark for 15 min at 25°C. Following
the addition of 400 μl binding buffer to each tube, the cells were
analyzed by FCM within 1 h. Cells that stained positive for
FITC-annexin V and negative for PI were considered to be in
apoptosis. Cells that stained positive for FITC-annexin V and PI
were considered to either be in necrosis or dead. Cells that
stained negative for FITC-annexin V and PI were considered to be
living. The FACSCalibur flow cytometer (BD Biosciences, Franklin
Lakes, NJ, USA) was used to conduct the FCM. Data were analyzed
using CellQuest software and the apoptosis rates were provided.
Statistical analysis
Statistical analysis of the results was carried out
by analysis of variance (ANOVA) using the Statistical Package for
the Social Sciences (SPSS; SPSS, Inc., Chicago, IL, USA). Either
the Student’s t-test or the Wilcoxon rank sum test were used,
depending on the normality of the data distribution. P<0.05 was
considered to indicate a statistically significant result.
Results
Cell viability
The influence of Aβ and the different concentrations
of AGEs on the viability of PC12 cells is presented in Table II. Optical density (OD) values
were determined by MTT assay. The cell viability was calculated
using the following formula: Cell viability = (ODsample
value/ODcontrol value × 100%. Statistical analysis
indicated that cell viability in the Aβ and Aβ+AGEs groups was
lower than that of the control group (P<0.01). The cell
viability decreased in the three groups of the different volumes of
AGEs compared with the Aβ group (P<0.01). The effects of AGEs on
Aβ-PC12 cell viability were dose dependent. As demonstrated in
Table III, the cell viability in
the trypsin group was greater than that of the non-trypsin group
(P<0.01).
| Table IIChanges in cell viability following
incubation with Aβ and AGEs. |
Table II
Changes in cell viability following
incubation with Aβ and AGEs.
| Group | OD | Viability (%) |
|---|
| Control | 0.8109±0.1826 | 100.00 |
| Aβ | 0.5847±0.1044a | 72.11 |
| Aβ+L-AGE | 0.3876±0.0781a,b | 47.80 |
| Aβ+M-AGE | 0.2758±0.0593a,b | 34.01 |
| Aβ+H-AGE | 0.0969±0.0800a,b | 11.95 |
| Table IIIChanges in cell viability following
incubation with trypsin and AGEs. |
Table III
Changes in cell viability following
incubation with trypsin and AGEs.
| Group | OD | Viability (%) |
|---|
| No trypsin | 0.1103±0.0451 | 13.01 |
| Trypsin | 0.4414±0.0357a | 52.05 |
RT-PCR
The expression of RAGE mRNA in the PC12 cells was at
408 bp in all groups. The levels of RAGE mRNA in the Aβ+L-AGE,
Aβ+M-AGE and Aβ+H-AGE groups were higher than in the Aβ group. The
expression value of RAGE mRNA increased with increasing AGE
concentration, as demonstrated in Table IV. The expression of NF-κB mRNA in
the PC12 cells was at 198 bp in all groups. The levels of NF-κB
mRNA in the Aβ+L-AGE, Aβ+M-AGE and Aβ+H-AGE groups were higher than
in the Aβ group. The expression value of NF-κB mRNA increased with
increasing AGE concentration, as presented in Table IV.
| Table IVExpression of RAGE mRNA and NF-κB mRNA
in PC-12 cells. |
Table IV
Expression of RAGE mRNA and NF-κB mRNA
in PC-12 cells.
| Group | RAGE | β-Actin | RAGE/β-actin | NF-κB | β-Actin | NF-κB/β-actin |
|---|
| Aβ | 113.27±4.16 | 290.83±8.47 | 38.94±0.60 | 121.20±8.21 | 278.80±15.33 | 43.47±5.02 |
| Aβ+L-AGE | 178.63±10.05 | 318.93±5.65 | 56.01±4.15a | 176.90±2.69 | 311.07±6.89 | 56.87±1.08a |
| Aβ+M-AGE | 308.80±7.89 | 362.60±12.11 | 85.16±2.11a,b | 265.83±13.01 | 354.20±4.83 | 75.05±3.31a |
| Aβ+H-AGE | 347.67±5.67 | 373.77±20.69 | 93.02±5.09a,b | 316.87±10.05 | 371.17±10.18 | 85.37±6.63a,b |
Apoptosis rate
Apoptosis was induced by Aβ and the different
concentrations of AGEs, and was quantified as the percentage of
apoptotic cells. The rate of apoptosis was calculated using the
following formula: Apoptosis rate = (number of apoptotic
cells/total number of cells) × 100%. Apoptosis rates in all groups
of Aβ-PC12 cells (16.06, 24.57, 36.89 and 43.85% in the Aβ,
Aβ+L-AGE, Aβ+M-AGE and Aβ+H-AGE groups, respectively) were higher
than in PC12 cells (2.01% in the control group; P<0.01). The
cell apoptosis rate significantly increased in the Aβ+M-AGE and
Aβ+H-AGE groups compared with the Aβ group (P<0.01). In the
Aβ+AGE groups, the cell apoptosis rate increased with increasing
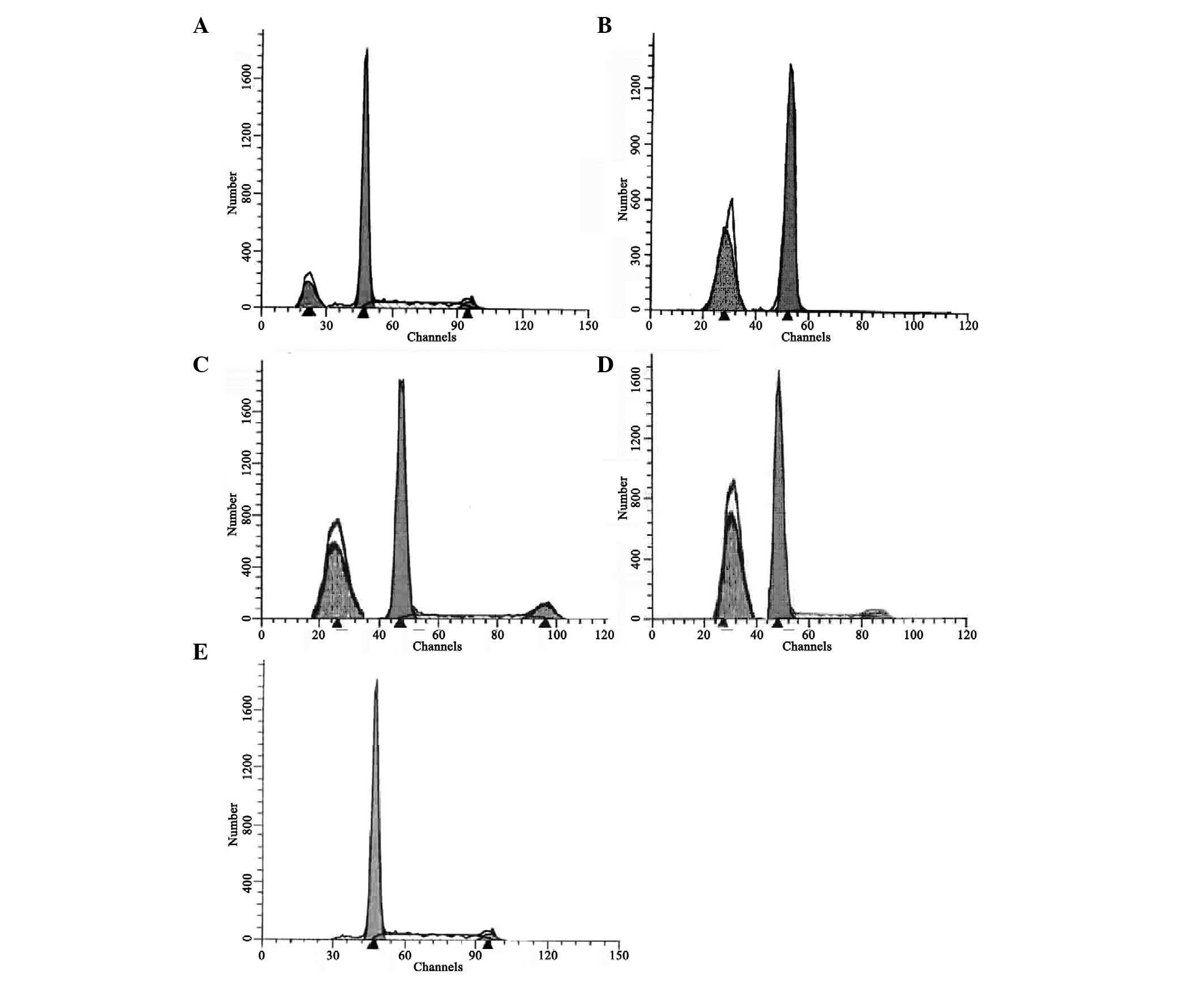
AGE concentration, as demonstrated in Fig. 1.
Discussion
AD is a neurodegenerative disorder characterized by
progressive degeneration and loss of neurons in the brain, which
has been correlated with the appearance of senile plaques, the
neuropathological hallmarks of AD. As the major component of senile
plaques, Aβ is considered to play a key role in the development and
progression of AD (15).
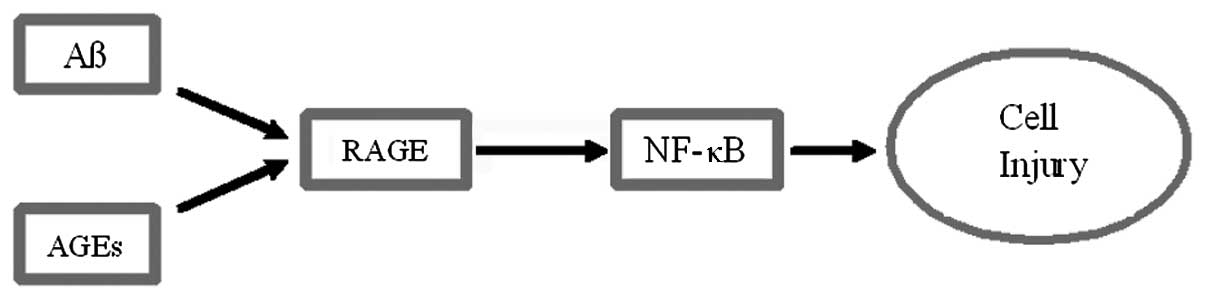
A potential mechanism for the effect of Aβ is
presented in Fig. 2. This
mechanism accounts for the fact that the increasing concentrations
of AGEs with age predispose to the injurious signal presented by
Aβ.
Aβ-PC12 cells, PC12 cells treated with Aβ25–35, have
been recognized as having the ability to mimic classical AD
pathology, such as inhibited cell multiplication, induced cell
metamorphosis, cell damage, functional loss and even cell death
(12). In vitro studies
have demonstrated that Aβ has nutritional value for cultured
hippocampal neurons, as well as being toxic to these cells. The
role that Aβ assumes depends on two factors, which include the
maturity of neurons and the concentration of Aβ (14,16).
A high concentration of Aβ triggers the toxic property, causing a
loss of mature neurons and inhibition of axon growth. The working
domain of Aβ has been confirmed to be the amino acid residues in
25–35 sites. In the present study, Aβ25–35 was added to PC12 cells
as a neurotoxin. This addition inhibited PC12 cell multiplication
and induced cell death, which was consistent with previous studies
(12,17).
AGEs are important agents in the proposed mechanism
of Aβ-mediated cell injury in AD. AGEs are a series of irreversible
polymers produced by the Maillard reaction, a non-enzymatic
glycation and oxidation reaction between carbohydrate-derived
carbonyl compounds and the free N-terminal of proteins, forming
brown fluorescent reaction end products (18). As these products auto-fluoresce, we
can use the fluorescence chromatogram to identify them at an
excitation wavelength of 350–399 nm and an emission wavelength of
440–470 nm.
RAGE is a 404-amino acid protein, which belongs to a
family of cell surface molecules with immunoglobulin folds. RAGE is
expressed in endothelial cells, mononuclear phagocytes (monocytes,
macrophages and mesangial cells), neurons and muscle cells. The
protein is expressed at a high level in the nervous system during
development. Induced expression is frequently associated with
pathological stages, such as diabetic endothelial damage and AD
(8,9,19).
Proteins modified by AGEs may lose their normal
functions. Studies have proposed that AGEs transmit their cell
toxicity signals through RAGE. When AGEs are combined with RAGE,
located at the cytomembrane of macrophages, these proteins may be
degraded and cleared. Thus, the modification of proteins by AGEs is
considered to be a signal participating in the procedure of
rebuilding and clearing aging tissues. The production of AGEs is
enhanced due to the glycometabolic disorder in patients with AD,
while the clearance of AGEs is inhibited.
AGE formation is normally slow. In humans, AGEs
normally accumulate with increasing age. In AD, AGEs accumulate on
β-amyloid plaques near microglia and astrocytes. Aβ, derived by
proteolytic cleavage of the amyloid precursor protein, is the major
protein component of senile plaques. In vitro experiments
have demonstrated that AGEs and Aβ, co-localized in the core of
senile plaques, attract additional Aβ to form aggregates (20). Several studies have proposed that
once AGEs have accumulated on β-amyloid plaques in the brains of
patients with AD, they may aggravate Aβ-mediated oxidative stress,
cell damage, functional loss and even neuronal cell death in the AD
brain via RAGE-dependent mechanisms.
Several different theories have been proposed for
the initial interaction between Aβ and the cell, as Aβ has a direct
toxic effect on neuronal cells. RAGE has been demonstrated to be
responsible for Aβ neurotoxicity (10). However, the mechanism whereby RAGE
is able recognize AGEs, if these synthetic peptides are not
glycated, remains unclear. In the present study, different
concentrations of AGEs were added to cultures of Aβ-PC12 cells to
determine whether RAGE was involved in Aβ toxicity. High
concentrations of AGEs accelerated the Aβ toxicity in PC12 cells.
The toxicity of AGEs and Aβ was decreased when the RAGE of the
cultured cell was inactivated by treatment with trypsin. Glycated
albumin bound to RAGE on the cell activated the Aβ toxic response
and increased the toxicity induced by Aβ in neural cells.
Inactivating RAGE may block neurocytotoxicity induced by Aβ. RAGE
mRNA was detected by PCR analysis when AGEs were added to cultures
of Aβ-PC12 cells. These results strongly suggested that RAGE was
involved in mediating the toxic effect of AGEs and Aβ on nerve
cells.
AGEs have been suggested to be inducers of chronic
inflammation and acute-phase responses in a variety of diseases
(4,11). However, this is not clear in brains
of patients with AD. Recent studies have determined that microglia
activated by AGEs are co-localized with AGE-modified β-amyloid
plaques (21,22). The present study and previous
studies have demonstrated that AGEs activate NF-κB and upregulate
NF-κB mRNA expression in Aβ-PC12 cells, and that NF-κB mRNA
expression increased with an elevation in AGE concentration.
Binding of Aβ to RAGE on neurons may cause cellular
perturbation due to the induction of oxidative stress and the
activation of the transcription factor, NF-κB. One of the
consequences of this interaction is the production of
microglia/macrophage growth factor and macrophage
colony-stimulating factor (M-CSF) (23). The promoter of the RAGE gene
contains two functional NF-κB binding sites, which provide a
mechanism whereby RAGE activation by Aβ results in increased
expression of the RAGE gene. Furthermore, RAGE-dependent NF-κB
activation by Aβ has other pro-inflammatory effects. For example,
RAGE-Aβ interactions lead to increased TNF-α secretion, and
increased M-CSF and vascular cell adhesion molecule expression by
neuroblastoma cells. Other RAGE ligands have also been determined
to induce NF-κB activation and contribute to inflammatory responses
(24).
In the present study, we inferred that an internal
cycle between Aβ, AGEs, oxidative stress and NF-κB exists; and that
this cycle may form a morbigenous network in AD pathogenesis. In
our findings, Aβ cooperated with AGEs, resulting in
concentration-dependent expression of NF-κB. However, the effects
of AGEs and Aβ were no longer evident if RAGE was blocked by
trypsin. This indicated that one of the pathways of cytotoxicity
caused by Aβ was dependent on RAGE on the surface of neurons and
glial cells for stimulating the release of reactive oxygen species
(ROS), as well as the expression of NF-κB and the series of
subsequent pathological processes.
Aβ may be involved in the etiology of AD through
oxidative stress. Aβ generates free radicals in a metal-catalyzed
reaction, which is able to induce neuronal cell death by a
ROS-mediated process, and is able to damage neuronal membrane
lipids, proteins and nucleic acids. Several studies have
demonstrated that necrotic and apoptotic mechanisms are implicated
in Aβ-mediated neurotoxicity (25). Apoptosis is induced by micromolar
concentrations of Aβ in cultured neurons (26,27).
Our experiment demonstrated that the rate of PC12 cell apoptosis
was higher in the Aβ+AGEs groups than in the Aβ group, and that the
apoptosis rate increased with increasing AGE concentration.
Additionally, RAGE may be involved in the pathogenesis of AD cells
as one of the surface receptors mediating apoptosis. Further
research is required to clarify this mechanism.
References
|
1
|
Jellinger KA and Attems J: Prevalence of
dementia disorders in the oldest-old: an autopsy study. Acta
Neuropathol. 119:421–433. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tam JH and Pasternak SH: Amyloid and
Alzheimer’s disease: inside and out. Can J Neurol Sci. 39:286–298.
2012.
|
|
3
|
Schmidt AM, Yan SD, Yan SF and Stern DM:
The biology of the receptor for advanced glycation end products and
its ligands. Biochim Biophys Acta. 1498:99–111. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yan SD, Chen X, Fu J, et al: RAGE and
amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature.
382:685–691. 1996.
|
|
5
|
Bartus RT and Dean RL 3rd: Pharmaceutical
treatment for cognitive deficits in Alzheimer’s disease and other
neurodegenerative conditions: exploring new territory using
traditional tools and established maps. Psychopharmacology (Berl).
202:15–36. 2009.
|
|
6
|
Lue LF, Walker DG, Brachova L, et al:
Involvement of microglial receptor for advanced glycation
endproducts (RAGE) in Alzheimer’s disease: identification of a
cellular activation mechanism. Exp Neurol. 171:29–45.
2001.PubMed/NCBI
|
|
7
|
Yan SD, Bierhaus A, Nawroth PP and Stern
DM: RAGE and Alzheimer’s disease: a progression factor for
amyloid-beta-induced cellular perturbation? J Alzheimers Dis.
16:833–843. 2009.
|
|
8
|
Yamagishi S: Role of advanced glycation
end products (AGEs) and receptor for AGEs (RAGE) in vascular damage
in diabetes. Exp Gerontol. 46:217–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Srikanth V, Maczurek A, Phan T, et al:
Advanced glycation endproducts and their receptor RAGE in
Alzheimer’s disease. Neurobiol Aging. 32:763–777. 2011.
|
|
10
|
Kojro E and Postina R: Regulated
proteolysis of RAGE and AbetaPP as possible link between type 2
diabetes mellitus and Alzheimer’s disease. J Alzheimers Dis.
16:865–878. 2009.PubMed/NCBI
|
|
11
|
Schwedler S, Schinzel R, Vaith P and
Wanner C: Inflammation and advanced glycation end products in
uremia: simple coexistence, potentiation or causal relationship?
Kidney Int Suppl. 78:S32–S36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pike CJ, Burdick D, Walencewicz AJ, Glabe
CG and Cotman CW: Neurodegeneration induced by beta-amyloid
peptides in vitro: the role of peptide assembly state. J Neurosci.
13:1676–1687. 1993.PubMed/NCBI
|
|
13
|
Makita Z, Vlassara H, Cerami A and Bucala
R: Immunochemical detection of advanced glycosylation end products
in vivo. J Biol Chem. 267:5133–5138. 1992.PubMed/NCBI
|
|
14
|
Weldon DT, Maggio JE and Mantyh PW: New
insights into the neuropathology and cell biology of Alzheimer’s
disease. Geriatrics. 52(Suppl 2): S13–S16. 1997.
|
|
15
|
Maczurek A, Shanmugam K and Münch G:
Inflammation and the redox-sensitive AGE-RAGE pathway as a
therapeutic target in Alzheimer’s disease. Ann NY Acad Sci.
1126:147–151. 2008.PubMed/NCBI
|
|
16
|
Perrone L, Sbai O, Nawroth PP and Bierhaus
A: The complexity of sporadic Alzheimer’s disease pathogenesis: the
role of RAGE as therapeutic target to promote neuroprotection by
inhibiting neurovascular dysfunction. Int J Alzheimers Dis.
2012:7349562012.
|
|
17
|
Mucke L and Selkoe DJ: Neurotoxicity of
Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring
Harb Perspect Med. 2:a0063382012.
|
|
18
|
Visentin S, Medana C, Barge A, Giancotti V
and Cravotto G: Microwave-assisted Maillard reactions for the
preparation of advanced glycation end products (AGEs). Org Biomol
Chem. 8:2473–2477. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li JJ, Dickson D, Hof PR and Vlassara H:
Receptors for advanced glycosylation endproducts in human brain:
role in brain homeostasis. Mol Med. 4:46–60. 1998.PubMed/NCBI
|
|
20
|
Cuevas E, Lantz SM, Tobón-Velasco JC, et
al: On the in vivo early toxic properties of A-beta 25–35 peptide
in the rat hippocampus: involvement of the Receptor-for-Advanced
Glycation-End-Products and changes in gene expression. Neurotoxicol
Teratol. 33:288–296. 2011.PubMed/NCBI
|
|
21
|
Wilkinson K and El Khoury J: Microglial
scavenger receptors and their roles in the pathogenesis of
Alzheimer’s disease. Int J Alzheimers Dis. 2012:4894562012.
|
|
22
|
Lue LF, Kuo YM, Beach T and Walker DG:
Microglia activation and anti-inflammatory regulation in
Alzheimer’s disease. Mol Neurobiol. 41:115–128. 2010.
|
|
23
|
Llano DA, Li J, Waring JF, et al:
Cerebrospinal fluid cytokine dynamics differ between Alzheimer
disease patients and elderly controls. Alzheimer Dis Assoc Disord.
26:322–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhu C, Xiong Z, Chen X, et al: Artemisinin
attenuates lipopolysaccharide-stimulated proinflammatory responses
by inhibiting NF-κB pathway in microglia cells. PLoS One.
7:e351252012.PubMed/NCBI
|
|
25
|
Fang F, Lue LF, Yan S, et al:
RAGE-dependent signaling in microglia contributes to
neuroinflammation, Abeta accumulation, and impaired learning/memory
in a mouse model of Alzheimer’s disease. FASEB J. 24:1043–1055.
2010.PubMed/NCBI
|
|
26
|
Neniskyte U, Neher JJ and Brown GC:
Neuronal death induced by nanomolar amyloid β is mediated by
primary phagocytosis of neurons by microglia. J Biol Chem.
286:39904–39913. 2011.
|
|
27
|
Cai Z, Zhao B and Ratka A: Oxidative
stress and β-amyloid protein in Alzheimer’s disease. Neuromolecular
Med. 13:223–250. 2011.
|